
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid separation and identification of beer spoilage bacteria by inertial microfluidics and MALDI-TOF mass spectrometry
Mark R.
Condina†
 a,
Brooke A.
Dilmetz†
a,
Sajad
Razavi Bazaz
b,
Jon
Meneses
c,
Majid
Ebrahimi Warkiani
*bd and
Peter
Hoffmann
*a
a,
Brooke A.
Dilmetz†
a,
Sajad
Razavi Bazaz
b,
Jon
Meneses
c,
Majid
Ebrahimi Warkiani
*bd and
Peter
Hoffmann
*a
aFuture Industries Institute, University of South Australia, Adelaide, Australia. E-mail: Peter.Hoffmann@unisa.edu.au
bSchool of Biomedical Engineering, University of Technology Sydney, Australia. E-mail: majid.Warkiani@uts.edu.au
cCoopers Brewery Ltd., Adelaide, Australia
dInstitute of Molecular Medicine, Sechenov University, Moscow 119991, Russia
First published on 17th May 2019
Abstract
Matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF MS), in combination with Biotyper software, is a rapid, high-throughput, and accurate method for the identification of microbes. Microbial outbreaks in a brewery present a major risk for companies as it can lead to cost-intensive recalls and damage to the brand reputation. MALDI-TOF MS has been implemented into a brewery setting for quality control practices and the identification of beer spoilage microorganisms. However, the applicability of this approach is hindered by compatibility issues associated with mixed cultures, requiring the use of time-consuming selective cultivation techniques prior to identification. We propose a novel, low-cost approach based on the combination of inertial microfluidics and secondary flows in a spiral microchannel for high-throughput and efficient separation of yeasts (Saccharomyces pastorianus and Saccharomyces cerevisiae) from beer spoilage microorganisms (Lactobacillus brevis and Pediococcus damnosus). Flow rates were optimised using S. pastorianus and L. brevis, leading to separation of more than 90% of the L. brevis cells from yeast. The microorganisms were then identified to the species level using the MALDI-TOF MS platform using standard sample preparation protocols. This study shows the high-throughput and rapid separation of spoilage microorganisms (0.3–3 μm) from background yeast (5 μm) from beer, subsequent identification using MALDI Biotyper, and the potential applicability of the approach for biological control in the brewing industry.
Introduction
Biological quality control in a brewery is required to maintain high-quality beer and customer satisfaction. Beer is considered as an unfavourable environment for the growth of spoilage and pathogenic microorganisms due to associated inhibitory factors; 1) high ethanol content (0.5–10% v/v), 2) high acidity, 3) low dissolved oxygen content, 4) high carbon dioxide content, and 5) nutrient availability.1–3 Beer production processes, such as wort boiling, pasteurisation, sterile filtration, and cool temperatures also reduce the potential for growth of microorganisms.2,4 Microorganisms, including bacteria and wild yeasts, can thrive in these conditions and form undesirable flavours, aromas, hazes, and sediments.5 Contamination of beer with spoilage microorganisms can occur during all stages of brewing, from raw materials to fermentation, conditioning, and bottling stages. The most common beer spoilage microorganisms accounting for 75% of consumer complaints and spoilage incidents are the facultative anaerobic microorganisms Lactobacillus brevis (L. brevis), Lactobacillus lindneri (L. lindneri), and Pediococcus damnosus (P. damnosus).6 Lactic acid bacteria (LAB) produce lactic acid and other acids from simple sugars during fermentation, while Pediococcus infections lead to a diacetyl formation and a ‘buttery’ aroma. These microorganisms compete with yeast during fermentation for nutrients, which results in reduced ethanol content, changes in turbidity, colour, and the final organoleptic profile of the beer. Beer spoilage can lead to significant economic losses and potentially damage the brand reputation.Current microorganism detection in breweries relies on classical cultivation techniques using selective media, staining, and physiological-biochemical tests.1,7 Several molecular techniques for beer spoilage detection are available, including polymerase chain reaction (PCR),8,9 ribotyping,10,11 16S ribosome sequencing,12 and immunoassays.13 Despite these advances, traditional cultivation techniques are the most widely used in breweries due to the monetary burden and time-consuming nature of these techniques. Recently, matrix-assisted laser desorption ionisation time-of-flight mass spectrometry (MALDI-TOF MS) has emerged as a rapid and cost-effective tool for the identification of microorganisms that can be implemented in brewery settings for contamination detection.14–17 MALDI-TOF MS analysis uses highly abundant ribosomal proteins extracted from intact cells, which results in high spectral reproducibility.18 Extracted proteins are used to generate a mass spectrum profile (MSP) for the microorganism to the species level, which is subsequently matched to an MSP in a proprietary database.19 While the initial capital investment is high (around US$150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), identification of isolates can occur in minutes at a low cost per sample (typically less than US$1 per sample), which is significantly faster and cheaper than other typically used identification techniques, including traditional cultivation techniques and PCR.20,21 Using PCR for identification (the initial capital investment $US25000 (ref. 22)) typically requires a pre-enrichment step, which requires between 3–7 days, along with 2 hours for the PCR itself. For Coopers Brewery, selective plating costs US$5 per sample, while PCR costs US$4 per sample (including pre-enrichment steps). To minimise cost of analysis, PCR is only performed after growth is detected. Regular sampling of the process and final product is required to ensure potentially contaminated products are identified prior to shipment to the customer base.
000), identification of isolates can occur in minutes at a low cost per sample (typically less than US$1 per sample), which is significantly faster and cheaper than other typically used identification techniques, including traditional cultivation techniques and PCR.20,21 Using PCR for identification (the initial capital investment $US25000 (ref. 22)) typically requires a pre-enrichment step, which requires between 3–7 days, along with 2 hours for the PCR itself. For Coopers Brewery, selective plating costs US$5 per sample, while PCR costs US$4 per sample (including pre-enrichment steps). To minimise cost of analysis, PCR is only performed after growth is detected. Regular sampling of the process and final product is required to ensure potentially contaminated products are identified prior to shipment to the customer base.
Successful species level identification using the MALDI Biotyper platform requires analysis of an isolated colony, as overlapping MSPs from more than one microorganism cannot be distinguished by the software. Methods to generate an isolated colony, such as cultivation on selective media, significantly increases the time required to obtain a positive identification. This is because cultivation techniques typically require several days and can result in the distribution of beer prior to obtaining a microbial outcome.23 Developing methods that can rapidly separate spoilage microorganisms from background yeast prior to MALDI Biotyper would open the possibility to use this approach for routine final product testing. Obtaining species level identification is important to determine the most appropriate clean in place (CIP) regime, hygiene practices and understand the potential damage the spoilage microorganism can do to the beer.
Considerable literature has recently grown around the theme of microfluidics as an efficient strategy for cell separation.24–31 Generally, microfluidics is defined as the precise control and manipulation of fluid through microchannels.32 These miniaturised devices have multiple practical applications, including particle/cell separation,33 fluid mixing,34 and droplet generation.35 Among them, microfluidic separation, which is considered as a low-cost and high-throughput technique for the separation of particles and cells, has gained much more attention.36 As such, these devices can be an ideal candidate for the rapid, high-throughput separation of yeast and bacteria. Cell separation and isolation methods using microfluidics are categorized as an active or passive mode.37,38 Active technologies require electric, acoustic, or magnetic forces to allow cell separation.39 Passive technologies are broadly dependent on the intrinsic properties of the cells (e.g., size, shape, and deformability) and rely on the channel's geometry and hydrodynamic forces for cell separation.37,39 The small sample/reagent volume, rapid separation time (within minutes24), and ease of operation have resulted in these devices being successfully implemented in many applications such as cell sorting,24,30,37,40,41 cell culturing42 and cell lysis.43 Khoshmanesh et al. successfully demonstrated the applicability of microfluidics for the separation of Lactobacillus and yeast using di-electrophoresis.44 However, the requirement of di-electrophoresis on the conductivity of the media to suppress Joule heating effects and undesirable chemical reactions have hindered its high-throughput capacity and use for large-scale applications.25,45 An alternative method is needed to meet all mentioned requirements.
