
All-in-one microfluidic device for on-site diagnosis of pathogens based on an integrated continuous flow PCR and electrophoresis biochip†
Zhenqing
Li
 a,
Ruixue
Ju
a,
Shinichi
Sekine
b,
Dawei
Zhang
*a,
Songlin
Zhuang
a and
Yoshinori
Yamaguchi
*cd
a,
Ruixue
Ju
a,
Shinichi
Sekine
b,
Dawei
Zhang
*a,
Songlin
Zhuang
a and
Yoshinori
Yamaguchi
*cd
aEngineering Research Centre of Optical Instrument and System, Ministry of Education, Key Lab of Optical Instruments and Equipment for Medical Engineering, Ministry of Education, Shanghai Key Lab of Modern Optical System, University of Shanghai for Science and Technology, Shanghai, China. E-mail: dwzhang@usst.edu.cn
bDepartment of Preventive Dentistry, Graduate School of Dentistry, Osaka University, Osaka, Japan
cInstitute of Photonics and Bio-medicine (IPBM), Graduate School of Science, East China University of Science and Technology, Shanghai, China
dDepartment of Applied Physics, Graduate School of Engineering, Osaka University, Osaka, Japan. E-mail: yoshi.yamaguchi@ap.eng.osaka-u.ac.jp
First published on 27th June 2019
Abstract
Current continuous flow polymerase chain reaction (CF-PCR) microfluidic chips require external precision syringe pumps and off-line methods (e.g., electrophoresis and hybridization) to detect PCR products, resulting in complex operations and possible cross-contamination and consequently CF-PCR is still confined to laboratories. Herein, a portable all-in-one microfluidic device is fabricated for rapid diagnosis of pathogens based on an integrated CF-PCR and electrophoresis biochip. A new method was proposed for automatic sample injection into the chip which can substitute the costly external precision syringe pump. It not only achieves rapid DNA amplification and on-site PCR product detection, but also realizes automatic sample injection. As an application, three periodontal pathogens (e.g., Porphyromonas gingivalis, Treponema denticola and Tannerela forsythia) were successfully amplified in the device. Treponema denticola was amplified in as short as 2′31′′, and detection of PCR products was completed within 3′43′′. The minimum number of bacteria that can be amplified was 125 cfu per μl. The all-in-one device has the potential to be applied in point-of-care nucleic acid testing for diseases.
Introduction
Polymerase chain reaction (PCR)1 is widely employed in genomic analysis applications, e.g. infectious and hereditary diseases, and detection of various bacterial pathogens. The repeated temperature cycling for PCR is conventionally performed by inserting a tube containing the PCR chemical mixture into the heating block of the thermocycler device. Such a device usually needs about 60 min to complete amplification due to the small temperature ramp rate.Recently, various methods were proposed as alternatives to traditional PCR thermocyclers, including stationary chamber PCR chips,2 natural convection PCR,3–5 continuous flow PCR (CF-PCR),6–15 loop-mediated isothermal amplification (LAMP),16 and droplet digital PCR (ddPCR).17,18 Amongst these, a small chamber is difficult and expensive to fabricate and its size limits the sample volume. Furthermore, large evaporation at high temperatures would become a serious problem for such a small sample volume.
Although LAMP needs only one temperature zone and has high amplification efficiency, the primer design is complicated and false positives easily arise. ddPCR cannot distinguish the false positives from the positives by fluorescence. Wittwer's group developed a prototype instrument for amplification in 15–60 seconds by rapidly changing the samples between two water baths with annealing/extension temperatures of 62–76 °C and denaturation temperatures of 85–92 °C, respectively, but the system is quite massive.19 Amongst these, CF-PCR may be the most promising technology for fast analysis, high sensitivity, and low-cost PCR. However, it needs external precision syringe pumps to control the flow rate of the PCR solution and off-line electrophoresis6–11,13–15 or hybridization12 to detect PCR products. External pumps may lead to a low automation and costly CF-PCR device, and even complex and difficult operations. Meanwhile, off-line detection methods not only increase the possibility of sample cross-contamination, but also prolong the time for analysis. Thus, CF-PCR is still confined to laboratories.
To address these problems, we introduced an all-in-one portable device based on a microfluidic chip, which achieved automatic sample injection, rapid DNA amplification, and on-site PCR product detection in sequence. This device dispensed with the costly external precision syringe pump, and the pathogens can be diagnosed within a short time by adding the sample into the inlet of the chip (Scheme 1).
