
Remotely controlled nanofluidic implantable platform for tunable drug delivery†
Nicola
Di Trani
 ab,
Antonia
Silvestri
ac,
Giacomo
Bruno
ac,
Thomas
Geninatti
a,
Corrine Ying Xuan
Chua
a,
April
Gilbert
a,
Giulia
Rizzo
ac,
Carly S.
Filgueira
a,
Danilo
Demarchi
c and
Alessandro
Grattoni
*ade
ab,
Antonia
Silvestri
ac,
Giacomo
Bruno
ac,
Thomas
Geninatti
a,
Corrine Ying Xuan
Chua
a,
April
Gilbert
a,
Giulia
Rizzo
ac,
Carly S.
Filgueira
a,
Danilo
Demarchi
c and
Alessandro
Grattoni
*ade
aNanomedicine Department, Houston Methodist Research Institute, Houston, TX, USA. E-mail: agrattoni@houstonmethodist.org; Tel: +1 (713) 441 7324
bUniversity of Chinese Academy of Science (UCAS), Shijingshan, 19 Yuquan Road, Beijing 100049, China
cDepartment of Electronics and Telecommunications, Politecnico di Torino, Turin 10024, Italy
dDepartment of Surgery, Houston Methodist Hospital, 6550 Fannin Street, Houston, TX 77030, USA
eDepartment of Radiation Oncology, Houston Methodist Hospital, 6550 Fannin Street, Houston, TX 77030, USA
First published on 3rd June 2019
Abstract
Chronic diseases such as hypertension and rheumatoid arthritis are persistent ailments that require personalized lifelong therapeutic management. However, the difficulty of adherence to strict dosing schedule compromises therapeutic efficacy and safety. Moreover, the conventional one-size-fits-all treatment approach is increasingly challenged due to the intricacies of inter- and intra-individual variabilities. While accelerated technological advances have led to sophisticated implantable drug delivery devices, flexibility in dosage and timing modulation to tailor precise treatment to individual needs remains an elusive goal. Here we describe the development of a subcutaneously implantable remote-controlled nanofluidic device capable of sustained drug release with adjustable dosing and timing. By leveraging a low intensity electric field to modify the concentration driven diffusion across a nanofluidic membrane, the rate of drug administration can be increased, decreased or stopped via Bluetooth remote command. We demonstrate in vitro the release modulation of enalapril and methotrexate, first-line therapeutics for treatment of hypertension and rheumatoid arthritis, respectively. Further, we show reliable remote communication and device biocompatibility via in vivo studies. Unlike a pulsatile release regimen typical of some conventional controlled delivery systems, our implant offers a continuous drug administration that avoids abrupt fluctuations, which could affect response and tolerability. Our system could set the foundation for an on-demand delivery platform technology for long term management of chronic diseases.
Introduction
The escalating burden of chronic diseases constitutes a major public health problem, accounting for approximately two-thirds of all deaths and 75% of healthcare expenditures.1,2 With a protracted course of disease and requirement for continuous therapeutic management, chronic conditions constitute a complex, long-term challenge for patients and healthcare systems alike. Moreover, healthcare systems are typically focused on acute episodic care rather than structured continuous management of the disease, which increases the economic burden.3 Most chronic diseases such as hypertension are often accompanied by various comorbidities, thus requiring attentive vigilance in disease management, specifically compliance to therapeutic interventions. However, only approximately 50% of individuals with chronic diseases adhere to treatment regimens, posing significant risks of complications and even mortality.4 While the advent of implantable sustained release platforms5,6 has significantly improved treatment regimen of certain pathologies, not all chronic diseases benefit from constant drug administration. Considering that physiologic variables such as blood pressure are typically predictably synchronized with circadian activity,7 the conventional paradigm of constant drug delivery to target these parameters is unnecessary and often associated with undesirable problems of resistance, tolerability and side effects.8 For instance, anti-inflammatory drugs for rheumatoid arthritis are most effective during the 4–5 hour window of night-time immune hyperactivity, rendering daylong systemic immunosuppression needless and unwarranted.9,10 More importantly, advances in precision medicine have challenged the conventional one-size-fits-all blanket treatment approach due to the intricacies of inter- and intra-individual variabilities. This motivated the need for technological innovations in drug delivery that meet the demand for customizable therapy to achieve maximal treatment efficacy with minimal side effects.Thus far, implantable delivery systems that provide drug administration at a defined time and dose or rate constitute a desirable but unmet clinical need. Recent advances have led to developments of such technologies, some of which include polymeric, pump, and microelectromechanical-based systems (MEMS) for either pulsatile or continuous release.11,12 Biodegradable polymeric systems13,14 allow for multiple-pulse drug release, whereas propellant infusion pumps15 offer a steady release over time; however both lack dosing control after implantation. While peristaltic implantable pumps such as those from Synchromed Medtronic16,17 offer a zero-order release manner with externally modifiable dosing, these benefits could be overlooked due to prohibitive costs and bulky sizes. Research efforts have also focused on trigger-dependent systems based on reservoirs covered by membranes with tunable permeability to activate modulated drug release in a burst manner.18,19 However, a point of consideration for feasibility and convenience of implementation is that these devices usually require external stimuli such as electromagnetic fields or near-infrared light throughout the duration of the release. As such, this could be cumbersome considering that individuals would need to be subjected to the stimuli for long periods of time, not to mention the possibility of injury due to repeated exposure to stimuli (i.e., thermal injury).18 Another development of note is the use of remote communication to control drug release, as represented by MEMS-based implants such as Microchip20 and ChipRx.21 Microchip is a silicon chip with twenty 600 nanoliter drug reservoirs that are individually opened via a wireless command. This system completed the first clinical trial in Europe for assessing the efficacy in treating women with osteoporosis (EudraCT, number 2010-020040-35).22 Albeit promising, limitations in the reservoir size and quantity present challenges for long-term disease management. ChipRx (Lexington, KY, USA) uses a single electronic signal to open micrometric-sized holes to release drugs. However, the development was suspended due to undisclosed reasons. Moreover, it is important to note that pulsatile release systems such as the aforementioned MEMS-based implants cause abrupt fluctuations of drug concentrations, which may affect clinical response and tolerability.23
That said, an ideal programmable delivery system should entail: 1) zero-order release kinetics, 2) modulation capabilities for dosing-on-demand, tunable dose, rate and time, 3) wireless or remote communication capabilities for convenience of monitoring or telemedicine intents, 4) release stoppage when not needed (i.e., hypertension drug release only during the sleep cycle) or when unexpected side effects occur, and 5) compact size dimensions for feasibility of discrete implantation. With this in mind, we developed a subcutaneously implantable delivery device for dosing modulation via remotely controlled application of a low-intensity electric field across a nanofluidic membrane. With no application of an electric field, the implant releases drugs through diffusion, driven by a concentration gradient between the reservoir and subcutaneous space. Therefore zero-order passive release is achieved without the use of mechanical parts by tailoring of the size of channels to the size and properties of drug molecules (net charge, molecular weight, and hydrophobicity). By applying a small electric field across the membrane via two platinum electrodes, ionic species are forced to redistribute or are electrophoretically driven across the nanoconfined space, thus effectively actively modulating the drug release rate. As such, our implant offers a continuous drug administration for smooth transition between different release phases. This allows for avoidance of peak-to-trough plasma fluctuations that affect drug administration efficiency and potentially cause adverse effects.24
In this work, we first demonstrate the drug modulation capability by leveraging the electric field across a nanofluidic membrane through in vitro studies. Next, we incorporated this technology into a drug delivery implant and characterized its capacity for reliable Bluetooth communication and release rate modulation. Finally, we validated the telemetric capacity and biocompatibility of our remotely controlled drug delivery implant in rats and a non-human primate. Overall, we present the proof of concept of a telemetry-enabled delivery system, which could eventually allow physician autonomy to access data and remotely adjust drug release and dosing schedule according to individual patients' needs. Our wirelessly programmable nanofluidic approach holds promise for the development of a drug delivery platform capable of maximizing therapeutic efficacy and minimizing side effects towards improving patient adherence, quality of life and treatment of chronic diseases.
Materials and methods
Fabrication of silicon nanofluidic membranes
Silicon membranes were manufactured using standard semiconductor fabrication techniques described in detail elsewhere25 (Fig. 1A). Briefly, slit-nanochannels were obtained starting from a deposited silicon nitride (SiN) thin film (1.7 μm thick) on a silicon (Si) wafer (700 μm thick). The inlet and outlet microchannels were perpendicularly etched on the silicon wafer with a cross-section of 1 μm × 3 μm (Fig. 1B). The nanochannels were obtained by removing a sacrificial tungsten layer, which lies between the Si and the SiN, with hydrogen peroxide (H2O2) (Fig. 1B and C). The configuration of slit-nanochannels parallel to the surface ensures high structural stability and mechanical robustness, while the addition of a TaN/SiC layer coating to the membrane provides bioinertness.26 This study utilized nanofluidic membranes of 13 nm and 200 nm, which feature a geometrically organized array of 340![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 252 densely packed slit-nanochannels.
252 densely packed slit-nanochannels.