To date, the use of inertial microfluidic separation techniques, as a rapid and high-throughput approach, has not been applied to the separation and isolation of beer spoilage bacteria and yeast. In this work, for the first time, we demonstrate the use of inertial microfluidics in a spiral microchannel for the rapid separation and isolation of beer spoilage bacteria and yeast, followed by their identification using MALDI-TOF MS. A mixed culture of Saccharomyces pastorianus (S. pastorianus), Wyeast Kölsch, and the common beer spoilage microorganisms, L. brevis and P. damnosus, were used as model systems to assess identification of spoilage microorganisms in the presence of a background yeast in beer.
Experimental
Microfluidic channel: design and fabrication
For the separation of a yeast (S. pastorianus) from a beer spoilage microorganism (L. brevis), a spiral chip was used. This chip was fabricated via a softlithography process where PDMS pre-polymer and the curing agent (Sylgard 184 from Dow Corning, MI, USA) with the ratio (W/W) of 10:1 were mixed, degassed in a vacuum chamber for 15 min, and baked 2 h in oven at 60 °C after pouring onto an aluminum mould. The aluminum mould utilized in this study was fabricated by the milling process. Thereafter, the PDMS was peeled-off from the mold, inlets and outlets were punched, and PDMS-based microchannel was bonded onto a PDMS layer by plasma activation. For the aim of better separation and focusing of particle, a trapezoidal cross section instead of a rectangular one was used. The flow direction in this study was backward where the solution was injected from the centre of spiral. The spiral microchannel used in this study had 4 loops with a width of 400 μm and the inner and outer walls of 100 μm and 40 μm, respectively.
Microorganism cultivation and sample preparation
The facultative anaerobic beer spoilage microorganism L. brevis (DSM20054) was purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (Braunschweig, Germany). S. pastorianus (bottom fermenting yeast, Coopers lager yeast) streak plates were collected and provided by Coopers Brewery Ltd. (Adelaide, Australia). Liquid yeast Wyeast Kölsch (Wyeast 2565, top fermenting yeast, ale yeast) (Wyeast, Odell, OR, USA) and P. damnosus (LAB sold as Wyeast 5733 Pediococcus) were purchased from BeerBelly Brewing Equipment (Adelaide, Australia).L. brevis are polydisperse, with average major and minor diameters of roughly 2.8 ± 1 μm and 0.75 ± 0.25 μm, respectively. S. pastorianus are elongated and elliptical in shape, with average major and minor diameters of approximately 5 ± 0.5 μm and 4.3 ± 0.5 μm, respectively. Both L. brevis and P. damnosus (typical cell diameter between 0.36–1.43 μm (ref. 46)) were cultivated in a flask containing 50 mL of de Man, Rogosa, Sharpe (MRS; ThermoFisher Scientific, Waltham, USA) medium under anaerobic conditions at 37 °C and 27 °C with gentle agitation, respectively. Coopers S. pastorianus and Wyeast Kölsch were cultivated in a 100 ml flask containing yeast-peptone D-glucose medium (YPD) broth under aerobic conditions at 30 °C for 24 h. The YPD broth contained the following constituents: D-glucose, 2 gL−1; peptone, 22 gL−1; yeast extract, 11 gL−1.
The microfluidic device was optimised using cultures of L. brevis and S. pastorianus in phosphate buffered saline (PBS; Sigma-Aldrich, St. Louis, USA) and the optimal flow rate determined by measuring the number of yeast cells using an automated cell counter. To measure bacteria, an optical density UV3600 spectrophotometer (Shimadzu, Kyoto, Japan) was used and the values compared against a calibration curve. To generate the calibration curve, L. brevis was diluted into different concentrations (colony forming units per mL; CFU per mL). The cells were harvested by centrifugation at 8000 × g for 2 minutes and re-suspended into PBS (1 mL). The samples were then analysed using the OD spectrophotometer and a calibration curve was generated. After optimisation using L. brevis and S. pastorianus in PBS, the device was used to separate L. brevis from S. pastorianus, as well as P. damnosus (another common beer spoilage microorganism) from Wyeast Kölsch, in beer. This was done to ensure the optimisation was applicable for multiple spoilage microorganisms directly in beer as the matrix.
For the assessment of spoilage microorganism separation from yeast in beer, the cell numbers of L. brevis and P. damnosus were counted using a CFU assay on the agar plates of MRS media. The concentration of yeast cells (i.e. S. pastorianus and Wyeast Kölsch) were determined by cell counting using an automated TC20 cell counter (Bio-Rad, Hercules, USA).
Experimental setup and procedure
Experiments were first performed on separate cultures of L. brevis and S. pastorianus in PBS. For the aim of finding the best operational situation for efficient separation, flow rates of 1, 1.5 and 2.0 mL min−1 were tested. Before loading of the samples, the system was flushed with PBS (5 mL), and between samples, the system was flushed with 3% (v/v) sodium hypochlorite (5 mL) (ChemSupply, Port Adelaide, Australia) and PBS (5 mL) for sterilisation of the microchannels and tubing. For optimisation of the spoilage microorganism separation, L. brevis (9.00 × 105 CFU per mL) and S. pastorianus (2.00 × 106 cells per mL) were re-suspended in PBS. A plastic syringe (20 mL, Terumo Luer Lock, ThermoFisher Scientific) was filled with the suspension containing bacteria or yeast. The chip was connected to the syringe through the associated tubing's. A 100 legacy syringe pump (kdScientific, Holliston, USA) was used to control the flow rate and volume. The fractions were collected into 15 mL tubes and the cells harvested by centrifugation at 3260 × g for 5 minutes. To improve the separation efficiency, the inner outlet tube containing S. pastorianus was collected and subsequently separated twice more. The schematic illustration of the device is shown in Fig. 1.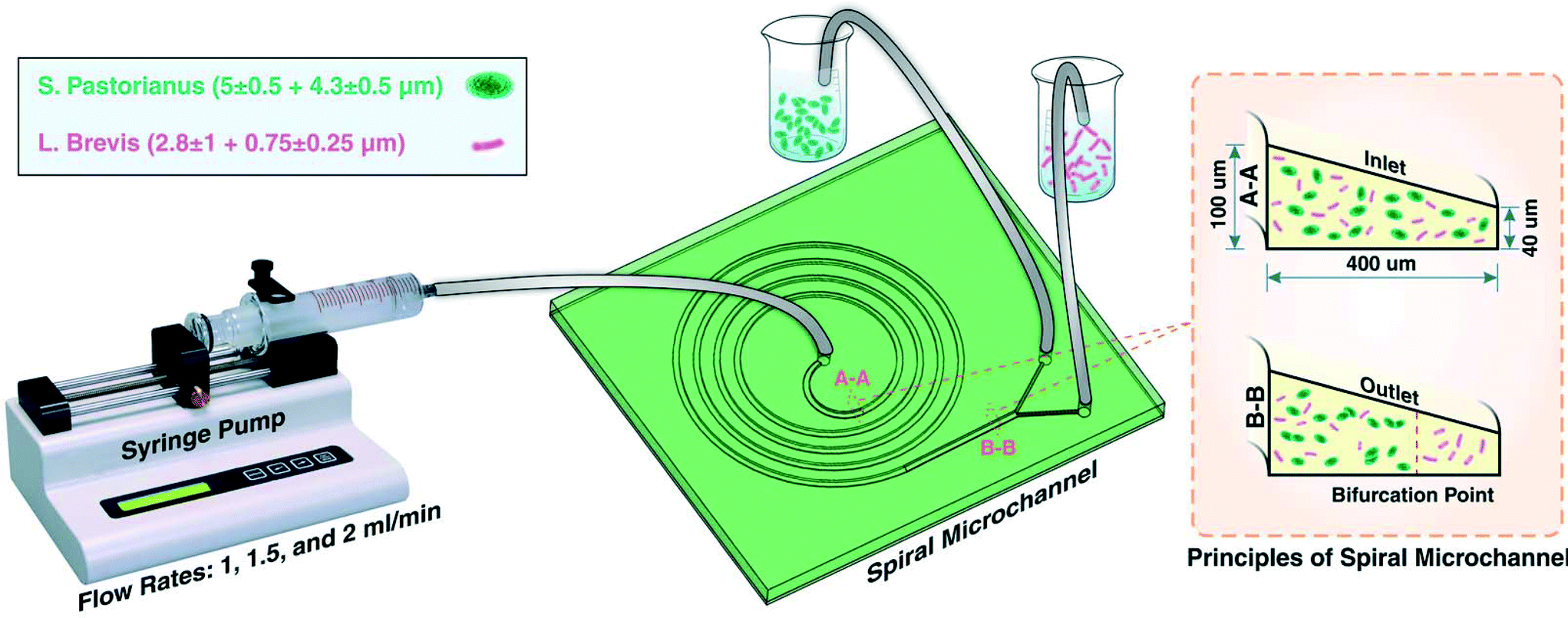 | ||
| Fig. 1 Schematic illustration of the experimental setup for S. pastorianus and L. brevis separation. At the inlet (as identified by A-A cross section), S. pastorianus and L. brevis are randomly dispersed. After inertial fractionation (as recognized by B-B cross section), S. pastorianus are focused along the inner wall of the channel, and by placing appropriate outlets at the bifurcation point, these cells can be separated. | ||
After determining the optimal flow rate, mixed cultures of L. brevis and S. pastorianus in Coopers lager beer (10 mL) were separated as described above. As for the initial experiments, S. pastorianus was suspended at 2.00 × 106 cells per mL with varying concentrations of L. brevis.