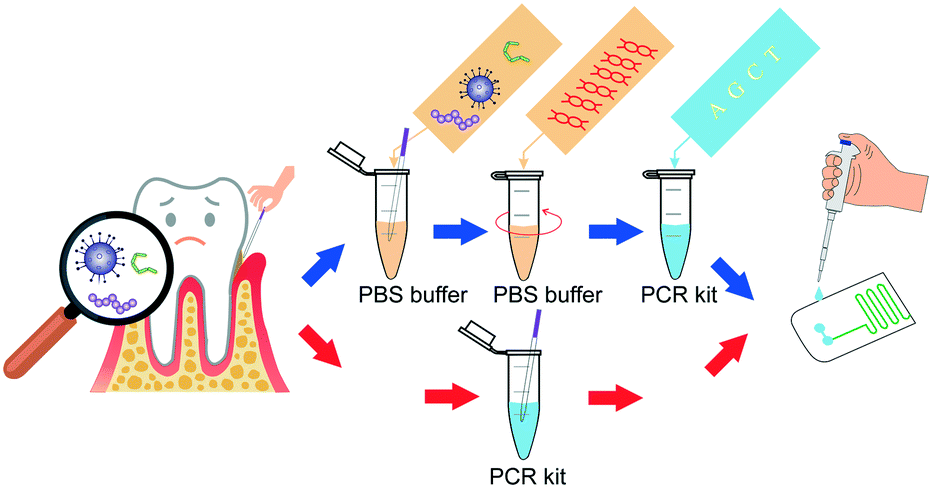 | ||
| Scheme 1 The schematic for the diagnostic process of pathogens in the all-in-one device. | ||
Materials and methods
Materials
A 100 bp DNA ladder was purchased from Takara (Shiga, Japan). Hydroxyethyl cellulose (HEC, 1300 k) was bought from Sigma-Aldrich (Saint Louis, USA). 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× SYBR Green I and 10× TBE (1× TBE = 89 mM Tris/89 mM boric acid/2 mM EDTA, pH = 8.4) buffers were bought from Solarbio (Beijing, China). Polyvinyl pyrrolidone (PVP) and Tween 20 were obtained from Aladdin (Shanghai, China).
000× SYBR Green I and 10× TBE (1× TBE = 89 mM Tris/89 mM boric acid/2 mM EDTA, pH = 8.4) buffers were bought from Solarbio (Beijing, China). Polyvinyl pyrrolidone (PVP) and Tween 20 were obtained from Aladdin (Shanghai, China).
PCR amplification of periodontal pathogens
Bacterial strains of Porphyromonas gingivalis (P.g) (ATCC 33277), Treponema denticola (T.d) (ATCC 35405) and Tannerela forsythia (T.f) (ATCC 43037) were obtained from Microbiologics Inc. The PCR solution contained 1.0 μl template, 5.0 μl 10× Fast Buffer I, 4.0 μl dNTP mixture (2.5 mM), 2 μl primer solution (10 μM forward and reverse primer) (FASMAC Co., Ltd., Kanagawa, Japan), 0.25 μl SpeedSTAR HS DNA Polymerase (5 U μl−1) (Takara, Japan), 4.0 μl PVP (0.085 mM), 2.0 μl Tween 20 with a final concentration of 0.16% (v/v), and 31.75 μl ultra-pure water. Then the PCR solution was injected into the channel for reaction with different velocities. Conventional PCR was performed in a T100 thermal cycler (Bio-Rad, USA). The thermo cycling program consisted of 40 cycles of 95 °C for 10 s (denaturation) and 64 °C for 30 s (annealing and extension) with an initial cycle of 95 °C for 2 min. For the negative control, the template was not added to the PCR solution.DNA preparations from real samples
Two hours after a meal, the gingival crevicular fluid was collected from the periodontal pocket of the upper central incisor. The volunteers were asked to clean their mouth by chewing gum. Then a sterile paper point was carefully inserted into the target site and held in place for 1.0 min. Finally, the paper point was transferred into 100 μl phosphate-buffered saline (Fluka, Switzerland) for 3.0 min, and then was centrifuged in a Chibitan-II personal centrifuge (Hitachi, Tokyo, Japan) at 10000 rpm for 10 min and the final supernatant was saved for DNA precipitation.All-in-one device construction
The system (Fig. 1a) mainly consists of an innovative pumping unit, two aluminum heaters and an integrated CF-PCR and electrophoresis microfluidic chip. The pumping unit is composed of a Z-axis lifting device, which is driven by a stepping motor. The integrated biochip was made of polycarbonate (PC), and it was fabricated by plastic injection molding. A 0.3 mm PC film was glued by ultrasonic bonding for 2.0 s to cover the PCR channel. It contained sample reservoirs (4.93 mm, internal diameter), a 94.55 cm long meandering microchannel (100 μm × 13.01 mm × 100 μm, width–length–depth), and a straight channel (1.55 mm × 41.46 mm × 2.21 mm, width–length–depth) for