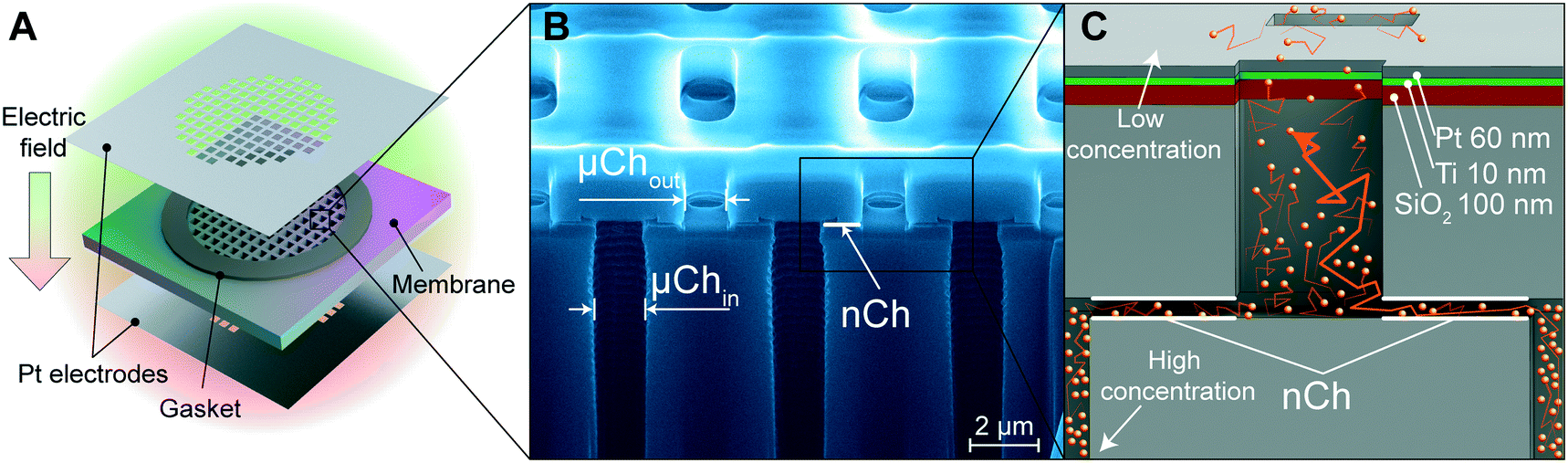 | ||
| Fig. 1 Nanofluidic membrane and platinum electrodes. (A) Schematic of the nanofluidic membrane with platinum (Pt) foil electrodes separated by gaskets. (B) FIB-SEM image of the nanofluidic membrane without electrodes. Inlet and outlet microchannels are denoted as μChin and μChout, respectively. Nanochannels (nCh) are perpendicular to the μCh. (C) Schematic of the deposited electrode stack achieved through the second method of electrode integration (sputtering). Order of deposition (bottom to top): silicon dioxide (SiO2; 100 nm, red layer), titanium (Ti; 10 nm, green layer), platinum (Pt; 60 nm, grey layer). Orange particles represent the diffusing drug molecules. Orange arrows represent the diffusing path. | ||
Incorporation of platinum electrodes onto membrane
Platinum (Pt) electrodes were integrated onto the silicon membrane using two different techniques, as previously described.27 The first method uses 100 μm thick Pt foil (Sigma Aldrich, St. Louis, MO, USA) which was laser cut (A-Laser, Inc., CA, USA) to fit the membrane in a 6 mm × 6 mm square shape and affixed to the membrane with UV epoxy (OG116, Epoxy Technologies, Inc.). UV epoxy was cured overnight with a UV lamp (UVL-18, UVL) (Fig. 1A). To prevent epoxy leakage into the channels, a silicone gasket was positioned between the Pt foil on each side of the membrane (Fig. 1A). The second method is the chemical vapor deposition (sputtering) of the SiO2–Ti–Pt stack on the top and bottom surface of the membrane (Fig. 1C). First, the SiO2 (250 nm) substrate was deposited on the SiN surface, and the presence of argon plasma ensured a defect-free dielectric layer. A Ti adhesion layer was deposited prior to the final Pt film. To avoid channel clogging and achieve equal layer thickness perpendicular to the membrane, Ti (10 nm) and Pt (60 nm) layers were deposited at an ∼45° angle with respect to the surface (Fig. 1C). Insulated 36 AWG wires were connected to the electrodes with electrically conductive silver epoxy (H20E, Epoxy Technology, MA) and cured at 130 °C for 15 min. Pt foil was wired using a lead-free solder (SN99, MG Chemicals, BC, Canada). Additional details are available elsewhere.27Focused ion beam (FIB) – scanning electron microscopy (SEM) imaging
Silicon nanofluidic membranes were evaluated using a dual-beam ion-beam (FIB) system FEI 235 at the Nanofabrication Facility of the University of Houston, Texas. The membranes were imaged at an angle of 52° using scanning electron microscopy (SEM) in the FIB system.In vitro controlled release from membranes
An enalapril in vitro release test was performed with 13 nm nanochannels and Pt foil electrodes, while 200 nm membranes with sputtered electrodes were used for methotrexate. Membranes were first immersed in isopropyl alcohol to ensure proper nanochannel wetting, then rinsed with deionized (DI) H2O, and inserted in a dual-reservoir diffusion apparatus.28 The source chamber contained either 200 μL of 10 mg mL−1 enalapril (Cayman Chemical, Ann Arbor, MI) or 200 μL of 3 mg mL−1 methotrexate in 50 mM NaCl. Both drugs are negatively charged, −1q (= −1.6 × 10−19 C) for enalapril and −2q (= −3.2 × 10−19 C) for methotrexate. UV-cuvettes loaded with 4.45 mL of 50 mM NaCl in DI H2O were employed as sink reservoirs. The assembled custom cuvette systems were loaded onto a robotic carousel29 of a Cary 50 UV-vis spectrophotometer (Agilent Technologies). Electric potentials were applied to the membrane electrodes using a DC power supply (E3643A, Agilent Technologies) in three phases with the following sequence: 0 V, −1.5 V, 0 V, and +1.5 V. Each phase was run for 8 hours for the methotrexate study and 24 hours for enalapril. UV absorption measurements of the sink solution were automatically performed using the robotic carousel every 7.5 minutes. Wavelengths used for detection were 287 nm for methotrexate and 219 nm for enalapril.Printed circuit board (PCB) and remote communications
The printed circuit board (PCB) was designed for the control of therapeutic release. The system-on-chip (SoC) CC2541 (Texas Instrument) was used for the Bluetooth low energy (BLE) communication since it provides ultra-low energy consumption. A commercially available discoidal CR2016 (VARTA) battery was used as a power supply for the board. The battery has a total capacity of 90 mAh. A step-down voltage converter (LM3670, Texas Instrument) was used to lower the battery voltage from 3 V to 1.5 V. Two of the available general-purpose input–output (GPIO) pins (P1 and P2) were connected to the electrodes used for drug modulation. P1 was connected to the converter for the 1.5 V output, while P2 was connected directly to the GPIO for 3 V. The wireless communication with a remote PC was implemented through a USB Bluetooth dongle CC2540 (Texas Instrument) and MATLAB® script.Power consumption evaluation
The power consumption of the PCB was evaluated considering the instant current absorbed by the PCB. An oscilloscope (U2702A, Agilent Technologies) was used to monitor the voltage across a resistor of 50 Ω, emulating the internal load of the battery. The power supply (3 V) was provided by a power generator (E3643A, Agilent Technologies).Implant assembly
The implant capsule made of polyether ether ketone (PEEK) was machined at the machine shop core of the Houston Methodist Hospital. The capsule hosts two separate compartments: one for the membrane that also serves as a drug reservoir and the other for the battery and electronics. The reservoir was sealed with a PEEK lid where two loading/venting ports were incorporated. Ports were sealed with implantable self-sealing silicone glue (Nusil MED3-4213, Nusil Technology). Biocompatible thermal epoxy (Epotek 302-3M, Epoxy Technology) was centrifuged at 5000g for 2 minutes to remove bubbles. The battery and electronics were embedded in the implant capsule with thermal epoxy under a sterile laminar flow hood and cured in a sterile container at 65 °C for 3 hours. The membrane was glued in place with biocompatible UV epoxy (OG116, Epoxy Technologies, Inc.) and cured overnight with a UV lamp (UVL-18, UVL). Finally, the lid was sealed with UV epoxy.Implant leakage test
Assembled implants (n = 6) were loaded through the loading ports with 550 μL of 1 mg mL−1 rhodamine B (sigma) in PBS using two 3 mL syringes with 25G needles, one for loading and the other for venting. The silicone loading ports were then sealed with UV epoxy to avoid any leakage. The implants were placed in a borosilicate bottle containing 20 mL of PBS and kept in a 37 °C incubator under constant agitation to ensure sample homogeneity for the 12 day duration of the experiment. 200 μL samples were collected daily and replaced with a fresh PBS solution. The sample concentration was analyzed with a high precision spectrofluorimeter (PC1, ISS) using an excitation wavelength of 525 nm and an emission of 550 nm.In vitro communication stability
Connection between the assembled implants (n = 5) and the computer was performed at increasing distances from 30 cm to 180 cm. We measured the received signal strength indication (RSSI) as a measure of communication quality. Communication was performed under two conditions, with the implants in air or submerged in water as a surrogate for the subcutaneous implantation. The communication test was performed over 60 min and repeated three times for each circuit.In vitro drug release from assembled implants