To quantify the degree of cell sorting in beer, cells before and after (inner and outer outlets) separation were harvested by centrifugation at 3260 × g for 5 minutes. The cell concentration of L. brevis was determined by plating onto MRS agar, and S. pastorianus concentration was determined by cell counting using an automated TC20 cell counter (Bio-Rad). The performance of the microfluidic device and its efficiency at cell sorting was determined by the percentage of L. brevis focusing to the outer outlet as well as the percentage of S. pastorianus focusing to the inner outlet.
To determine the efficiency of separation of L. brevis from S. pastorianus and P. damnosus from Wyeast Kölsch, mixed cultures were prepared in Coopers premium lager (Coopers Brewery Ltd.) and Coopers light (Coopers Brewery Ltd.), respectively. Prior to use, the Coopers beers were pre-treated with degassing for 2 hours as these were carbonated. The respective beers were spiked with S. pastorianus or Wyeast Kölsch (2 × 106 cells per mL) and L. brevis or P. damnosus colonies and subsequently separated using the microfluidic device. The cells from the inner and outer outlets were collected by centrifugation at 3260 × g for 5 minutes, and the proteins were extracted for MALDI Biotyper identification.
Protein extraction
In order to use the MALDI Biotyper MS platform for data analysis, proteins were extracted from cells in the inner and outer outlets obtained from the microfluidic device. The samples were washed three times in 75% (v/v) ethanol (400 μL) (ChemSupply) with centrifugation at 14000 × g for 2 minutes. The pellet was allowed to dry at room temperature for 5 minutes to remove residual ethanol. Cells were lysed by the addition of 70% (v/v) formic acid (LC-MS Grade; Sigma Aldrich) and mixed thoroughly by pipetting and vortexing, followed by the addition of 100% (v/v) acetonitrile (ACN) (LC-MS Grade; Merck, Darmstadt, Germany). Samples were centrifuged at 14000 × g for 2 minutes, and the cleared protein lysates (supernatant) were transferred to fresh tubes, analysed and stored at 4 °C.
MALDI-TOF MS analysis
An aliquot of the sample (1 μL) was spotted onto an MTP 384 polished steel target plate (Bruker Daltonik) and allowed to dry at room temperature. Bacterial test standard (BTS) (1 μL) in 50% (v/v) ACN (LC-MS Grade, Merck) containing 2.5% (v/v) trifluoroacetic acid (TFA) (LC-MS Grade; ThermoFisher Scientific) was spotted and used as an external calibrant and allowed to dry at room temperature. All samples were overlaid with α-cyano-4-hydroxycinnamic acid (HCCA) (1 μL) matrix (10 mg mL−1) and allowed to crystallise at room temperature. MS data was acquired on an AutoFlex Speed MALDI-TOF/TOF mass spectrometer (Bruker Daltonik) operated in linear positive mode under MALDI Biotyper 3.0 Real-time Classification (version 3.1, Bruker Daltonik) and FlexControl (version 3.4, Bruker Daltonik) software. Spectra were acquired in the mass range of 2000 to 20000 Da with variable laser power, and a total of 1200 sum spectra were collected in 40 shot steps. The sample spectra were identified against an MSP database library (5989 MSP entries including 28 additional entries of custom MSPs representing brewing yeasts and beer spoilage microorganisms). Identification scores of 2.300–3.000 indicate highly probable species identification, scores of 2.000–2.299 indicate secure genus identification and probable species identification, scores of 1.700–1.999 indicate probable genus identification, and a score of ≤1.699 indicates that the identification is not reliable.
Results
Flow rate optimisation
L. brevis is the most commonly encountered beer spoilage microorganism in the brewery and accounts for 50% of contamination outbreaks in the brewery.47 As such, the flow optimisation was completed on L. brevis, and then evaluated using the other common spoilage microorganism for beer, P. damnosus. The ability to rapidly isolate and detect L. brevis in beer is imperative to prevent the distribution of contaminated beer. For this aim, inertial microfluidics within a spiral microchannel was used as a high-throughput approach for particle/cell isolation.48 Particles and cells in a spiral microchannel experience two major forces which are inertial lift force and drag force, each of which can be calculated by eqn (1) and (2).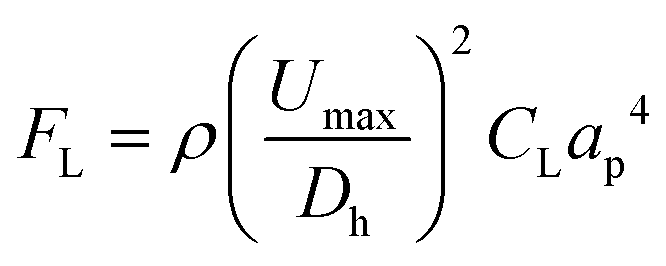 | (1) |
| Fd = 5.4 × 10−4πμDe1.63ap | (2) |
Contrarily, small particles were more influenced by drag forces generated by Dean flow and small particles were dispersed in the microchannel. Hydraulic diameter of the microchannel as well as confinement ratio (CR), which is the ratio between particle diameter to hydraulic diameter of the channel, play a crucial role in focusing behaviour of particles.49 The microchannel used in this study was carefully designed which enjoyed trapezoidal cross section, where the inner wall (100 μm) was bigger than the outer wall (40 μm) with width of 400 μm. This device was chosen to account for the small difference in size observed between S. pastorianus and L. brevis (Fig. 2A).