electrophoresis. A 100 μm thick elastic film was glued below the sample reservoirs, and thus the PCR solution was propelled into the microchip by the deformation of the film, which was induced by the stepping motor (see the ESI,† Movie S1). Between the meandering microchannel and the channel for electrophoresis, there is a 2.90 mm × 2.21 mm (ID-depth) outlet for sample collection. The microchip with 35 cycles was placed on two parallel heating blocks, which can be separately set at different temperatures for denaturation and annealing/extension by self-developed software. Fig. 1(b and c) show photographs of the microchip and the prototype system. The size of the prototype system is 20 × 14 × 7 (cm, width–length–height). The custom-made all-in-one device is shown in Fig. 1d, and the size is about 26 × 25.2 × 25 (cm, width–length–height), which includes the prototype, a cooling fan for heat dispersion, a CCD camera for imaging, and a USB interface for data transmission between the all-in-one device and the self-made software.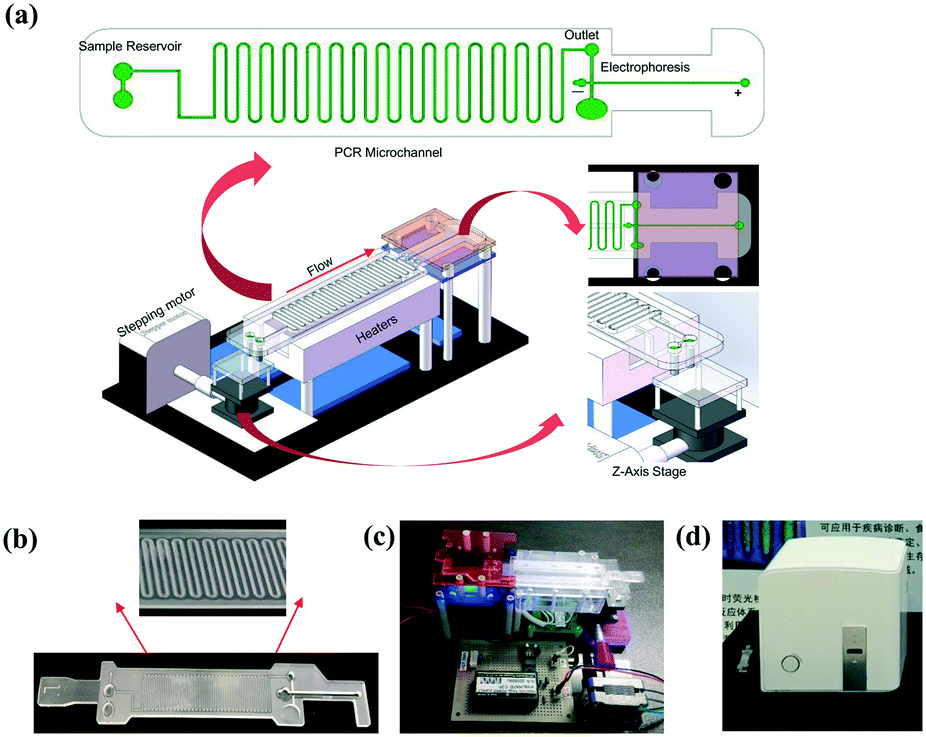 | ||
| Fig. 1 The schematic for the (a) integrated CF-PCR-electrophoresis microfluidic chip and the all-in-one device. The photos of the (b) microfluidic chip, (c) prototype system, and (d) all-in-one device. | ||
Capillary electrophoresis
We built a custom-made capillary electrophoresis (CE) system, which was reported elsewhere.5,20,21 The system consisted of a high-voltage power supply (MODEL 610E, TREK, Medina, NY, USA), a microscope with epi-illumination (BX51, Olympus, Tokyo, Japan), a photomultiplier tube (R928, Hamamatsu Photonics, Japan), and data acquisition and instrument control software (LabView 2016, National Instrument). A certain length fused-silica capillary (75 μm/365 μm, internal diameter/outside diameter) was covalently coated with polyacrylamide.22,23 The entire detection system was enclosed in a black box. The DNA sample was electrokinetically introduced into the capillary at 100 V cm−1 for 1.0 s. After each run, the capillary was flushed with sterilized water using a pump for 1.0 min. All separations were performed at 26 °C in a clean room controlled by an air-conditioner.Results and discussion
Temperature and flow rate measurement
To evaluate the stability of the heaters, we firstly measured two certain points on the surface of the heaters using an IR camera (Testo 865, Testo, Inc., and Germany). Fig. 2(a and b) depict time-dependent IR images, which correspond to the low and high temperatures, respectively. The temperature measured on the surface of the aluminium heaters is deemed to be the temperature inside the microchannel, since the PC film between the heaters and the microfluidic chip is only 0.3 mm thick. So, the heat can effectively transfer into the PCR solution without great loss, although the thermal conductivity of PC (0.22 W K−1 m−1) is not high. The data in Fig. 2(c and d) reveal that it took about 9 min and 15 min for the aluminium surface to reach the predetermined temperature, respectively. The temperature of the lower temperature heater surface was measured to be close to 65.63 ± 0.63 °C for the annealing and extension zone, and 90.64 ± 1.51 °C for the denaturation zone.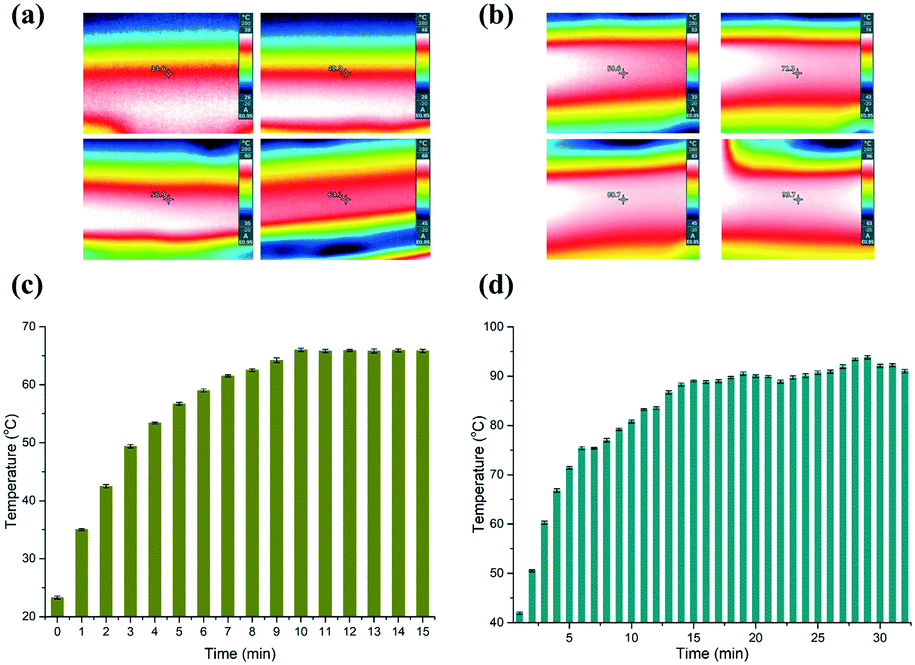 | ||