A customized cuvette was used for the release study, specifically designed to host the implant for measurement in the UV-vis spectrophotometer carousel. The body of the cuvette is composed of two parts: a cuvette and a PEEK container to host the implant glued together with UV epoxy (OG116, Epoxy Technologies, Inc.). A 13 nm nanochannel membrane with sputtered electrodes was embedded in the implant. The implant reservoir was filled with 550 μL of methotrexate (500 μg mL−1 in 50 mM NaCl). The sink solution of 7 mL 50 mM NaCl was under continuous agitation via magnetic stirring to ensure sample homogeneity. Under our experimental conditions (pH 7), methotrexate has a net charge of −2q. Drug concentration measurement in the sink solution was performed every 7.5 minutes via UV-vis spectroscopy at a wavelength of 287 nm. In the first 16 hours, no voltage (0 V) was applied to the electrodes, followed by a voltage of +1.5 V applied for 9 h, and then 0 V for 14 h and +1.5 V for 25 h. The total duration of the experiment was 100 h.In vivo biocompatibility and communication testing in rats
10 week-old male Wistar-Kyoto rats (n = 6) from Charles River Laboratories (Wilmington, MA) were used in this study. The animals were housed at the Houston Methodist Research Institute (HMRI) Comparative Medicine animal facility in standard cages. All animal experiments conducted were approved by the Institutional Animal Care and Use Committee (IACUC) of HMRI and performed in accordance with the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals. Each implant was assembled as previously described, and the reservoir was loaded with sterile PBS. Using an aseptic technique, a 2 cm incision was made on the dorsum of the animal. The implant was positioned in a subcutaneous pocket with circuitry facing the epidermis and the membrane facing the dermis. The incision was closed with metal wound clips. Bluetooth communication with the implant was performed every 5 minutes using the MATLAB® script running on a computer positioned 2 meters away from the animal. Connections to the implant were performed at regular intervals of 24 h for the whole duration of the experiment. The applied phases were: passive, active (1.5 V), passive, and active (−1.5 V), each for 5 days. Communication reliability was assessed by quantifying the number of advertisement packets that were lost during the discovery routine of the MATLAB® script running on the computer resulting in a non-successful discovery of the implant. After 21 days, the animals were euthanized and the implants were explanted for analysis.In vivo biocompatibility and communication testing in a non-human primate (NHP)
A 4 year-old rhesus macaque (Macaca mulatta) was used in a non-terminal study setting, to assess the biocompatibility and communication performances in a most relevant animal model in view of potential clinical translation. The NHP experiments were conducted at the AAALAC-I accredited Michale E. Keeling Center for Comparative Medicine and Research, The University of Texas MD Anderson Cancer Center (UTMDACC), Bastrop, TX. Experiments were performed in accordance with the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals. All procedures were performed in accordance with a protocol (00001183-RN00) approved by the Institutional Animal Care and Use Committee at UTMDACC. The implant was assembled as described above with Pt foil electrode membranes and the reservoir was filled with sterile PBS. Using an aseptic technique, a 2 cm skin incision was made in the animal dorsum, mid-thorax parallel to the spine, approximately 3 cm to the left of the spine. The subcutaneous space was bluntly dissected ventrally to create a pocket for the implant, positioned approximately 2 cm ventral from the incision. The implant was positioned with the circuitry facing the epidermis and the membrane facing the dermis. The subcutaneous layer was closed with a continuous absorbable monofilament suture (4–0 PDS). The skin was closed in a continuous intradermal pattern with the same suture.Following the minimally invasive implantation of the device, the animal was monitored for any sign of discomfort or unusual behavior. Bluetooth communication with the implant was performed every 5 minutes using the MATLAB® script running on a computer positioned 3 meters away from the animal. Connections to the implant were performed at regular intervals of 1 h for the first 50 h, then every 6 h thereafter. Communication reliability was assessed as in the rat experiment. The implant was explanted after 10 days.
Histological analysis
Tissues were fixed in 10% neutral-buffered formalin and embedded in paraffin. Five (5 μm) slices were cut and stained with hematoxylin & eosin (H&E) at the Research Pathology Core of Houston Methodist Research Institute (HMRI), Houston, TX, USA. Slides were assessed by a pathologist at the HMRI.Statistical analysis
Graphs were plotted and statistical data analysis was performed using GraphPad Prism 7 (version 7.0a; GraphPad Software, Inc., La Jolla, CA). Data are represented as mean ± SD. Statistical significance was determined using one-way analysis of variance (ANOVA). The release rate for each phase of a cumulative release was considered as the angular coefficient of the first order polynomial fit calculated with the MATLAB® polyfit (fitting errors were always under 2%).Results and discussion
The key feature of our platform involves leveraging the electric field for modulated control of drug release through nanofluidic membranes. We investigated two types of electrode integration to determine which could offer better performance in terms of drug release modulation via the application of a trans-membrane electric field. In one method, we affixed two platinum (Pt) foil electrodes on the top and bottom of the membrane (Fig. 1A), while the second method consists of a Pt electrode stack layer sputtered directly onto the membrane surface27 (Fig. 1C).Mechanism of drug diffusion in nanofluidic channels
Fluidic behavior at the nanoscale is different from those at the micro and macroscale.30 When charged particles diffuse through the nanochannels, they electrostatically interact with the electrical double layer (EDL), which is a counter-ion distribution present at the interface of the charged walls in our silicon nanofluidic membranes. The ratio δ = λ/h between the Debye length (λ), a characteristic measure of the extension of the EDL, and the nanochannel size (h) represents how much the charged molecule diffusion is influenced by the EDL. When no voltage is applied to the nanofluidic membrane (passive phase), the concentration difference between a high concentration region (drug reservoir) and a low concentration region (sink solution) drives diffusion of the drug molecules through the nanochannels. However, in the passive phase, when δ is large and the size of molecules approaches h in order of magnitude, steric and electrostatic hindrance effects imposed by the nanochannels act as a bottleneck for free diffusion, generating a saturated diffusive transport that is quasi-independent from the concentration gradient.31,32In all our experiments, the solution used was 50 mM, resulting in a Debye length of λ = 1.3 nm. We hypothesized that for small δ (i.e., δ = 0.0065 for h = 200 nm) channels with the application of a small transmembrane electric field (active phase), the active transport of charged molecules would be driven by electrophoresis. In contrast, for larger δ (i.e., δ = 0.1 for h = 13 nm) channels which display a charge-selective behavior, we hypothesized that the application of an electric field would generate a redistribution of ionic species at the micro–nanochannel interface. Therefore, we postulated that the active transport of molecules would be affected by ionic depletion and enrichment consistent with the beginning of transition to ionic concentration polarization (ICP).