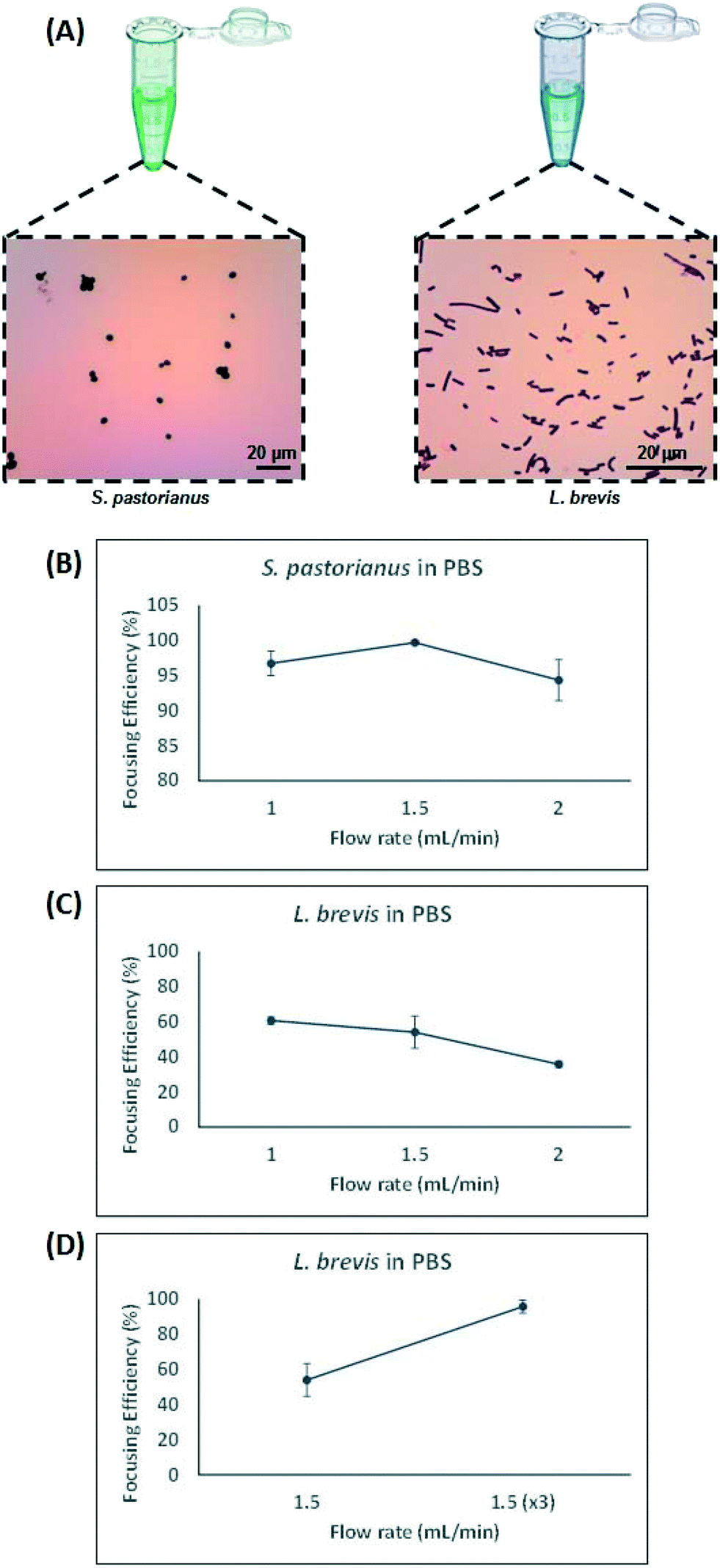 | ||
| Fig. 2 Optimisation of separation for S. pastorianus and L. brevis. A) Micrographs of gram-stained S. pastorianus and L. brevis cells. B) Results for flow rate optimization of S. pastorianus separation using cell counts and L. brevis based on OD measurements (C and D). The optimal fractionation was found to occur at 1.5 mL per min for S. pastorianus and L. brevis. All values were analysed in biological triplicate. | ||
In this study, larger particles (S. pastorianus) was focused at the inner wall while smaller particles (L. brevis) were dispersed in the microchannel. In addition, in order to dedicate extra space to cells to focus at the inner wall, we designed the inner wall bigger than the outer wall. To determine the flow rate (Q) that results in the efficient focussing of these microorganisms, preliminary experiments assessed S. pastorianus and L. brevis at 2 × 106 cells per mL and 9 × 105 CFU per mL in PBS, respectively. Samples were separated at either Q = 1.0, 1.5 or 2 mL min−1. The focussing efficiency is the percentage of cells exiting the inner and outer outlet divided by the total number of cells exiting the device. For S. pastorianus, the focussing efficiency (%) to the inner outlet was based on cell counts and was above 90% for all flow rates (Fig. 2B). The highest efficiency was obtained at Q = 1.5 mL min−1. The focussing efficiency of L. brevis to the outer outlet was based on OD measurements and compared against a calibration curve from known CFU per mL concentrations of L. brevis in PBS. For L. brevis, the highest focussing efficiency was obtained at 1.5 mL min−1 as flow rate was increased from 1.0 to 2.0 mL min−1 (Fig. 2C). As Fig. 2C illustrates, the focussing efficiency of L. brevis was more than 50% for 1 and 1.5 mL min−1, but dropped to less than 40% for 2 mL min−1. To increase the focussing efficiency of L. brevis, the sample was separated three consecutive times at Q = 1.5 mL min−1 (Fig. 2D). Adopting this strategy yielded a focussing efficiency of >90%. This flow rate was subsequently used as it provided the highest focussing efficiency of S. pastorianus, which ensures the highest fractionation from potentially contaminating spoilage microorganisms. The separation strategy was then evaluated using cultures of S. pastorianus and L. brevis re-suspended in Coopers premium lager beer at 2 × 106 cells per mL and 9 × 105 CFU per mL, respectively. The samples were separated at either Q = 1.0, 1.5, 2 or 3 × 1.5 mL min−1. For S. pastorianus, the focussing efficiency (%) to the inner outlet was based on cell counts and was above 90% for all flow rates (Fig. 3A).