| Fig. 2 IR camera images showing the temperature at (a) the blocks of lower temperature for annealing and extension and (b) high temperature for denaturation. Time-dependent temperature profiles measured on the blocks of (c) lower temperature and (d) higher temperature. | ||
Next, we measured the velocity of the flow front in the micro-channel by changing the speed of the stepping motor. The PCR solution was driven by the deformation of the 100 μm PC film taped below the sample reservoirs. The theoretical time duration needed for the PCR solution to reach the outlet was marked with black color (see the ESI,† Fig. S1). The flow velocity was measured by tracing the position of the fluid front in the microchannel. It demonstrated that the real time duration slightly deviated from the theoretical value. The coefficients of variation (CV) corresponding to 3 min, 6 min, 9 min, 12 min, and 15 min are 7.78%, 1.94%, 1.48%, 1.67%, and 5.33%, respectively. The reason for the high CV corresponding to 3 min is possibly because the deformation of the film is not great enough to drive the PCR solution smoothly in the microchannel.
Amplification of periodontal pathogens in the chip
Gingivitis is a highly prevalent periodontal disease affecting 50–90% of adults worldwide.24P.g, T.f, and T.d were supposed to be the three major pathogens related to periodontal disease.25–27 Thus, PCR primers (Table 1) targeting the conserved regions of 16S rDNA were designed for these three periodontal pathogens, and the sizes of the amplified products for P.g, T.d, and T.f were 197 bp, 311 bp, and 641 bp, respectively. The specificity and selectivity of the primers have been evaluated in our previous work.20 The PCR solution with 35 cycles was collected using a pipettor from the outlet of the microchannel.| Target | Sequence 5′–3′ | Amplicon (bp) |
|---|---|---|
| P.g | Fw TGTAGATGACTGATGGTGAAAACC | 197 |
| Rv ACGTCATCCCCACCTTCCTC | ||
| T.d | Fw AAGGCGGTAGAGCCGCTCA | 311 |
| Rv AGCCGCTGTCGAAAAGCCCA | ||
| T.f | Fw GCGTATGTAACCTGCCCGCA | 641 |
| Rv TGCTTCAGTGTCAGTTATACCT |
The amplicons were confirmed by the self-built CE system in our lab. It showed that the PCR products of T.d were resolved within 55′′ (Fig. 3a). Moreover, the peaks of the primers were only observed from the CE of the PCR negative control (containing no DNA template) (data not shown). The data in Fig. 3a demonstrated that there was no peak when the fluid front took about 1′34′′, indicating that there were no PCR products of T.d. This is possibly because the PCR solution ran in the microchannel so quickly that the time duration for the DNA denaturation was not enough, while when the fluid front took about 2′31′′, the peak corresponding to the PCR products slightly appeared in the electropherogram. When the time of the flow front was up to 3′16′′, the peak intensity greatly increased. Furthermore, P.g and T.f were successfully amplified in this microfluidic chip (Fig. 3b). The raw data for Fig. 3 are listed in Table S1 (see the ESI†).