In vitro release modulation of methotrexate
We first investigated how the application of a low intensity electric field across the membrane with Pt foil electrodes could modify the release rate of methotrexate, an antifolate drug used for first-line treatment of rheumatoid arthritis. For analysis of modulated in vitro release, we alternated between 3 different phases: passive with no applied voltage and two active phases with either a negative or positive voltage. Upon application of a +1.5 V potential between the Pt electrodes, a consistent increase of the cumulative release rate with respect to the passive phase was observed (Fig. 2A). Conversely, the application of −1.5 V resulted in a decrease of the release rate compared to the previous phase. Regardless of the polarity of the previously applied potential, upon deactivation of the voltage, the release returned to a zero-order concentration driven diffusion. The release rate calculated for each phase is shown in Fig. 2A (bottom). To quantify the effect of externally applied potential on the release rate, we averaged the release rate of each phase (horizontal lines) and compared active release to passive release. We observed a 2.5-fold decrease when a negative voltage was applied and a 4.7-fold increase when a positive voltage was applied. When grouping the release rates by phase typology, we detected a statistically significant difference between each group, indicating effective modulation of the release rate through the application of the external potential.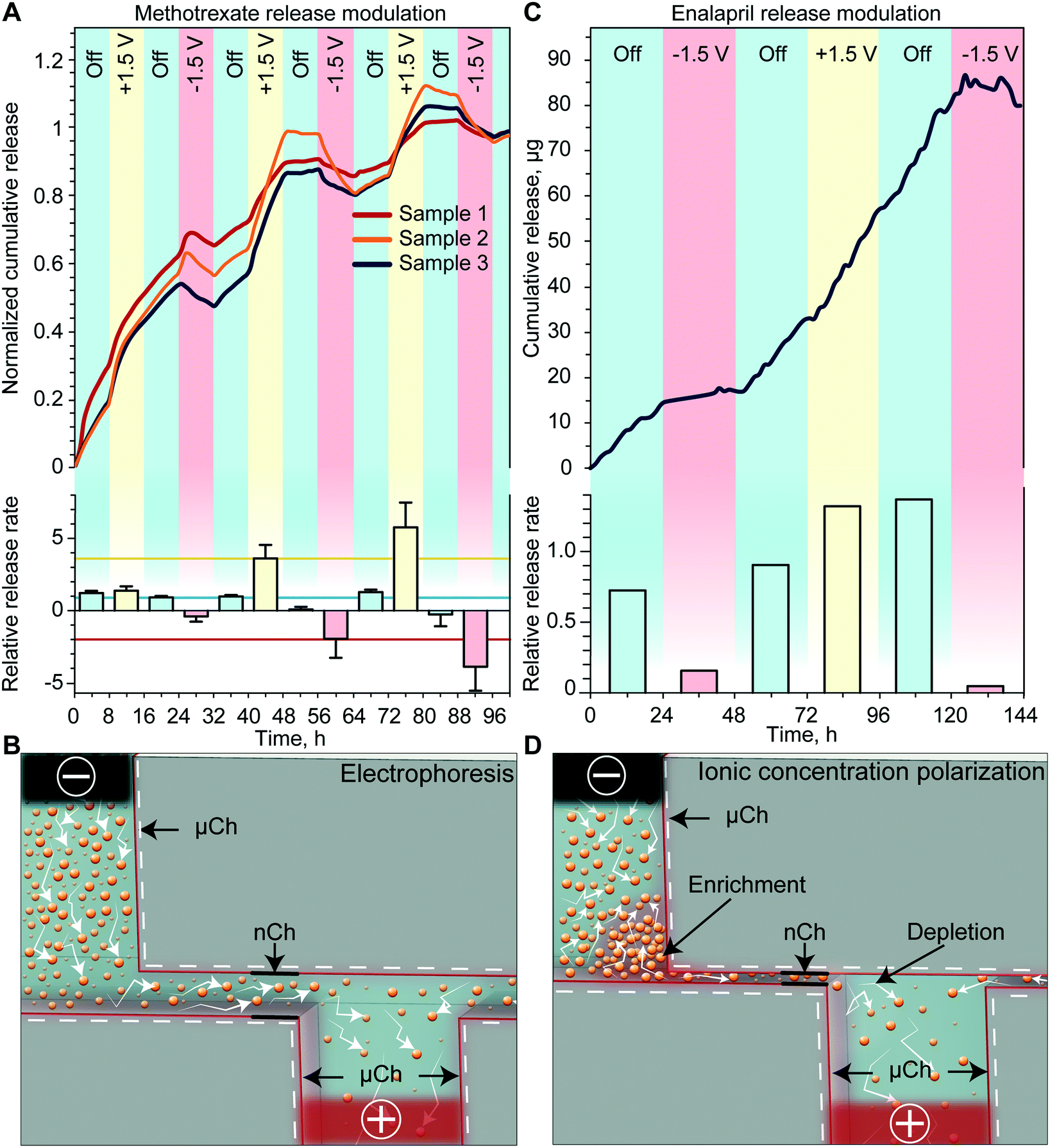 | ||
| Fig. 2 In vitro release rate of methotrexate and enalapril. (A) Methotrexate cumulative release (normalized to sink concentration at hour 100) (top) and release rate (bottom) for different applied voltages normalized to its passive release rate. Horizontal lines in the bar plot are the averages of the +1.5 V (yellow), passive (blue) and –1.5 V (red) release rates. Sputtered electrode configuration employed. (B) Schematic of electrophoretic transport in nanochannels. (C) Enalapril cumulative release (top) and release rate (bottom) for different applied voltages normalized to its passive release rate. Platinum foil electrode configuration employed. (D) Schematic of ionic concentration polarization in nanochannels, highlighting enrichment and depletion regions. | ||
The results obtained in this release study, which was performed with a 200 nm membrane, are consistent with the electrophoretic charge transport phenomenon (Fig. 2B). Specifically, charged molecules in the bulk have a negligible interaction with the EDL and are driven through the channel by the external electric field applied by the electrodes. Additional validation of electrophoretic transport is through the observation of a reduction in methotrexate cumulative release (negative release rate) upon application of a negative voltage. Since our testing setup is a closed system, it is expected that the drug molecule could be driven back into the reservoir if the external electric field overcomes the concentration gradient force. We expect that this phenomenon would not be observed in the subcutaneous space where the drug would be rapidly uptaken by surrounding tissues. These scenarios would instead result in a more pronounced reduction of the release rate.
In vitro release modulation of enalapril
To evaluate the drug modulation capability of the electric field across the membrane with sputtered Pt electrodes, we performed an in vitro release study with enalapril. Enalapril is an angiotensin-converting enzyme inhibitor used for management of hypertension. Fig. 2C shows the cumulative release achieved with enalapril when cycling through the three different phases. For each passive phase, a quasi-zero-order concentration driven diffusion was achieved with an average of 0.8 μg h−1 of enalapril released. With the application of a positive voltage, we observed a slight increase in the release rate, whereas the application of a negative voltage consistently led to a several-fold decrease in the release rate. When comparing the release rate of each phase to the passive rate, we observed that a negative applied voltage yielded on average a 7-fold decrease in the release rate, while a positive applied voltage resulted in a 1.8-fold increase of the release rate (Fig. 2C, bottom).As this study was performed with a smaller membrane (13 nm), the EDL spans over most of the nanochannel volume, which leads to the electrostatic exclusion of co-ions and enrichment of counter-ions (Fig. 2D).33 When an external tangential electric field is applied, co-ions will start to accumulate in the upstream enrichment region, while a depletion region will form downstream (Fig. 2D). The co-ion enriched region will create both a concentration gradient and an electric field against the externally applied potential, which will prevent additional drug molecules from entering the nanochannels.34 In fact for enalapril, the application of a negative voltage resulted in a more pronounced decrease in the release rate, while a positive voltage yielded a smaller increase in the release rate when compared to methotrexate. We attributed this phenomenon to the onset of ICP, which could lead to a reduction in the release rate when either a positive or negative potential is applied to the electrodes (Fig. 2D).
We have previously demonstrated that in even smaller nanochannels (5.7 nm), ICP can overcome other effects such as electrophoretic force and concentration gradient, leading to a decrease in the release rate with either positive or negative applied potentials.35 Here we observed slightly different passive release rates depending on the previously applied potential, which we attribute to the slow redistribution of ions generated by ICP. This relatively slow ion redistribution allows for a smooth transition of release between different phases. In the eventual need for a sharper transition, the sequence of applied potentials can be changed accordingly. Overall, we posit that ICP is an important phenomenon that should be taken into account when modulating the drug release rate through small nanochannels.
Influence of the electrode configuration on drug release modulation
Although the phenomena that lead to the release modulation of methotrexate and enalapril differ, we attribute the difference in reproducibility to the employment of different electrode configurations, specifically their relative distance. Pt foil electrodes glued onto the membrane result in a distance of ∼1 mm while sputtered electrodes are separated only by the membrane thickness (700 μm). Our results demonstrated that a smaller distance between Pt electrodes yielded a higher intensity of the applied electric field36 for the same applied potential. In particular, we estimated an electric field of 1.5 V mm−1 and 2.14 V mm−1 for Pt foil and sputtered electrodes, respectively. Therefore, we achieved a more reproducible control of methotrexate release modulation with respect to enalapril.On the other hand, regardless of the different inter-electrode distance in the two configurations, both electrode integration methods represent feasible approaches to achieve drug modulation via an external electric field. Further considerations for choosing the optimal electrode integration methods concern their fabrication technique. While Pt foil electrodes constitute an easier method, they are costly and could prove challenging to adapt to intricate fluidic structures. Conversely, sputter deposition allows for coating of complex surfaces with a fine control over the thickness of the deposited material. However, the poor adhesive properties of sputtered Pt to our substrate require the adoption of other materials such as silicon dioxide (SiO2) and titanium (Ti) to improve adhesion. Nevertheless, both electrodes are demonstrated to be biocompatible and bioinert27 and thus suitable for clinical translation.