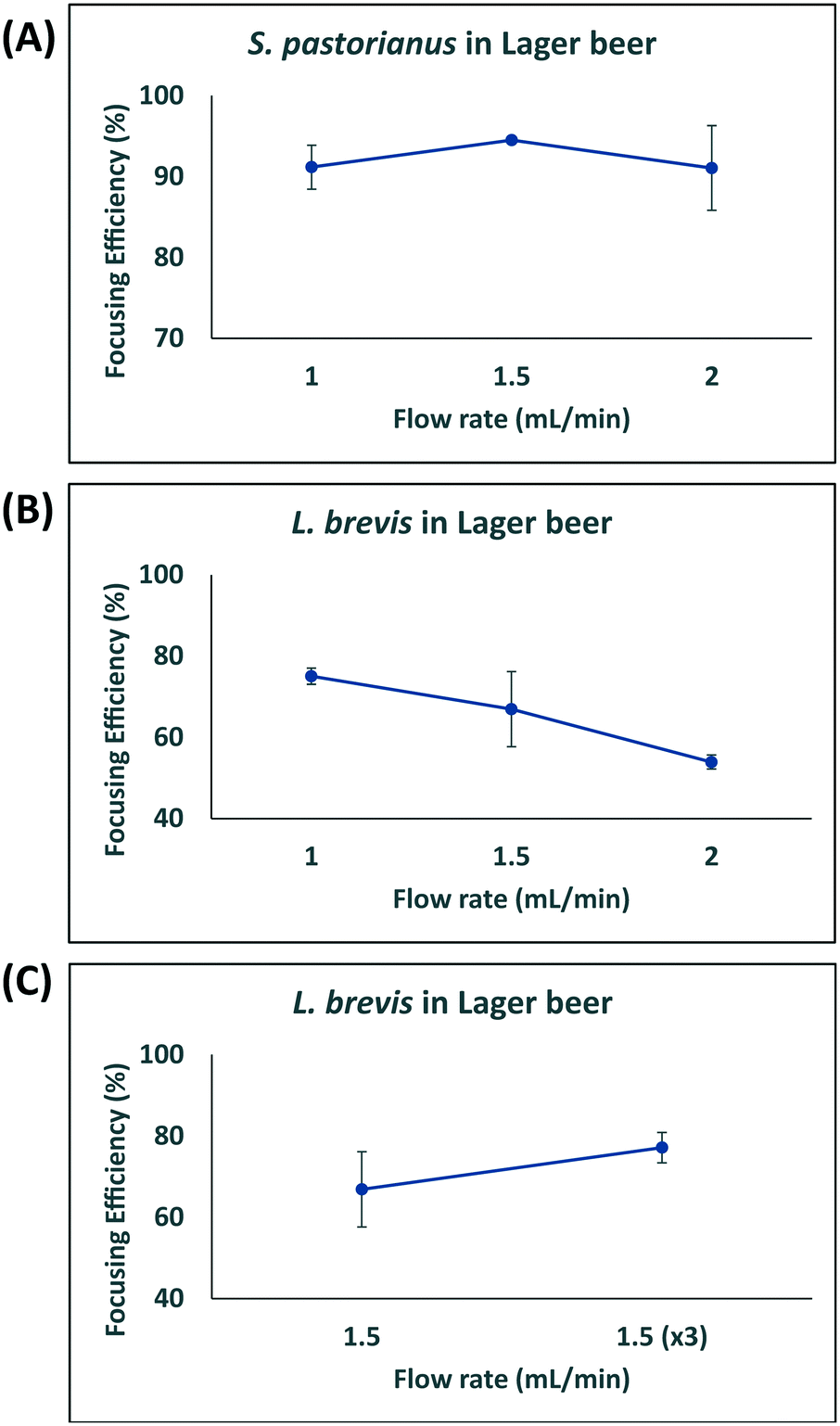 | ||
| Fig. 3 Optimisation of separation for S. pastorianus and L. brevis in beer. Results for flow rate optimization of S. pastorianus (A) separation from L. brevis (B and C) based on cell counts on YPD and MRS agar, respectively. All values were analysed in biological triplicate. | ||
The highest focussing efficiency for S. pastorianus was obtained at Q = 1.5 mL min−1. The focussing efficiency of L. brevis to the outer outlet was based on CFU counts plated onto agar and the highest focussing efficiency. As for L. brevis separation in PBS, the lowest focusing efficiency was obtained at 2.0 mL min−1 (Fig. 3B). Separation using Q = 1.5 mL min−1 3 consecutive times only slightly improved the focusing efficiency compared with Q = 1.0 or 1.5 mL min−1 (Fig. 3C). From the results obtained in PBS and beer (Fig. 2 and 3), it was decided that Q = 1.5 mL min−1 3 consecutive times should be used to ensure the separation approach can be used for multiple potential spoilage microorganisms.
Bacterial identification in beer
The MALDI Biotyper platform is currently available only for the identification of a single isolated colony. To demonstrate the applicability of using the inertial microfluidics with optimised flow rate and proper channel dimensions and cross-sections for the rapid separation of spoilage microorganisms in beer containing yeast, beer was spiked with a mix containing yeast cells with either L. brevis or P. damnosus. After microfluidic separation, the separated samples were identified using the MALDI Biotyper. The limit of detection (LOD) was defined as the ability to repeatedly obtain species level identification after separation. L. brevis, at a varying number of colonies, was mixed with S. pastorianus (2 × 106 cells per mL) into Coopers premium lager. Similarly, P. damnosus, at a varying number of colonies, was mixed with Wyeast Kölsch (2 × 106 cells per mL) into Coopers premium light. The amount of S. pastorianus and Wyeast Kölsch inoculated into the beer was based on the number of cells present in the final product of a typical ale style beer. The size of the L. brevis colonies selected was between 1–2 mm, whilst for P. damnosus colonies, those of 0.5 mm were selected. As it is illustrated in Table 1, as the number of L. brevis and P. damnosus colonies spiked into the beer per mL decreased, the ability of the Biotyper to detect these spoilage microorganisms at the species level decreased.| Beer type | Sample concentration | Biotyper ID | Biotyper score , | |
|---|---|---|---|---|
| Minimum | Maximum | |||
| a The best-matched microorganism identified using the Biotyper MSP database. b Scores for identified microorganism; threshold defined as >2.0 for species level, >1.7 for genus level and <1.7 for not reliable identification. c Three technical sum spectra were acquired for each sample and analysis was performed in biological duplicate. d Top identification of Wyeast Czech rather of Wyeast Kölsch – Biotyper cannot distinguish between them as they are the same species. | ||||
| Premium lager | 1 colony per mL | L. brevis | 2.070 | 2.218 |
| 1 colony/2 mL | L. brevis | 1.992 | 2.245 | |
| 1 colony/3 mL | Genus level yeast | 1.795 | 1.982 | |
| Premium light | 3 colonies per mL | Wyeast 5733 Pediococcus | 2.046 | 2.233 |
| 2 colonies per mL | Wyeast Czech | 1.888 | 2.069 | |
The limit of detection of the MALDI Biotyper to identify L. brevis and P. damnosus at the species level mixed with S. pastorianus in beer was determined to be 1 colony/2 mL and 3 colonies/1 mL for L. brevis and P. damnosus, respectively.
Utilising microfluidics to improve limit of detection of bacterial detection in beer
For samples containing yeast and a spoilage microorganism, which could not be successfully identified at species level using the MALDI Biotyper platform, the inertial microfluidic separation was performed prior to identification. The microorganisms isolated to the inner and outer outlets following separation were harvested and the proteins extracted prior to spotting onto the MALDI target plate in technical triplicates. The obtained spectra were subsequently searched and identified using our customised beer spoilage database, the results of which are illustrated in Table 2.| Beer type | Sample name | Outlet | Biotyper identificationa | Biotyper scoreb,c |
|---|---|---|---|---|
| a The best-matched microorganism identified using the Biotyper MSP database. b Scores for identified microorganism; threshold defined as >2.0 for species level, >1.7 for genus level and <1.7 for not reliable identification. c Three technical sum spectra were acquired for each sample and analysis was performed in biological duplicate. d Top identification of Coopers lager yeast rather of Wyeast Kölsch – Biotyper cannot distinguish between them as they are the same species. | ||||
| Premium lager | 1 colony/3 mL | Before Sep. | L. brevis | 1.781, 1.834, 1.866, 1.878, 1.898, 1.944 |
| Outer | L. brevis | 1.795, 2.058, 2.087, 2.220, 2.182, 2.386 | ||
| 1 colony/4 mL | Before Sep. | L. brevis | 1.728, 1.730, 1.735, 1.820, 1.830, 2.010 | |
| Outer | L. brevis | 1.990, 2.020, 2.020, 2.102, 2.192, 2.220 | ||
| Premium light | 1 colony/1 mL | Before Sep. | Genus level yeast | 1.775, 1.779, 1.818, 1.827, 1.902, 1.971 |
| Outer | P. damnosus | 1.777, 1.957, 2.043, 2.068, 2.164, 2.165 | ||
| 1 colony/2 mL | Before Sep. | Genus level yeast, Coopers lagerd | <0, 1.668, 1.711, 1.777, 1.853, 1.941, 2.171 | |
| Outer | Genus level yeast | 1.553, 1.572, 1.576, 1.734, 1.833, 1.942 | ||
A microbial mixture (consisting two or more organisms) leads to overlapping spectral patterns that the Biotyper software cannot distinguish, resulting in an ambiguous result. MALDI Biotyper analysis containing yeast and L. brevis resulted in identification of L. brevis to the species level at 1 colony/2 mL. The use of inertial microfluidics improved the identification such that L. brevis could be detected at the species level at 1 colony/4 mL. Similarly, for P. damnosus, the LOD for species level identification with the MALDI Biotyper decreased from 3 colonies per mL to 1 colony per mL. Representative MALDI-TOF MS spectra of the samples before and after separation (into the inner and outer channels) using microfluidics are shown in Fig. 4A and B.