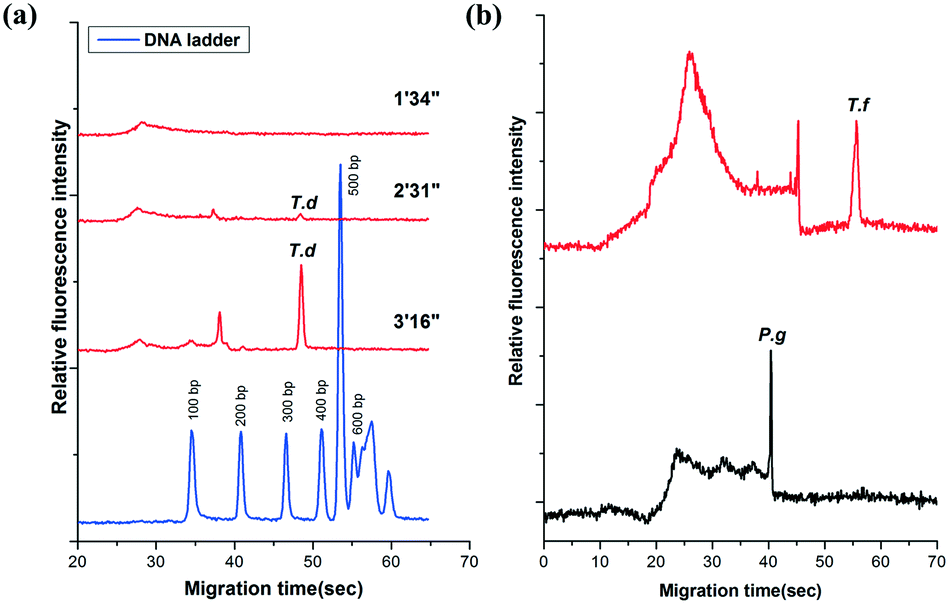 | ||
| Fig. 3 The electropherogram of (a) T.d with different CF-PCR times and a 100 bp DNA ladder for marker; (b) T.f and P.g PCR products. Capillary electrophoretic conditions: 0.5% HEC (1300 k) in 0.5× TBE solution; electric field strength: 240 V cm−1; sample injection (600 V, 1.0 s); total length and effective length of the capillary: 6.0 cm/2.0 cm. | ||
Limit of detection
To estimate the limit of detection (signal/noise = 3), we performed a series of T.d amplifications with different velocities in the microfluidic chip. The data in Fig. 4a demonstrated the relationship between the average relative peak intensities of the amplicon and the time for the flow front to reach the outlet. Results clearly showed that the amount of the PCR products dramatically increased with time. An effective fluorescence signal was measured when the time was above 2′31′′, while the relative fluorescence intensity increased by nearly 5.90 fold when the PCR solution ran in the channel for more than 3′31′′. This can be explained by the fact that the dwell time of the solution in each temperature region is sufficient for the PCR.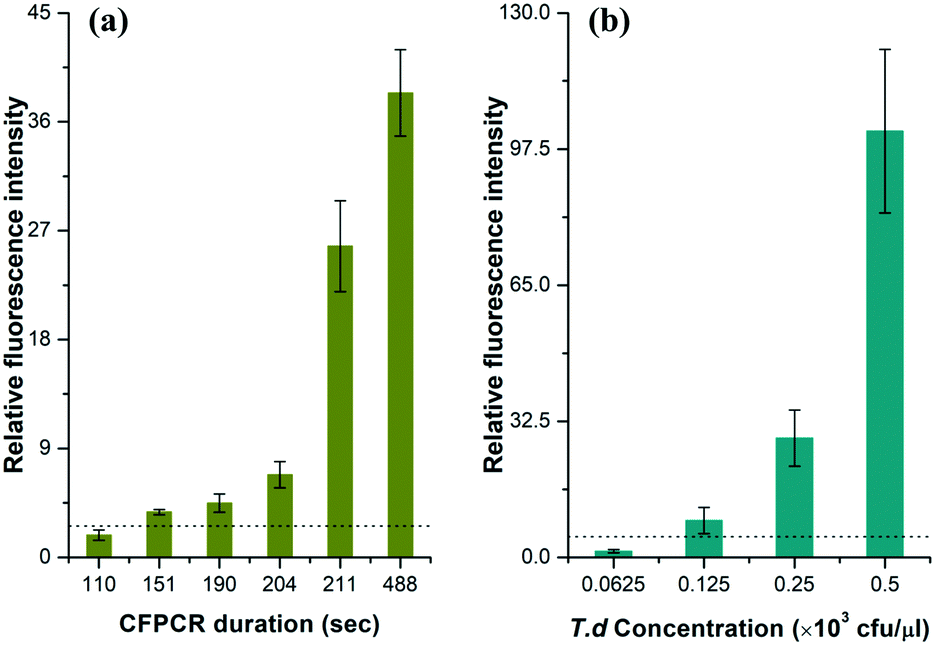 | ||
| Fig. 4 The relationship between the relative fluorescence intensity and (a) the CF-PCR duration and (b) the initial template concentration for PCR of T.d. CE conditions were the same as those in Fig. 3. The dotted line in the figure indicates that the signal/noise is 3.0. | ||
The minimum number of bacteria that can be amplified in a single reaction plays an important role in PCR. Here, we measured this indicator by diluting the same T.d template with different concentrations before mixing it with the PCR kit, and then forced the flow front to move in the channel for 20 min; finally we separated the positives by CE after PCR. The data in Fig. 4b showed that the DNA template was barely amplified when the initial bacterial concentration was lower than 62.5 cfu per μl, and the minimum number of bacteria that can be amplified was 125 cfu per μl. The raw data for Fig. 4 are listed in Tables S2 and S3 (see the ESI†).