Remote-controlled implant assembly, working configuration and leakage test
To create an implant which could be brought toward clinical translation, we selected PEEK as the fabrication material. PEEK is commonly used in orthopedic implants due to its mechanic robustness and bio-inertness.37 Importantly, PEEK avoids potential RF-shielding associated with metals such as titanium. The implant hosts two compartments: one for the membrane that also serves as a drug reservoir with 550 μl capacity (highlighted in red) and the other for the battery and electronics (highlighted in green) (Fig. 3A). The implant dimensions are 34 mm in length, 24 mm in width and 4.5 mm in thickness for a total volume of 2.8 cm3. The reservoir occupies 20% of the implant volume, while the circuitry chamber accounts for 40%. Notably, by comparing our implant to a relevant existing implant technology (Microchip), our implant hosts a drug reservoir ∼200 times larger, while maintaining an overall volume 5 times smaller than Microchip.20 This larger reservoir capacity could minimize excessive surgical procedures for implant replacement, which is a current challenge with existing implants of similar functions.20 An alternative to increasing reservoir capacity is through integration of refilling ports for drug replenishment, as previously demonstrated by our group.38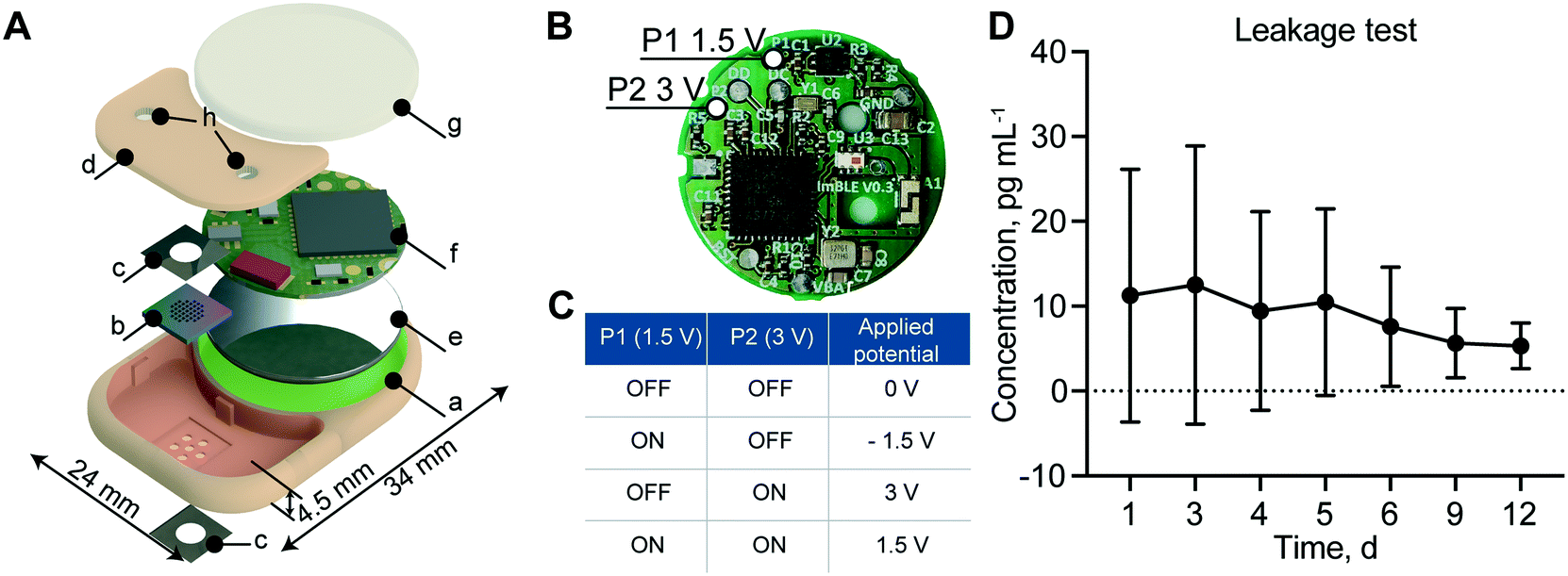 | ||
| Fig. 3 Implant components, configuration and leakage test. (A) Exploded view rendering of the implant: a. PEEK body; b. silicon nanofluidic membrane; c. platinum foil electrodes; d. PEEK lid used to seal the drug reservoir; e. battery; f. printed circuit board (PCB); g. epoxy sealant; h. silicone drug filling ports. Red and green highlighted areas are the reservoir compartment and the circuitry chamber, respectively. (B) The printed circuit board (PCB) with the two GPIO pins P1 and P2 highlighted, which can apply 1.5 V and 3 V to the electrodes, respectively. (C) Schematic of possible voltages that can be applied to the membrane. (D) Detection of rhodamine B concentration in sink solution over 12 days. Data are represented as mean ± SD (N = 3). | ||
In order to achieve different drug release rates, a different combination of P1 and P2 ON and OFF status on the PCB allows for the application of 4 different voltages, +1.5 V, −1.5 V, +3 V and 0 V (Fig. 3B). As summarized in Fig. 3C, when both P1 and P2 are ON, the applied voltage to the electrodes is +1.5 V; when P1 is ON and P2 is OFF, the applied voltage is −1.5 V; when P1 is OFF and P2 is ON, the applied voltage is +3 V; and when P1 and P2 are both OFF, the applied voltage is 0 V.
To ensure implant intactness and impermeability, we performed a leakage test with rhodamine B, a fluorescent dye used as a surrogate marker. The cumulative concentration of rhodamine B presented in Fig. 3D showed no significant change in concentration over the 12 days of analysis, confirming the implant intactness. We attributed the higher dye concentration observed initially to the residual dye deposited on the outside of the implant during the loading procedure. At the end of the study, after syringe withdrawal of the residual dye from the reservoir via the port, we examined the implant internally for permeability assessment. All internal components remained dry, confirming the implant intactness and impermeability.
Power consumption analysis
To estimate the lifespan of our implantable device, we assessed the different sources of power consumption of the PCB (Fig. 4A). Circuitry activity can be divided into background use, advertising and communication. Background processing and advertising (peaks in Fig. 4B) consumed 97.5 μW and 16.5 μW, respectively. Data transmission during a connection between the implant and server resulted in significant energy consuming activity spending 1.22 mW. Further, the current flow between the two electrodes for drug modulation also account for battery depletion. Specifically, current measurements during enalapril and methotrexate in vitro release studies (Fig. 2) ranged from a few μA to peaks of 100 μA (150 μW). In view of the contribution of these sources of battery drainage (capacity of 270 mWh), we estimated the lifetime of the implant in a high power consumption scenario to reach 20 days (ESI†).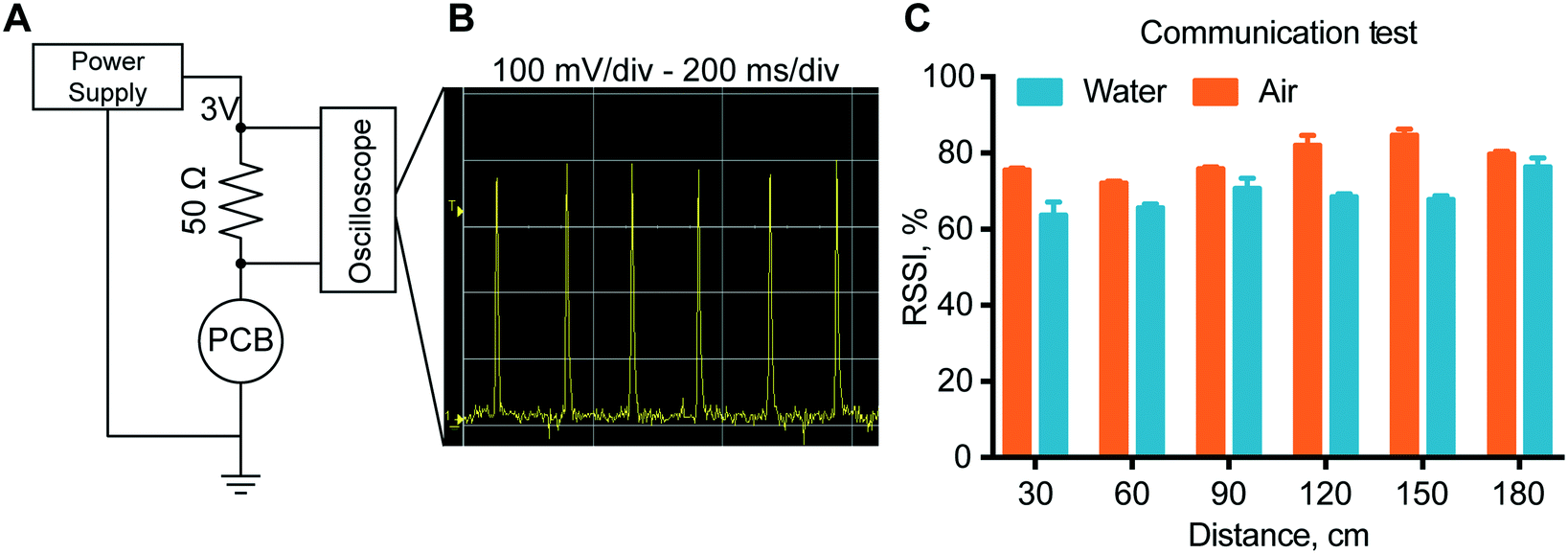 | ||
| Fig. 4 Power consumption and communication stability. (A) Experimental setup for the PCB power consumption test. (B) Oscilloscope measurements representing the amount of power required from the circuitry to advertise its presence via Bluetooth low energy. All the other PCB activities are completely obscured by the advertising phases. (C) Communication stability test, in open air vs. water container. Data are represented as mean ± SD (N = 15). | ||
We used different approaches to extend the lifetime of our implant. By reducing the advertising frequency from 100 ms to 10 s, the calculated lifespan was extended to 30 days without a considerable loss in connection stability. Similarly, in order to reduce the energy used by the electrodes, a possible solution is to investigate the application of a square or pulse wave with a duty cycle set in agreement with the characteristic response time of the system. A hypothetical reduction of current consumption to 1 μA could extend the implant lifespan to 3 months. Another approach to decrease power consumption is to reduce connections, for example exchanging the current state of the implant in a secure code via advertising (ESI†). In our current setting, the constant exchange of parameters represents a security measure, in order to avoid unexpected actions. For our current implant, this could translate to an extended lifespan of several months.