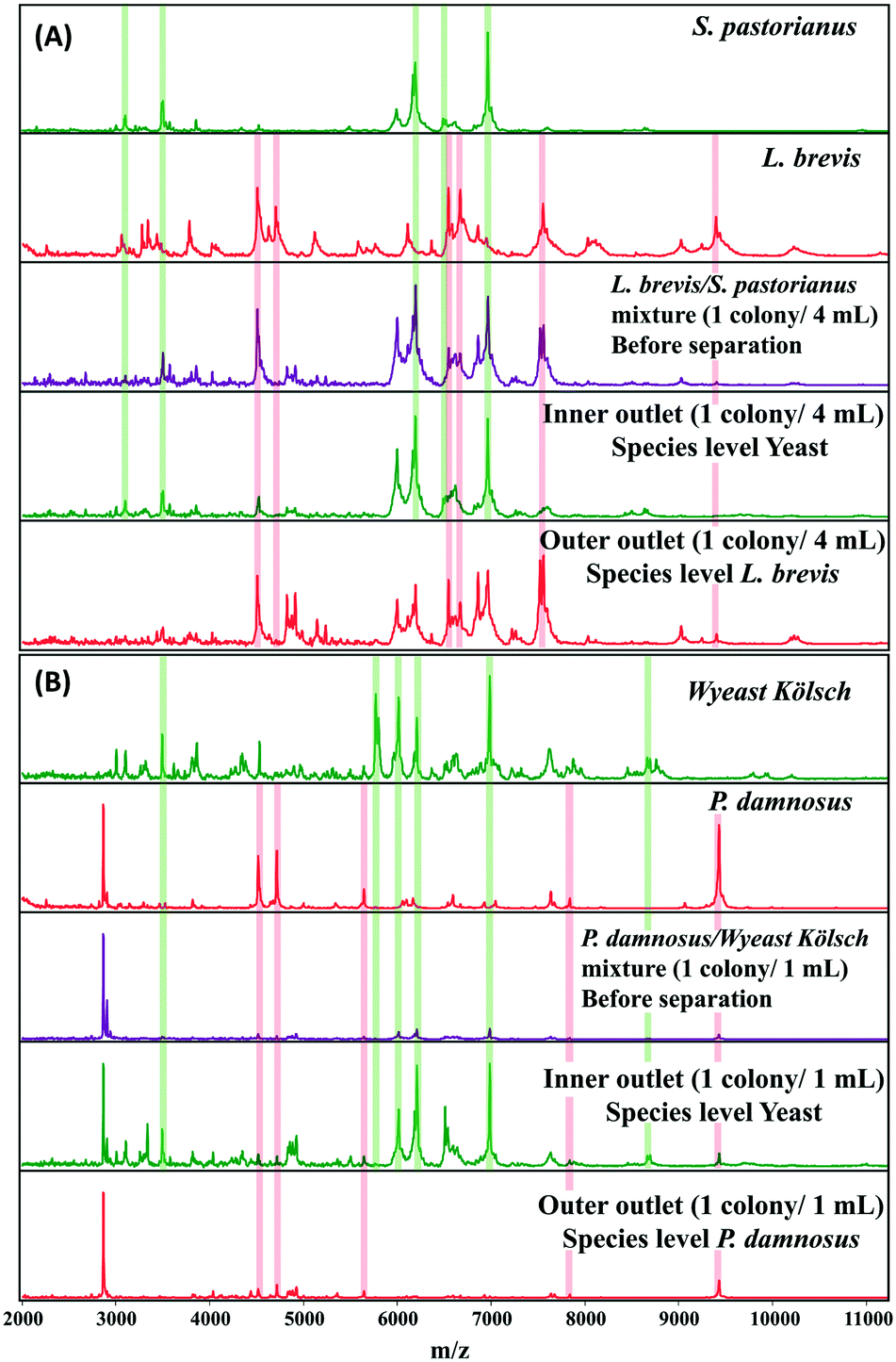 | ||
| Fig. 4 Representative MALDI-TOF MS spectra of L. brevis (4A) and P. damnosus (4B) before and after separation of background yeast in beer using inertial microfluidics. Proteins were extracted from microorganisms isolated from inner and outer outlets after separation. Sum spectra were acquired from six technical replicates, and representative spectra for microorganism before and after (inner and outer outlet) are shown. The green and red columns highlight some key peaks used for identification against the spectral database. Species level identification was obtained for L. brevis and P. damnosus from MALDI Biotyper analysis of the outer outlet. | ||
Discussion
The detection of microbial contamination in the food and beverage industry, specifically brewing, is vital for quality control purposes. Current routine microbiology quality control practices in the brewery use traditional cultivation on selective media or PCR for the identification of beer spoilage microorganisms. The monetary burden of PCR and the requirement of known primers to identify a specific and selective number of microorganisms has led to the continued use of cultivation on selective agar. Recently, the applicability of MALDI-TOF MS for routine microbial contamination identification in a local brewery was investigated and allowed the rapid, accurate, and cost-effective identification of beer spoilage microorganisms.15 Cultivation of single colonies is a pre-requisite for MALDI-TOF MS-based identification as spectra from poly-microbial cultures can lead to ambiguous results due to overlapping spectral patterns.50 The requirement for the cultivation of single colonies hinders its use in the brewery as a platform for high-throughput product testing, due to the increase in time required to cultivate an isolated colony for species level identification. As such, cost-effective and higher-throughput approaches for detection of spoilage microorganisms in the presence of yeast in beer would see a greater uptake of this platform across the industry, particularly for final product testing. To provide information regarding trace level contamination of products, the ability for quickly and near real-time monitoring is desirable to minimise delays in the product release.As highlighted in Table 1, the MALDI Biotyper approach has the capability to identify the presence of beer-spoilage bacteria at species level in the presence of background S. pastorianus, with a LOD of 1 colony/2 mL for L. brevis and 3 colonies/1 mL for P. damnosus. This was achieved without the need for any microfluidic separation. The concentration of yeast cells used was based on the typical concentration of yeast cells present in the final product of the beer samples. For bacteria, the final concentration of bacteria was placed at CFU per mL. From Coopers' experience, a concentration of 3 CFU per mL of bacteria will initiate spoilage, so the approach would require the ability to obtain species level identification at this concentration or lower. Achieving species-level identification of the spoilage microorganisms even in the presence of yeast is due to the fact that the spoilage bacteria lyse more efficiently than the yeast cells during the sample preparation for MALDI Biotyper. The rigid cell walls of yeast are difficult to break and generally requires combinations of detergents, alkaline conditions and glass beads.51 However, the sample preparation used for routine MALDI Biotyper avoids these steps to ensure compatibility with subsequent MS analysis. As such, the typical sample preparation approach is advantageous for the detection of these bacteria in final beer products that have observable and/or detectable issues. To improve detection of spoilage bacteria in beer that show no observable or detectable signs of contamination would, however, require the ability to improve the detection limit of species level identification in a cost-effective and straightforward manner.