Examination of periodontal pathogen cells from real samples
To further check the practicability of the device, we have collected the gingival crevicular fluid from a volunteer, then carried out PCR in the microfluidic chip and a conventional PCR thermocycler for comparison. The gingival crevicular fluid from the periodontal pocket was processed with and without PBS, respectively. In the conventional PCR thermocycler, the PCR was performed with 40 cycles which took about 60′1′′, while in the self-developed device, the PCR took about 14′30′′ (with PBS) and 13′20′′ (without PBS), respectively. Finally, the PCR products were directly analyzed by CE without dilution.From Fig. 5(a and b), we can find that PBS exerted no obvious effect on traditional PCR, since the peak intensity was nearly the same. This is possibly because the traditional PCR took a long time, and the cell membrane was broken by the high temperature. The peak intensity of the PCR products in the microfluidic chip (Fig. 5c) was nearly the same as that in the traditional one when the sample was treated with PBS and centrifuged in advance. In contrast, the peak intensity was evidently low (Fig. 5d) when we directly added the gingival crevicular fluid into the microchip for PCR, although the amplification time was nearly the same. The reason behind this is that the cell of the periodontal pathogen was not broken, and the DNA cannot be well mixed with the PCR kit; thus PCR for the real sample failed during such a short time. In a word, the self-developed device successfully realized the rapid amplification of periodontal pathogens from real samples. The raw data for Fig. 5 are listed in Table S4 (see the ESI†).
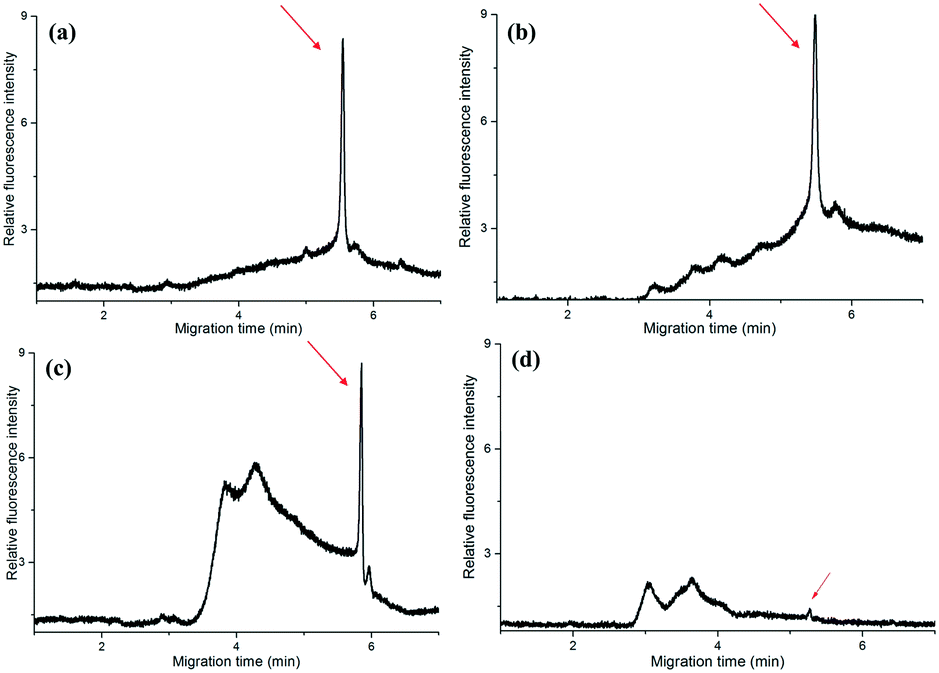 | ||
| Fig. 5 Electrophoresis of the amplicons of T.d from real samples. The gingival crevicular fluid was collected from the periodontal pocket of the upper central incisor. The fluid was processed with (a and c) and without (b and d) phosphate-buffered saline (PBS), and then PCR was performed in a traditional commercial PCR thermocycler (a and b) and the CF-PCR-electrophoresis microchip (c and d), respectively. Capillary electrophoretic conditions: 0.5% HEC (1300 k) in 0.5× TBE solution; electric field strength: 100 V cm−1; sample injection (800 V, 1.0 s); total length and effective length of the capillary: 8.0 cm/6.0 cm. | ||
On-site detection of PCR products in the microfluidic chip
To realize on-site detection of PCR products in the integrated microfluidic chip, we filled 2% agarose into the channel of the chip for electrophoresis. Two electrodes were inserted into the two ends of the channel for power supply. Below the chip, there was one LED light source, which was filtered to be 485–510 nm to match well with the central wavelength of the conjugate of DNA and SYBR Green I. And above the chip, the fluorescence emitted by the conjugate was filtered by another 515–575 nm bandpass filter, which was finally collected by a CCD.The PCR product was added to the well in agarose gel after it reached the outlet. To estimate the size of the PCR product, we firstly ran a 50 bp DNA ladder in the chip (Fig. 6a), and then we performed the electrophoresis of PCR products in the gel under the same electrophoretic conditions (Fig. 6(b and c)). It was found out that DNA ladders smaller than 500 bp were well resolved within 3′43′′. Size determination of the amplicon was based on the production of the calibration plot (Fig. 6d). Results showed that the migration distance (cm) and logarithm of DNA