In vitro communication stability analysis
To verify proper RF-communications, we performed a communication test, where a connection was initiated between the remote control and the implant. The results showed that with the increase of the distance between the implant and the receiver, there is no definite drop of the signal power up to 180 cm. In fact, the transmission power in air has an RSSI that is almost consistently 80% for all distances tested. In water, the RSSI is approximately 70% at every distance. In order to ensure a stable connection, the signal quality should be above 25–30%. Here we observed a signal quality above 60% for both in air and in water for every distance ensuring stable connections up to 180 cm with ample margin.In vitro modulated release analysis with the assembled nanofluidic implant
To assess the drug release modulation capability of the fully assembled implant, we performed an in vitro release study of methotrexate. The implant was positioned in a custom-made cuvette as depicted in Fig. 5A. The release rate and duration of each phase were controlled using the MATLAB® script running on a nearby computer. By sending a Bluetooth command to the immersed implant on a predetermined schedule, the script executed the application of an electric field across the nanochannels. This communication allowed the alternation of passive phases that resulted in a purely diffusive transport, with active phases that leveraged nano-electrofluidic transport to alter the release rate. Depending on the polarity of the generated electric field, an increase or decrease of the release rate was achieved (Fig. 5B). Upon deactivation of the applied potential after the active phases, the release reverted to zero-order concentration-driven diffusion.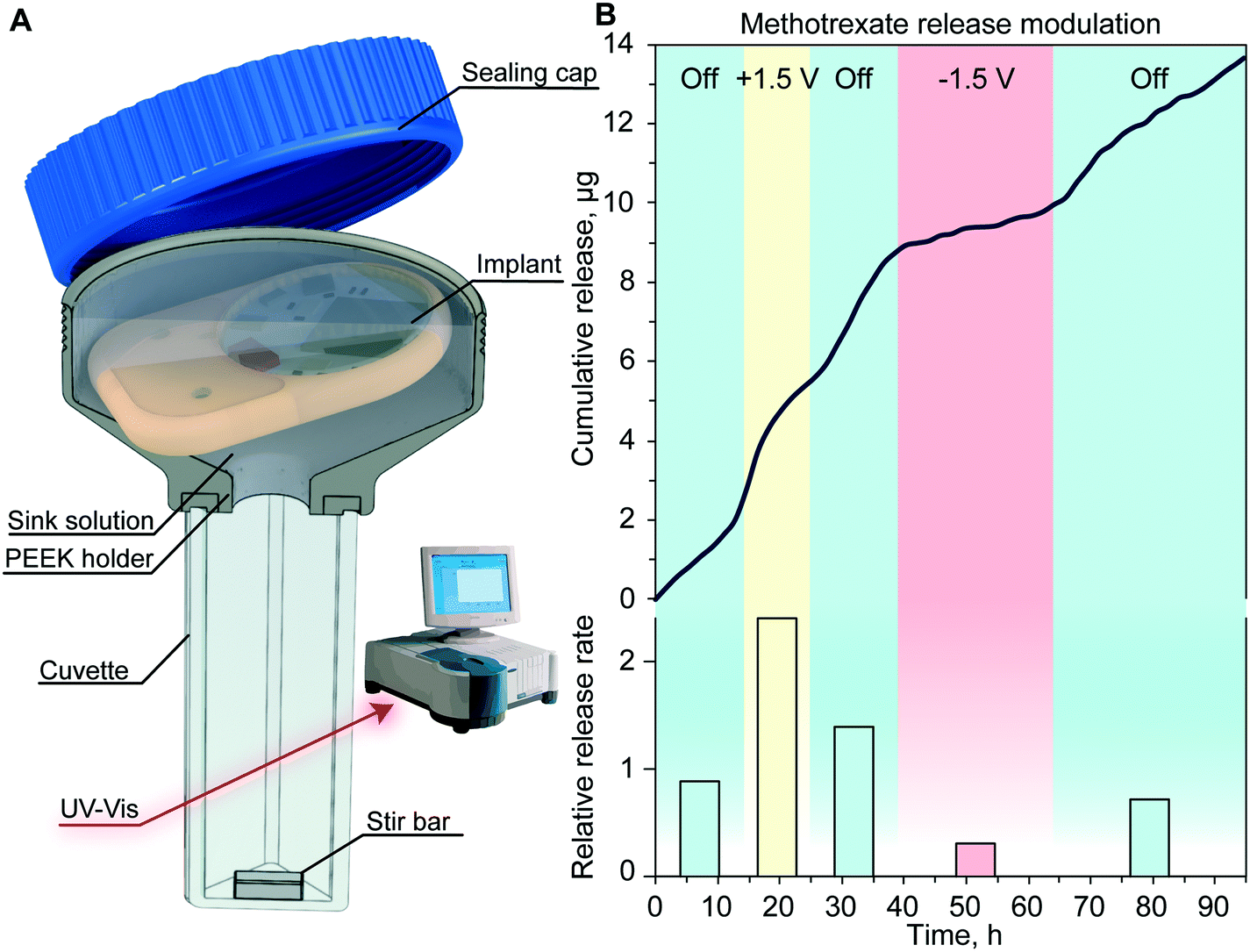 | ||
| Fig. 5 In vitro release of methotrexate with the assembled implant. (A) Experimental setup for the in vitro release. The implant is in a sealed cuvette, phases are set by remote communication with the nearby computer. Drug concentration in the sink solution is measured via UV-vis (Cary 50 Bio represented). (B) Methotrexate cumulative release (top) and release rate (bottom) for every applied voltage normalized to the passive release rate. | ||
When comparing the release rate of each phase to the average of the passive phase (Fig. 5B, bottom), we observed that the application of +1.5 V leads to a 2.4-fold increase of methotrexate release resulting in a release rate of 9.3 μg per day. Conversely, the application of −1.5 V resulted in a 3.24-fold decrease and a release rate of 1.2 μg per day. A similar dosage of methotrexate injected subcutaneously was demonstrated to be effective in treating rats without side effects.39
Throughout this experiment, we achieved a reliable signal strength of up to 3 meters with no communication errors and smooth transition between different release rates. The slight delay observed in the onset of a different release rate was attributed to the testing apparatus and the time that it takes for the stir bar to homogenize the sink solution concentration.29 Our results with the nanofluidic implant demonstrated fine control in timing and dose administration which could be tailored to the individual's specific pathology.
In vivo remote communication assessment in rats and the non-human primate
To validate the reliability of remote communication with our implant, we performed in vivo experiments in rats and in a non-human primate (NHP), in view of future clinical translational goals. Communication reliability was assessed by quantifying lost advertisement packets during the discovery routine of the MATLAB® script (Fig. 6A). In rats, the percentage of successful implant discovery was relatively stable for up to 11 days and thereafter showed a decreasing trend that we attributed to the slow discharge of the battery (Fig. 6B). In the NHP, we observed an average of ∼95% successful discoveries over 9 days (Fig. 6C).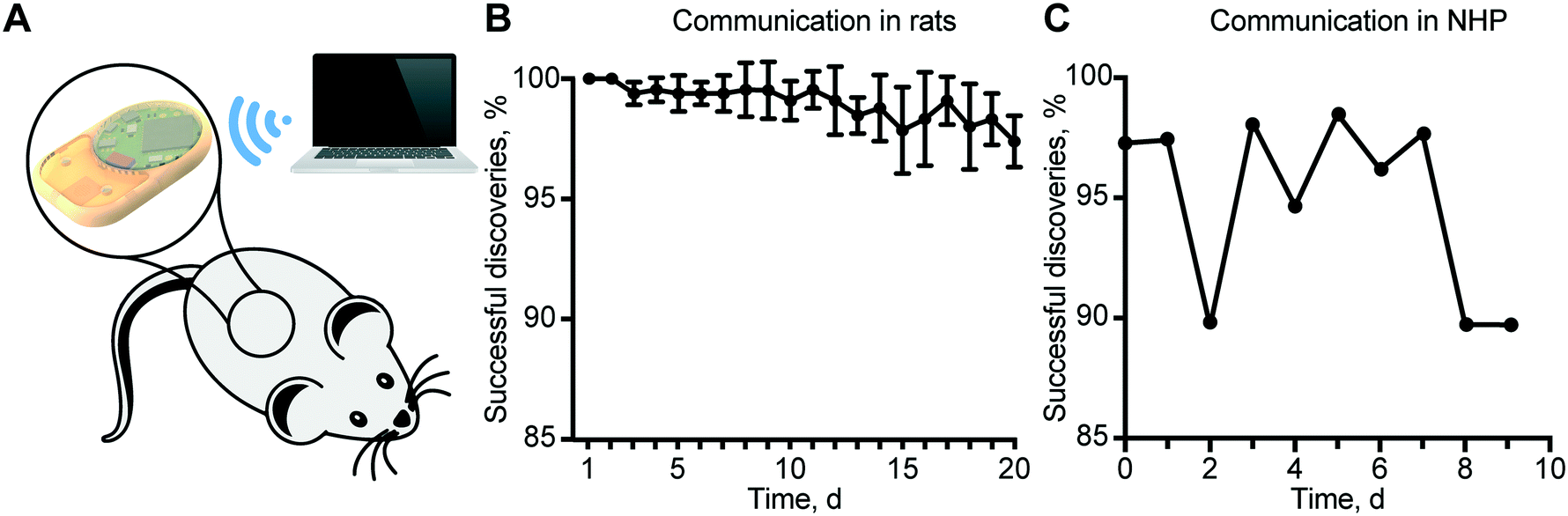 | ||
| Fig. 6 Communication reliability in rats and the non-human primate. (A) Custom MATLAB® script for the remote control of several implants at the same time. (B) Percentage of the successful discovery of implants (n = 6) subcutaneously implanted in rats. Data show mean ± SD. (C) Daily successful discoveries of the implant in the non-human primate. | ||
It is important to note that a non-successful discovery does not represent an error in communication, but the loss of advertisement packets. In the case of rats, battery discharge prevented brief but substantial burst of energy required for wireless communication, resulting in a reduced power to the antenna, and consequently, a shorter communication range. In the NHP, we ascribed the loss of advertisement packets to the considerable distance between the implant and external antenna due to the large confinement area of the animal. Therefore, a plausible solution is to employ a longer discovery interval in the server. Despite the loss of some packets during the advertisement phase, all the connections established were successful. In fact, the security routine that provides a feedback control loop and ensures that the read status is the same as the implemented was never triggered.
Remote connection via BLE offers high versatility allowing our implanted devices to interact in real-time with external servers, such as smartphones, smartwatches, or computers. We chose a completely automated MATLAB® script to perform all experiments since it obviates the need for operator intervention, which could be particularly useful in future clinical deployments. Additionally, we developed two other communication systems: an iOS application (Fig. S1†) and a web application that allows real-time monitoring of all active implants (Fig. S2†). These applications can be used as a patient monitoring method for physicians to remotely check or change the release regimen of the implanted devices. Remote communication and control can reduce recurring clinic visits, improve patient adherence and therapeutic effectiveness and set the pace for the future of telemedicine and patient care.
In vivo biocompatibility in rats and non-human primates
For preclinical safety assessment of our implant, we evaluated biocompatibility in rats as well as NHPs. NHPs provide high biological relevance especially in the context of immunotoxic side effects. In rats, throughout the duration of the 21 day study, the implants were well tolerated with no observed adverse skin reactions, infection or variation in their social behavior and mobility. Upon explantation, we observed fibrous encapsulation of the implant, indicative of foreign body reaction conventional to medical implants.40,41 Histological analysis was performed on the fibrotic capsule adjacent to the Pt electrode (Fig. 7A), epoxy (Fig. 7B) and PEEK (Fig. 7C). The side of the capsule facing the three aforementioned materials showed resolved mild inflammation and the presence of collagen, consistent with fibrosis formation. In particular, in areas near the Pt electrode and epoxy, minimal macrophage infiltration was detected. In areas near the PEEK material, we observed the presence of a minimal inflammation region in the final healing stages. Overall, there were no signs of significant active inflammatory reactions after 21 days of implantation, suggesting biocompatibility and tolerability of our implant.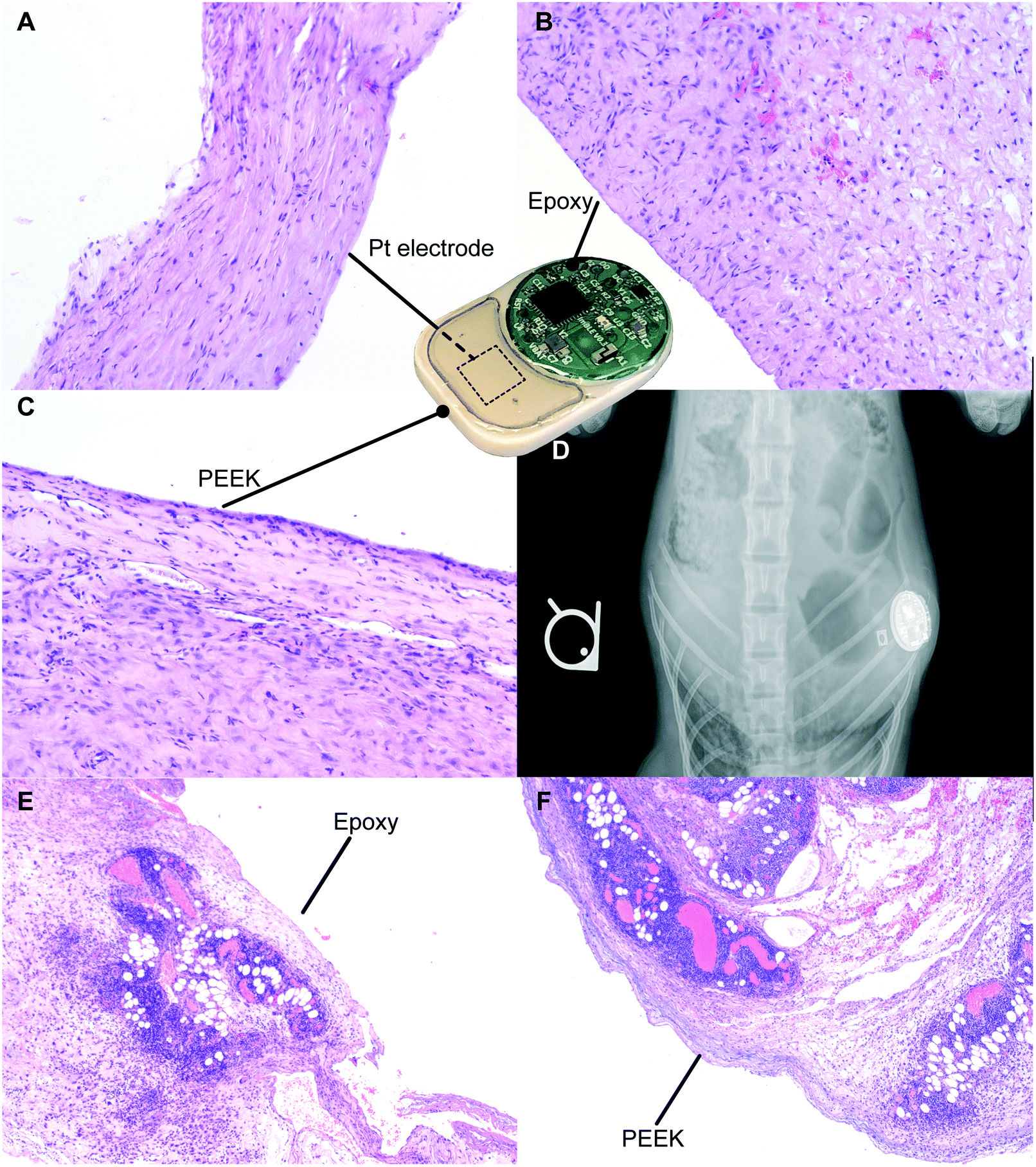 | ||
| Fig. 7 Biocompatibility test in rats and the non-human primate. Histology of the fibrotic capsule in contact with the platinum electrode (A), epoxy (B) and PEEK (C) in rats. (D) Post-surgery X-ray image of the implant to assess implant positioning. Histology of the surrounding tissues adjacent to the PEEK (F) and epoxy (E) sides of the implant in the non-human primate. | ||
In the NHP, X-ray imaging performed after the implantation surgery demonstrated implant positioning with the visible circuitry chamber and Pt electrodes (Fig. 7D). Histological assessment of tissues adjacent to the epoxy (Fig. 7E) and PEEK (Fig. 7F) sides of the implant both showed an ongoing inflammation characterized by the presence of leukocytes and macrophages. Based on the typical foreign body response, we posit that the inflammation observed is transient and associated with the recent surgical procedure for implantation of the device. Moreover, the acute inflammatory reaction observed was possibly augmented due to the young age of the animal, which typically has a robust immune system. Nonetheless, we observed regions with initial development of fibrous tissues, indicative of transition into the last phase of the foreign body response.
Conclusion and future outlook
Here we presented a proof of concept study of a remotely controlled implantable device that leverages electric field induced nanofluidic phenomena for tunable drug delivery. Our ongoing investigations are dedicated to minimization of power consumption for an extended implant lifespan. With a sufficiently longer implant lifespan, we could extend the release duration of each phase to better evaluate in vivo controlled drug delivery performance. Currently in vivo drug concentration measurements of the different release phases could be easily obscured by the drug pharmacokinetic tail, normal physiological fluctuation, or inherent animal to animal variability.While we anticipate that technological innovations of high-energy density batteries could soon be helpful, we are exploring alternative ways to control the applied electric field. In particular, we are investigating the application of a gate potential to modulate the EDL in the nanochannels as means to modify the release of drugs. With the development of a properly isolated gate electrode, electrostatic gating could in fact offer extremely low power consumption.34,42,43
Although further developments are needed, this proof of concept study sets the foundation for a generation of nanofluidic implants for tunable drug delivery. The ability to control and remotely administer therapy offers a feasible approach for precision medicine, chronotherapy or telemedicine purposes. More than that, remote-controlled drug delivery could increase patient compliance, in scenarios where conventional administration is painful or inconvenient, specifically for children, elderly or disabled individuals. Moreover, the potential of our technology resides in its versatility, in which numerous types of drugs can be released, allowing for treatment for a vast spectrum of pathologies. Future development could entail integration with other implantable sensors that could trigger drug release creating a close-feedback-loop to achieve the desired therapy.