MALDI-TOF MS analysis has been shown to be able to identify multiple beer-spoilage bacteria, highlighting the use of this platform for routine microbial quality control in the brewing industry.14 As such, developing a strategy to improve detection of these from background yeast is desirable. As highlighted in Fig. 2B and C, the use of inertial microfluidics in a spiral channel with trapezoidal cross section allows efficient focusing of the L. brevis and S. pastorianus using Q = 1.5 mL min−1. At this flow rate, S. pastorianus was focused to the inner outlet with an efficiency above 95%. For L. brevis, passing the outer component through the device 3 consecutive times improved the focusing efficiency to >95%. These preliminary analyses defined the focusing parameters for the separation of L. brevis and P. damnosus in a background of S. pastorianus and Wyeast Kölsch in beer, respectively. Using the device with beer as a matrix, a high separation efficiency for S. pastorianus was obtained at 1.5 mL min−1 (Fig. 2). For L. brevis, the separation efficiency at 1.5 mL min−1 was lower (70%) compared with the results from Fig. 3, and passing the outer component through the device 3 times did not improve this. The difference in focusing efficiency observed between Fig. 2 and 3 may be due to the effect of the matrix (beer) on focusing efficiency and/or loss of cells during focusing.
Coupling MALDI Biotyper with inertial microfluidics allows efficient separation of spoilage microorganisms in beer to improve species level identification using MALDI. This is highlighted in Table 2, where the species level LOD for two of the most common beer spoilage bacteria, L. brevis and P. damnosus, increased to 1 colony/4 mL and 1 colony per mL, respectively. In Coopers' experience, as little as 1 colony forming unit/100 mL of spoilage microorganism could lead to eventual spoilage, so improving the species level identification LOD is of critical importance. Achieving this low level of detection would require larger volumes of beer to be passed through the device, and may be coupled with a multiplexed series of microchannels for improved focusing efficiency. Species-level identification is essential to determine the potential impact on the final product as well as establish processes to minimise contamination.14 For instance, a microorganism classified as a potential spoilage (infections may take hold under specific conditions) focus efforts to deduce the cause of the conditions that allowed the growth (e.g. low alcohol content, absence of hops, elevated pH).52 This approach addresses a limiting factor for implementation of the MALDI-TOF MS for quality control, which is the need to enrich and isolate using conventional cultivation strategies.50,53,54
The use of an inertial microfluidics platform offers a solution that allows sampling of products from each fermentation batch for subsequent identification using the MALDI Biotyper, according to standard sample preparation protocols. Considering a starting volume between 10–50 mL, the time for efficient focusing and separation reduced the time frame of species level identification from 1–3 days via conventional cultivation with selective agar to less than 2 hours (usually between 20 min to 1.5 hours). The analyses performed here used two beer types, Coopers lager and light beer, which were pre-treated with degassing prior to use. The degassing is required for carbonated beer to avoid bubble formation. For routine testing, this would add an additional 2 hours to the time for analysis. However, sampling could occur prior to carbonation and this would eliminate the need for degassing. The cost of manufacture for the microfluidic chips used in this study are ∼US$4 each, which can be used multiple times. When coupled with the less than US$1 per sample for MALDI Biotyper, the workflow is a more cost effective on a cost/sample ratio than standard cultivation or PCR techniques. Furthermore, this approach does not require specific primers as per PCR for subsequent microbial detection. It is worthwhile to mentioning that the efficiency and the time required for separation can be further improved by designing a multiplex series of spiral microchannels within an automated microfluidic instrument for higher throughput and processing of larger volumes. This would further decrease the time required for sample preparation prior to identification using MALDI Biotyper of samples. The device also has the potential to be implemented in an online capacity such that the microfluidics system is attached to the production line with a multiplex series of spiral microchannels to separate yeast from the spoilage microorganisms. This would enable real-time monitoring of beer spoilage events and allow a fast response to contamination outbreaks in the brewery.
Conclusion
The present study demonstrated the ability to couple inertial microfluidics with MALDI-TOF MS for improving the limit of detection of common beer spoilage bacteria in beer samples. With a combination of inertial lift and Dean drag forces in a spiral channel with trapezoidal cross section, yeast cells migrated toward the inner wall of the outlet where the inner wall was bigger than the outer wall. Various flow rates were evaluated and flow rate of 1.5 mL min−1 was selected as the optimum one, and separation efficiency of more than 90% was achieved for S. pastorianus. Also, in order to increase separation efficiency L. brevis, it was recirculated three times thorough the spiral microchannel, leading to >90% separation efficiency. This approach improves the limit of detection for Biotyper species level identification without the need for time consuming cultivation on selective media. In summary, the combination of microfluidics separation and Biotyper identification provides a new, fast and sensitive method to detect microbial spoilage in beer which can be implemented in the quality control in breweries.Author contributions
Condina: conceptualization, methodology, investigation, data curation, writing – original draft, writing – review & editing, visualization, project administration.Dilmetz: conceptualization, methodology, investigation, data curation, writing – original draft, writing – review & editing, visualization, project administration.
Bazaz: resources, writing – original draft, writing – review & editing, visualization.
Meneses: funding acquisition, conceptualization, methodology, resources, writing – review & editing.
Warkiani: conceptualization, methodology, resources, writing – original draft, writing – review & editing, visualization.
Hoffmann: funding acquisition, conceptualization, methodology, supervision, resources, writing – original draft.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. E. W. would like to acknowledge the support of the Australian Research Council via Discovery Project Grants (DP170103704 and DP180103003). The research was supported by the Future Industries Accelerator Program.Notes and references
- W. Back, Colour Atlas and Handbook of Beverage Biology Carl Fachverlag, Hans, Nürburg, 2006 Search PubMed.
- M. K. Frank Vriesekoop, B. Hucker and G. Menz, J. Inst. Brew., 2013, 118, 335–345 CrossRef.
- K. Sakamoto and W. N. Konings, Int. J. Food Microbiol., 2003, 89, 105–124 CrossRef CAS PubMed.
- N. A. Bokulich and C. W. Bamforth, Microbiol Mol Biol Rev, 2013, 77, 157–172 CrossRef CAS PubMed.
- D. Briggs, P. A. Brookes, R. Stevens and C. A. Boulton, Brewing, Science and Practice, Woodhead Publishing, 1st edn., 2004 Search PubMed.
- K. Suzuki, in Brewing Microbiology: Managing Microbes, Ensuring Quality and Valorising Waste, ed. A. E. Hill, Woodhead Publishing, 2015, DOI:10.1016/B978-1-78242-331-7.00007-1, pp. 141–173.
- T. Takahashi, Y. Nakakita, J. Watari and K. Shinotsuka, Biosci., Biotechnol., Biochem., 2000, 64, 1032–1037 CrossRef CAS PubMed.
- J. Bergsveinson, V. Pittet and B. Ziola, Appl. Microbiol. Biotechnol., 2012, 96, 461–470 CrossRef CAS PubMed.
- R. Satokari, R. Juvonen, A. von Wright and A. Haikara, J. Food Prot., 1997, 60, 1571–1573 CrossRef CAS.
- R. Satokari, T. Mattila-Sandholm and M. L. Suihko, Microbiol. Mol. Biol. Rev., 2000, 88, 260–265 CAS.
- M. Barney, A. Volgyi, A. Navarro and D. Ryder, Appl. Environ. Microbiol., 2001, 67, 553–560 CrossRef CAS PubMed.
- K. Suzuki, K. Ozaki and H. Yamashita, J. Appl. Microbiol., 2004, 97, 712–718 CrossRef CAS PubMed.
- C. March, J. J. Manclus, A. Abad, A. Navarro and A. Montoya, J. Immunol. Methods, 2005, 303, 92–104 CrossRef CAS PubMed.
- A. D. Wieme, F. Spitaels, M. Aerts, K. De Bruyne, A. Van Landschoot and P. Vandamme, Int. J. Food Microbiol., 2014, 185, 41–50 CrossRef CAS PubMed.
- M. E. Turvey, F. Weiland, J. Meneses, N. Sterenberg and P. Hoffmann, Appl. Microbiol. Biotechnol., 2016, 100, 2761–2773 CrossRef CAS PubMed.