size had a good linearity (correlation coefficient R, 0.995) in the size range from 50 bp to 500 bp, and thus P.g (197 bp) and T.d (311 bp) were determined and marked in a red solid circle. We also performed the separation of D2000 plus (100 bp, 250 bp, 500 bp, 750 bp, 1000 bp, 1500 bp, 2000 bp, 3000 bp, and 5000 bp) and PCR products of P.g in the electrophoresis part of the chip (see the ESI,† Fig. S2). The electrophoretic conditions were as the follows: 1% agarose, 130 V, 5 min 36 s. It showed that the position of the product in the gel is between 100 bp and 250 bp.
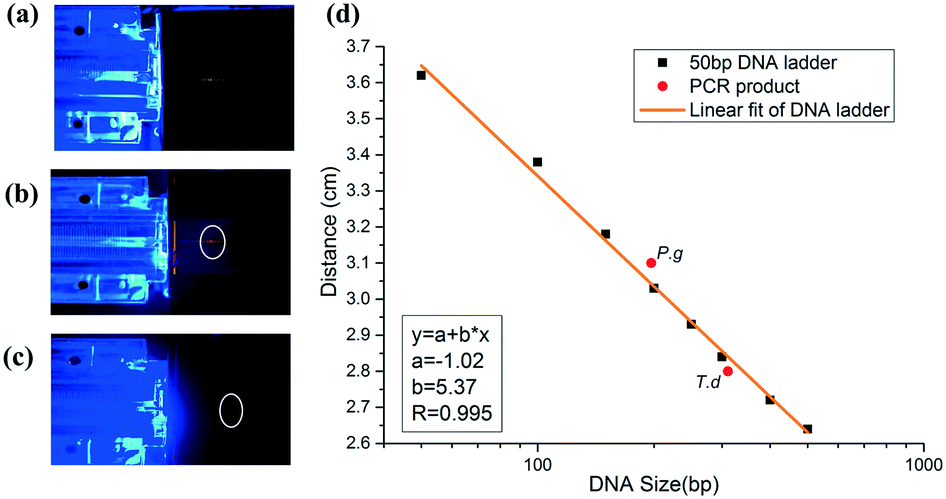 | ||
| Fig. 6 On-site detection of (a) the 50 bp DNA ladder and PCR product of (b) T.d and (c) P.g in the chip. (d) Dependence of the DNA migration distance on its size. Electrophoretic conditions: 2% agarose; electric field strength: 180 V. | ||
Moreover, we found that the fluorescence can be clearly observed (see the ESI,† Fig. S3) when the microchannel was filled with the PCR products of P.g and SYBR Green I mixture, indicating that such an all-in-one device can be easily improved for a real-time PCR instrument, which can not only distinguish the positives and the false-positives, but also measure the concentration of the PCR products.
Conclusions
We have designed and fabricated an all-in-one device, which realized automatic sample injection, rapid DNA amplification, and on-site PCR product detection in sequence. Through the proposed method for sample injection, such a device not only does away with the costly expense of the syringe pump, but also makes the CF-PCR microfluidic chip more practical. To validate the feasibility of the device, we performed amplification of 16S rDNA of periodontal pathogens and on-site detection of the PCR products. Results indicated that DNA can be amplified in as short as 2′31′′ and detection of PCR products was finished within 3′43′′, and the minimum number of bacteria that can be amplified was 125 cfu per μl. Our study therefore demonstrates the great potential of microfluidic chip-based pathogen detection, and we believe that this technology will facilitate the development of pathogen diagnosis and genetic research and advance the CF-PCR chip from the lab to real-world applications, such as point-of-care nucleic acid testing for diseases.Author contributions
Zhenqing Li: project administration, supervision, funding acquisition, and writing; Ruixue Ju: methodology and data acquisition; Shinichi Sekine: methodology and periodontal pathogen extraction; Songlin Zhuang: project administration and supervision; Dawei Zhang: project administration, supervision, and review & editing; Yoshinori Yamaguchi: data analysis, review & editing, conceptualization and supervision of the whole work.Statement
All experiments were performed in accordance with the Guidelines of Human & Animal Welfare, and approved by the ethics committee at the University of Shanghai for Science and Technology. Study participants were fully informed regarding the purposes of the study and consent was obtained.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFD0500603), Science and Technology Commission of Shanghai Municipality (China) (No.18441900400) and National Natural Science Foundation of China (No. 61775140). We also gratefully acknowledge financial support from the University of Shanghai for Science and Technology (No.2017KJFZ049).References
- R. K. Saiki, D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis and H. A. Erlich, Science, 1988, 239, 487–491 CrossRef CAS PubMed.
- N. B. Trung, M. Saito, H. Takabayashi, H. V. Pham, E. Tamiya and Y. Takamura, Sens. Actuators, B, 2010, 149, 284–290 CrossRef.
- M. Krishnan, V. M. Ugaz and M. A. Burns, Science, 2002, 298, 793–793 CrossRef PubMed.
- N. Agrawal, Y. A. Hassan and V. M. Ugaz, Angew. Chem., Int. Ed., 2007, 46, 4316–4319 CrossRef CAS.
- Z. Li, Y. Zhao, D. Zhang, S. Zhuang and Y. Yamaguchi, Sens. Actuators, B, 2016, 230, 779–784 CrossRef CAS.
- M. U. Kopp, A. J. Mello and A. Manz, Science, 1998, 280, 1046–1048 CrossRef CAS PubMed.
- Y. Schaerli, R. C. Wootton, T. Robinson, V. Stein, C. Dunsby, M. A. Neil, P. M. French, A. J. Demello, C. Abell and F. Hollfelder, Anal. Chem., 2008, 81, 302–306 CrossRef PubMed.
- K. D. Dorfman, M. Chabert, J. H. Codarbox, G. Rousseau, P. de Cremoux and J. L. Viovy, Anal. Chem., 2005, 77, 3700–3704 CrossRef CAS PubMed.
- P. J. Obeid, T. K. Christopoulos, H. J. Crabtree and C. J. Backhouse, Anal. Chem., 2003, 75, 288–295 CrossRef CAS PubMed.
- N. Park, S. Kim and J. H. Hahn, Anal. Chem., 2003, 75, 6029–6033 CrossRef CAS PubMed.
- H. Tachibana, M. Saito, K. Tsuji, K. Yamanaka, L. Q. Hoa and E. Tamiya, Sens. Actuators, B, 2015, 206, 303–310 CrossRef CAS.
- X. Jiang, N. Shao, W. Jing, S. Tao, S. Liu and G. Sui, Talanta, 2014, 122, 246–250 CrossRef CAS PubMed.
- K. T. L. Trinh and N. Y. Lee, Talanta, 2018, 176, 544–550 CrossRef CAS.
- Y. Li, C. Zhang and D. Xing, Microfluid. Nanofluid., 2011, 10, 367–380 CrossRef CAS.
- B. Shu, C. Zhang and D. Xing, Microfluid. Nanofluid., 2013, 15, 161–172 CrossRef CAS.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- C. M. Hindson, J. R. Chevillet, H. A. Briggs, E. N. Gallichotte, I. K. Ruf, B. J. Hindson, R. L. Vessella and M. Tewari, Nat. Methods, 2013, 10, 1003–1005 CrossRef CAS PubMed.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed.
- J. S. Farrar and C. T. Wittwer, Clin. Chem., 2015, 61, 145–153 CrossRef PubMed.
- Z. Li, S. Chen, C. Liu, D. Zhang, X. Dou and Y. Yamaguchi, J. Chromatogr. A, 2014, 1361, 286–290 CrossRef CAS PubMed.
- Z. Li, B. Yang, S. Sekine, S. Zhuang, D. Zhang and Y. Yamaguchi, Sens. Actuators, B, 2018, 265, 110–114 CrossRef CAS.
- S. Hjertén, J. Chromatogr. A, 1985, 347, 191–198 CrossRef.
- D. Schmalzing, C. A. Piggee, F. Foret, E. Carrilho and B. L. Karger, J. Chromatogr. A, 1993, 652, 149–159 CrossRef CAS PubMed.
- B. L. Pihlstrom, B. S. Michalowicz and N. W. Johnson, Lancet, 2005, 366, 1809–1820 CrossRef.
- J. J. Zambon, Ann. Periodontol., 1996, 1, 879–925 CrossRef CAS PubMed.
- P. J. Ezzo and C. W. Cutler, Periodontol. 2000, 2003, 32, 24–35 CrossRef.
- P. M. Loomer, Periodontol. 2000, 2004, 34, 49–56 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00305c |
| This journal is © The Royal Society of Chemistry 2019 |