Author contribution
NDT: conceptualization, data curation, formal analysis, investigation, software, visualization, and writing – original draft. AS: formal analysis, visualization, and writing – original draft. GB: conceptualization, data curation, investigation, and software. TG: conceptualization, data curation, and investigation. CYXC: validation and writing – original draft. AprilG: investigation. GR: investigation. CSF: conceptualization and formal analysis. DD: conceptualization. AlessandroG: conceptualization, funding acquisition, project administration, and validation.Conflicts of interest
All the authors declare no conflicts of interest.Acknowledgements
The membranes were provided by NanoMedical Systems. Funding support from the Houston Methodist Research Institute, NIH R21GM111544, NIH-NIGMS R01GM127558.References
- I. Poureslami, L. Nimmon, I. Rootman and M. J. Fitzgerald, Health Promot. Int., 2016, 32, 743–754 Search PubMed.
- W. Raghupathi and V. Raghupathi, Int. J. Environ. Res. Public Health, 2018, 15, 431 CrossRef PubMed.
- R. Reynolds, S. Dennis, I. Hasan, J. Slewa, W. Chen, D. Tian, S. Bobba and N. Zwar, BMC Fam. Pract., 2018, 19, 11 CrossRef PubMed.
- L. Peeples, Nature, 2018, 556, 290–292 CrossRef CAS PubMed.
- A. Kumar and J. Pillai, in Nanostructures for the Engineering of Cells, Tissues and Organs, ed. A. M. Grumezescu, William Andrew Publishing, 2018, pp. 473–511 Search PubMed.
- G. Tiwari, R. Tiwari, B. Sriwastawa, L. Bhati, S. Pandey, P. Pandey and S. K. Bannerjee, Int. J. Pharm. Invest., 2012, 2, 2–11 CrossRef PubMed.
- F. C. Bartter, C. S. Delea, W. Baker, F. Halberg and J. K. Lee, Chronobiologia, 1976, 3, 199–213 CAS.
- S.-Y. Lin and Y. Kawashima, J. Controlled Release, 2012, 157, 331–353 CrossRef CAS PubMed.
- G. Kaur, C. Phillips, K. Wong and B. Saini, Int. J. Clin. Pharm., 2013, 35, 344–358 CrossRef PubMed.
- F. Lévi, Lancet Oncol., 2001, 2, 307–315 CrossRef.
- F. P. Pons-Faudoa, A. Ballerini, J. Sakamoto and A. Grattoni, Biomed. Microdevices, 2019, 21, 47 CrossRef PubMed.
- A. Santos, M. S. Aw, M. Bariana, T. Kumeria, Y. Wang and D. Losic, J. Mater. Chem. B, 2014, 2, 6157–6182 RSC.
- L. Claes and A. Ignatius, Chirality, 2002, 73, 990–996 CAS.
- H. Tian, Z. Tang, X. Zhuang, X. Chen and X. Jing, Prog. Polym. Sci., 2012, 37, 237–280 CrossRef CAS.
- G. V. Gill, D. J. Husband, P. D. Wright, G. Sharpe, R. Taylor, S. Walford, S. M. Marshall and K. G. Alberti, Diabetes Res., 1986, 3, 135–137 CAS.
- B. Yue, R. Brendel, A. Lukitsch, T. Prentice and B. Doty, Neuromodulation, 2017, 20, 397–404 CrossRef PubMed.
- A. Michael, E. Buffen, R. Rauck, W. Anderson, M. McGirt and H. V. Mendenhall, Pain Med., 2012, 13, 175–184 CrossRef PubMed.
- B. P. Timko, M. Arruebo, S. A. Shankarappa, J. B. McAlvin, O. S. Okonkwo, B. Mizrahi, C. F. Stefanescu, L. Gomez, J. Zhu, A. Zhu, J. Santamaria, R. Langer and D. S. Kohane, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1349–1354 CrossRef CAS PubMed.
- T. Hoare, B. P. Timko, J. Santamaria, G. F. Goya, S. Irusta, S. Lau, C. F. Stefanescu, D. Lin, R. Langer and D. S. Kohane, Nano Lett., 2011, 11, 1395–1400 CrossRef CAS PubMed.
- J. T. Santini, M. J. Cima and R. Langer, Nature, 1999, 397, 335–338 CrossRef CAS PubMed.
- M. Staples, K. Daniel, M. J. Cima and R. Langer, Pharm. Res., 2006, 23, 847–863 CrossRef CAS PubMed.
- R. Farra, N. F. Sheppard, L. McCabe, R. M. Neer, J. M. Anderson, J. T. Santini, M. J. Cima and R. Langer, Sci. Transl. Med., 2012, 4, 122ra21–122ra21 Search PubMed.
- J. J. Sheehan, K. R. Reilly, D.-J. Fu and L. Alphs, Innov. Clin. Neurosci., 2012, 9, 17–23 Search PubMed.
- A. Ahmad and W. R. Garnett, Clin. Drug Invest., 2005, 25, 669–673 CrossRef CAS PubMed.
- D. Fine, A. Grattoni, S. Hosali, A. Ziemys, E. D. Rosa, J. Gill, R. Medema, L. Hudson, M. Kojic, M. Milosevic, L. B. Iii, R. Goodall, M. Ferrari and X. Liu, Lab Chip, 2010, 10, 3074–3083 RSC.
- C. Coletti, M. J. Jaroszeski, A. Pallaoro, A. M. Hoff, S. Iannotta and S. E. Saddow, Conf Proc IEEE Eng. Med. Biol. Soc., 2007, 2007, 5850–5853 CAS.
- T. Geninatti, G. Bruno, B. Barile, R. L. Hood, M. Farina, J. Schmulen, G. Canavese and A. Grattoni, Biomed. Microdevices, 2015, 17, 24 CrossRef PubMed.
- A. Grattoni, D. Fine, E. Zabre, A. Ziemys, J. Gill, Y. Mackeyev, M. A. Cheney, D. C. Danila, S. Hosali, L. J. Wilson, F. Hussain and M. Ferrari, ACS Nano, 2011, 5, 9382–9391 CrossRef PubMed.
- T. Geninatti, E. Small and A. Grattoni, Meas. Sci. Technol., 2014, 25, 027003 CrossRef.
- R. B. Schoch, J. Han and P. Renaud, Rev. Mod. Phys., 2008, 80, 839–883 CrossRef CAS.
- G. Bruno, N. Di Trani, R. L. Hood, E. Zabre, C. S. Filgueira, G. Canavese, P. Jain, Z. Smith, D. Demarchi, S. Hosali, A. Pimpinelli, M. Ferrari and A. Grattoni, Nat. Commun., 2018, 9, 1682 CrossRef PubMed.
- N. Di Trani, P. Jain, C. Y. X. Chua, J. S. Ho, G. Bruno, A. Susnjar, F. P. Pons-Faudoa, A. Sizovs, R. L. Hood, Z. W. Smith, A. Ballerini, C. S. Filgueira and A. Grattoni, Nanomedicine, 2019, 16, 1–9 CrossRef CAS PubMed.
- A. Plecis, R. B. Schoch and P. Renaud, Nano Lett., 2005, 5, 1147–1155 CrossRef CAS PubMed.
- S. Kim, E. I. Ozalp, M. Darwish and J. A. Weldon, Nanoscale, 2018, 10, 20740–20747 RSC.
- G. Bruno, T. Geninatti, R. L. Hood, D. Fine, G. Scorrano, J. Schmulen, S. Hosali, M. Ferrari and A. Grattoni, Nanoscale, 2015, 7, 5240–5248 RSC.
- Y. Eckstein, G. Yossifon, A. Seifert and T. Miloh, J. Colloid Interface Sci., 2009, 338, 243–249 CrossRef CAS PubMed.
- S. M. Kurtz and J. N. Devine, Biomaterials, 2007, 28, 4845–4869 CrossRef CAS PubMed.
- C. Y. X. Chua, P. Jain, A. Susnjar, J. Rhudy, M. Folci, A. Ballerini, A. Gilbert, S. Singh, G. Bruno, C. S. Filgueira, C. Yee, E. B. Butler and A. Grattoni, J. Controlled Release, 2018, 285, 23–34 CrossRef CAS PubMed.
- D.-Y. Liu, H.-K. Lon, Y.-L. Wang, D. C. DuBois, R. R. Almon and W. J. Jusko, Biopharm. Drug Dispos., 2013, 34, 203–214 CrossRef CAS PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- M. Kastellorizios, N. Tipnis and D. J. Burgess, in Immune Responses to Biosurfaces, ed. J. D. Lambris, K. N. Ekdahl, D. Ricklin and B. Nilsson, Springer International Publishing, Cham, 2015, vol. 865, pp. 93–108 Search PubMed.
- G. Bruno, G. Canavese, X. Liu, C. S. Filgueira, A. Sacco, D. Demarchi, M. Ferrari and A. Grattoni, Nanoscale, 2016, 8, 18718–18725 RSC.
- D. Fine, A. Grattoni, E. Zabre, F. Hussein, M. Ferrari and X. Liu, Lab Chip, 2011, 11, 2526 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00394k |
| This journal is © The Royal Society of Chemistry 2019 |