- H. Wang, Y. Y. Fan, T. Kudinha, Z. P. Xu, M. Xiao, L. Zhang, X. Fan, F. R. Kong and Y. C. Xu, J. Clin. Microbiol., 2016, 54, 1376–1380 CrossRef CAS PubMed.
- D. A. Wilson, S. Young, K. Timm, S. Novak-Weekley, E. M. Marlowe, N. Madisen, J. L. Lillie, N. A. Ledeboer, R. Smith, J. Hyke, C. Griego-Fullbright, P. Jim, P. A. Granato, M. L. Faron, J. Cumpio, B. W. Buchan and G. W. Procop, Am. J. Clin. Pathol., 2017, 147, 623–631 CrossRef CAS PubMed.
- V. Ryzhov and C. Fenselau, Anal. Chem., 2001, 73, 746–750 CrossRef CAS PubMed.
- T. Maier and M. Kostrzewa, Chemistry Today, 2007, vol. 25, pp. 68–71 Search PubMed.
- N. Dhiman, L. Hall, S. L. Wohlfiel, S. P. Buckwalter and N. L. Wengenack, J. Clin. Microbiol., 2011, 49, 1614–1616 CrossRef CAS PubMed.
- M. C. Ge, A. J. Kuo, K. L. Liu, Y. H. Wen, J. H. Chia, P. Y. Chang, M. H. Lee, T. L. Wu, S. C. Chang and J. J. Lu, J. Microbiol., Immunol. Infect., 2017, 50, 662–668 CrossRef CAS PubMed.
- J. M. Perkel and P. A. Fung, 2016 PCR Buyer's Guide: Proven Methodology Expanding Life Science Discoveries and Applications, https://www.biocompare.com/185163-Proven-Methodology-Expanding-Life-Science-Discoveries-and-Applications/, (accessed 05/03.2019, 2019) Search PubMed.
- M. Manzano, C. Giusto, I. Bartolomeoli, S. Buiatti and G. Comi, J. Inst. Brew., 2005, 111, 203–208 CrossRef CAS.
- X. Zhang, Z. Zhu, N. Xiang, F. Long and Z. Ni, Anal. Chem., 2018, 90, 4212–4220 CrossRef CAS PubMed.
- X. C. Xuan, J. J. Zhu and C. Church, Microfluid. Nanofluid., 2010, 9, 1–16 CrossRef.
- L. Wang and D. S. Dandy, Adv. Sci., 2017, 4, 1700153 CrossRef PubMed.
- J. Son, R. Samuel, B. K. Gale, D. T. Carrell and J. M. Hotaling, Biomicrofluidics, 2017, 11, 054106 CrossRef PubMed.
- C. W. t. Shields, C. D. Reyes and G. P. Lopez, Lab Chip, 2015, 15, 1230–1249 RSC.
- C. W. t. Shields, K. A. Ohiri, L. M. Szott and G. P. Lopez, Cytometry, Part B, 2017, 92, 115–125 CrossRef PubMed.
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P. I. Chen, B. Morgan, J. Trautwein, A. Kimura, S. Sengupta, S. L. Stott, N. M. Karabacak, T. A. Barber, J. R. Walsh, K. Smith, P. S. Spuhler, J. P. Sullivan, R. J. Lee, D. T. Ting, X. Luo, A. T. Shaw, A. Bardia, L. V. Sequist, D. N. Louis, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Sci. Transl. Med., 2013, 5, 179ra47 Search PubMed.
- J. M. Martel and M. Toner, Annu. Rev. Biomed. Eng., 2014, 16, 371–396 CrossRef CAS PubMed.
- M. Rasouli, A. A. Mehrizi, M. Goharimanesh, A. Lashkaripour and S. R. Bazaz, Chem. Eng. Process., 2018, 132, 175–186 CrossRef CAS.
- A. Kulasinghe, T. H. P. Tran, T. Blick, K. O'Byrne, E. W. Thompson, M. E. Warkiani, C. Nelson, L. Kenny and C. Punyadeera, Sci. Rep., 2017, 7, 42517 CrossRef CAS PubMed.
- S. R. Bazaz, A. A. Mehrizi, S. Ghorbani, S. Vasilescu, M. Asadnia and M. E. Warkiani, RSC Adv., 2018, 8, 33103–33120 RSC.
- A. Lashkaripour, A. A. Mehrizi, M. Goharimanesh, M. Rasouli and S. R. Bazaz, J. Mech. Med. Biol., 2018, 18, 1850002 CrossRef.
- S. Sofela, S. Sahloul, M. Rafeie, T. Kwon, J. Han, M. E. Warkiani and Y. A. Song, Lab Chip, 2018, 18, 679–687 RSC.
- H. Ramachandraiah, H. A. Svahn and A. Russom, RSC Adv., 2017, 7, 29505–29514 RSC.
- C. W. Shields, C. D. Reyes and G. P. Lopez, Lab Chip, 2015, 15, 1230–1249 RSC.
- C. W. Shields, K. A. Ohiri, L. M. Szott and G. P. Lopez, Cytometry, Part B, 2017, 92, 115–125 CrossRef PubMed.
- J. Cruz, T. Graells, M. Wallden and K. Hjort, Lab Chip, 2019, 19, 1257–1266 RSC.
- J. Cruz, S. H. Zadeh, T. Graells, M. Andersson, J. Malmstrom, Z. G. Wu and K. Hjort, J. Micromech. Microeng., 2017, 27, 084001 CrossRef.
- T. Osaki, V. Sivathanu and R. D. Kamm, Sci. Rep., 2018, 8, 5168 CrossRef PubMed.
- C. P. Jen, J. H. Hsiao and N. A. Maslov, Sensors, 2012, 12, 347–358 CrossRef CAS PubMed.
- K. Khoshmanesh, S. Baratchi, F. J. Tovar-Lopez, S. Nahavandi, D. Wlodkowic, A. Mitchell and K. Kalantar-zadeh, Microfluid. Nanofluid., 2012, 12, 597–606 CrossRef CAS.
- M. Li and R. K. Anand, Anal. Bioanal. Chem., 2018, 410, 2499–2515 CrossRef CAS PubMed.
- H. König and J. Fröhlich, Biology of Microorganisms on Grapes, in Must and in Wine, Springer, 2nd edn., 2017 Search PubMed.
- A. D. Paradh, W. J. Mitchell and A. E. Hill, J. Inst. Brew., 2011, 117, 498–506 CrossRef CAS.
- J. Zhang, S. Yan, D. Yuan, G. Alici, N. T. Nguyen, M. E. Warkiani and W. H. Li, Lab Chip, 2016, 16, 10–34 RSC.
- N. Xiang, Z. G. Shi, W. L. Tang, D. Huang, X. J. Zhang and Z. H. Ni, RSC Adv., 2015, 5, 77264–77273 RSC.
- P. Mahe, M. Arsac, S. Chatellier, V. Monnin, N. Perrot, S. Mailler, V. Girard, M. Ramjeet, J. Surre, B. Lacroix, A. van Belkum and J. B. Veyrieras, Bioinformatics, 2014, 30, 1280–1286 CrossRef CAS PubMed.
- T. T. Zhang, J. Lei, H. J. Yang, K. Xu, R. Wang and Z. Y. Zhang, Yeast, 2011, 28, 795–798 CrossRef CAS PubMed.
- M. E. Turvey, F. Weiland, E. J. Keller and P. Hoffmann, J. Inst. Brew., 2017, 123, 373–387 CrossRef CAS.
- G. C. Conway, S. C. Smole, D. A. Sarracino, R. D. Arbeit and P. E. Leopold, J. Mol. Microbiol. Biotechnol., 2001, 3, 103–112 CAS.
- C. C. Kern, R. F. Vogel and J. Behr, Food Microbiol., 2014, 40, 18–24 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |