
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A microwell array platform to print and measure biomolecules produced by single cells†
Fikri
Abali
 a,
Joska
Broekmaat
b,
Arjan
Tibbe
b,
Richard B. M.
Schasfoort
a,
Leonie
Zeune
a and
Leon W. M. M.
Terstappen
*a
a,
Joska
Broekmaat
b,
Arjan
Tibbe
b,
Richard B. M.
Schasfoort
a,
Leonie
Zeune
a and
Leon W. M. M.
Terstappen
*a
aDepartment of Medical Cell BioPhysics, University of Twente, Hallenweg 23, Enschede, 7522 NH, The Netherlands. E-mail: l.w.m.m.terstappen@utwente.nl
bVyCAP, Deventer, The Netherlands
First published on 1st May 2019
Abstract
Here we describe a combined method to monitor the secretion of molecules produced by single cells, followed by a method to isolate the individual cells that produced these molecules. The method is based on a self-sorting microwell chip that is connected to an activated membrane that collects the produced molecules. The produced molecules are printed by diffusion in small spots onto the membrane. The location of the printed spots can be correlated to the microwell number and the cell that produced these molecules. To demonstrate the method, we used the EpCAM antibody producing hybridoma cell line VU1D9 and a genetically engineered CHO cell-line producing Her2. VU1D9 cells produced 4.6 ± 5.6 pg (mean ± SD) of EpCAM antibody per 24 h and CHO cells 6.5 ± 8.2 pg per 24 h of Herceptin antibody.
Introduction
The ability to measure biomolecules such as cytokines, hormones and antibodies secreted by individual cells over a period of time under various conditions increases our understanding of complex biological processes. Here we present a technology that measures the production of biomolecules secreted by individual cells.Monoclonal antibodies that recognize specific antigens of interest were used as therapeutic agents or as tools for biomedical research. The majority of antibodies currently used are manufactured using mammalian cell systems.1,2 To select specific cells producing the desired proteins at high rate, the cells can be subjected to cloning using various methods. Clonality is a requirement for ensuring that the produced antibody is monoclonal.3
A traditional method for establishing cell lines for antibody production is limiting dilution.4–6 Cells are diluted and seeded at one or less than one cell per well.3,7,8 Subsequently, cells are allowed to grow for several days (5–10 days) before the supernatant is screened for antibody production using enzyme linked immune sorbent assay (ELISA). The procedure of cloning is repeated until monoclonality is achieved. This results in a cell line secreting unique monoclonal antibodies.9–12 However, although this method is simple and low in costs it is labor intensive and time consuming with several rounds of sub-cloning and selection and only limited number of cells can be screened.7,8 Therefore, the assay is performed when secreted proteins reach a detectable concentration.
An improvement to this method is the use of fluorescence-activated cell sorting (FACS). FACS is a high-throughput alternative that has the capability to measure single cells.8 To detect secreted biomolecules, cells are encapsulated in gel microdroplets or affinity matrixes to capture the secreted proteins near the cell.13,14 After incubation, the immobilized protein is detected using a fluorescent-labeled antibodies.3,7,8 Positive droplets, are then selected and sorted by a flow cytometer according to their level of production.3,8 However, FACS based methods, submit the cells to high shear stress which affects cell viability, cell growth and the short period of time used to detect the secretory potential of a cell may not be highly reflective.7 Other methods make use of semisolid medium to immobilize the cells and retains the secreted antibodies near the cells (e.g. Clonepix FL).3,15 In practice, a suspension of single cells is suspended in solid medium and allowed to grow into colonies before the cell secreted antibodies reach measurable concentrations; fluorescently labeled antibody are then used to detect the cell product near the colonies. High producing colonies can then be directly picked and grown for further evaluation.15 The potential advantage of this technique is that it offers a more cell-friendly environment. However, to verify clonality, microscopic observations of each culture well are performed during the incubation period that can last several weeks. For ClonePix FL, detection of antibody from single cells is not possible and therefore monoclonality is not always ensured.
Finally, there are lab-on-a-chip based approaches that use microwells to sort single cells and measure the secreted products.16–18 Single cells are sorted into individual wells and their secreted products were captured onto glass slides coated with antibodies against the secreted proteins. However, for recovery the cells are subjected to enzymatic digestion and a micromanipulator to retrieve the cells. This makes the procedure challenging in terms of throughput since only a limited number of cells can be obtained.19
The microwell printing method we present here, overcomes many of these disadvantages. The method comprises of three simple steps i.e. 1) distribute single cells in individual wells with high cell capture efficiency,20 2) measure the molecule secretion rate and quality of the secreted product at the single cell level, 3) cells of interest can be isolated by punching and clonally expanded.
The microwell chip is composed of 6400 individual wells and each microwell contains a pore at the bottom that enables the sorting of cells into wells as well the diffusion of cell secreted proteins towards a capturing membrane.20 After an overnight incubation, the membrane is separated from the microwells. While the cells in the microwells are left in the incubator the molecules captured on the membrane are analyzed with immunofluorescence techniques. Here, we show the feasibility of the method by measuring secreted antibodies from cells sorted in the microwell chip. The secreted antibodies are spotted as microarray onto a membrane and location of the spots is correlated with the microwell number that contains the cell that secreted these molecules. Interesting cells can be isolated and retrieved and expanded into a new cell line.
Results
Single cell distribution
The microwell array comprises 6400 individual microwells contained within a surface of 8 × 8 mm. Each microwell has a height of 360 μm, a diameter of 70 μm and a total volume of 1.4 nl. The optically transparent SiN bottom contains a single pore with a diameter of 5 μm.20 The distribution process is illustrated in Fig. 1. A small negative pressure 10–20 mBar was applied across the microwells to enable a flow though the microwells. Fluidic forces drag the single cell into the microwells. The fluid exists through the pore in the bottom of the microwell. Once a cell has landed onto the pore the flow is blocked and no other cell will be forced into the same microwell. The remaining cells in the cell suspension will be forced towards the next available microwell. In general, the cell density is chosen such that between 60–90% of the microwells do contain a single cell. Fig. S1a† show a typical image of cells in the after seeding. In addition, the microwell chip can be imaged after the printing assay to identify divided cells (Fig. S1b†). Therefore, using the before after images can clearly show the amount of cells in each well which ensures monoclonality.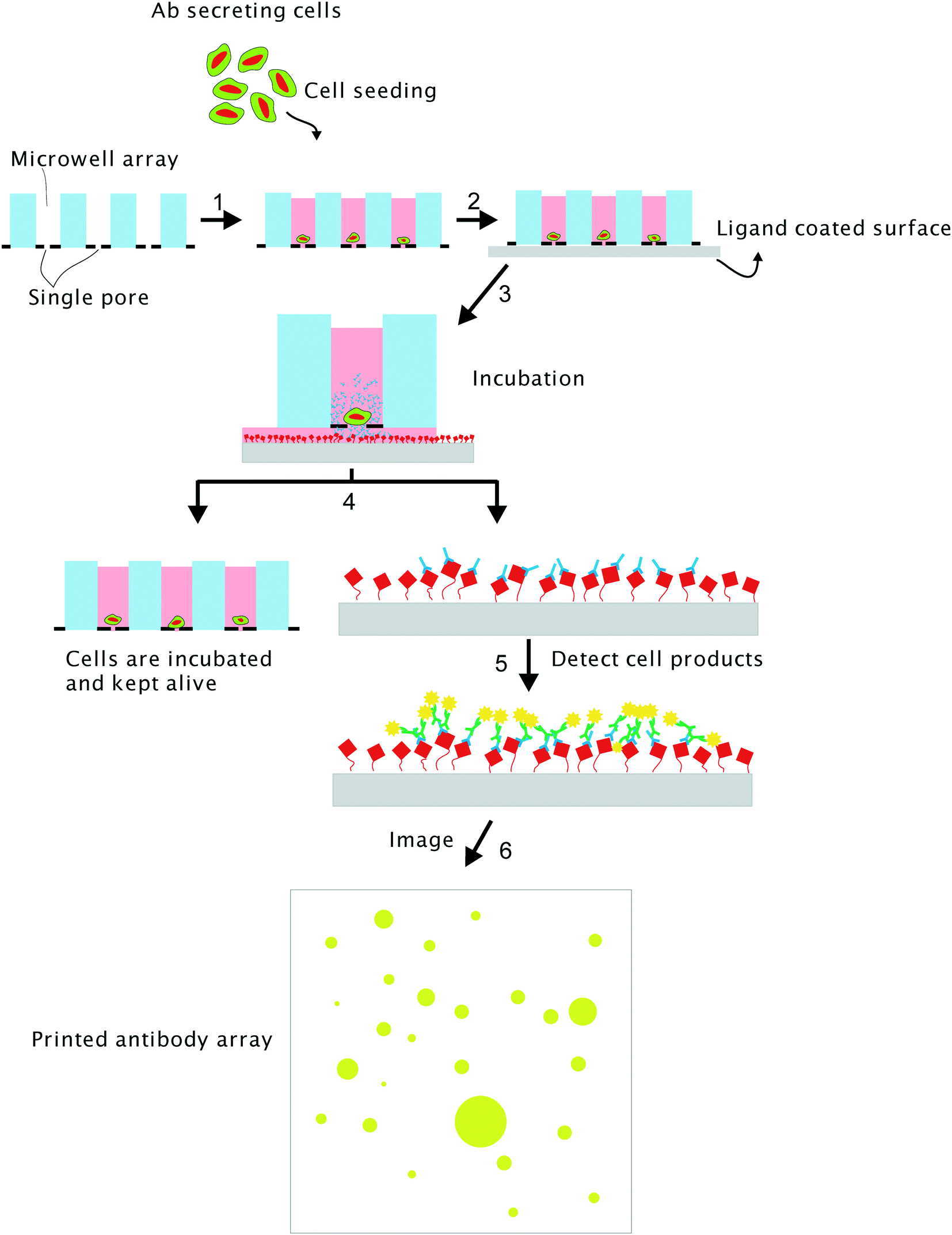 | ||
| Fig. 1 Principle of the self-sorting microwell platform to measure cell excretion. The microwell array (1 × 1 cm) contains 6400 wells each having a diameter of 70 μm with a height of 350 μm and are closed with a SiN bottom that contains a single pore with a diameter of 5 μm. After seeding the single cells are distributed in the individual wells (1), the microwells are brought in contact with a ligand coated PVDF membrane surface (2). During an incubation period, the antibodies secreted by the cell are captured on the PVDF membrane (3). The microwells are detached from the PVDF membrane and can be kept in an incubator (4), while analyzing the capturing surface (5). The antibodies secreted by the individual cells are visualized and by immunofluorescence staining and fluorescence microscopy (6). | ||
Printing antibodies
After the single cells have been distributed across the individual wells the bottoms of the microwells are connected to a capturing surface. A force of 10 N is used to establish good connection between the bottom of the microwells and the capturing surface. As a capturing surface we used a low fluorescence PVDF membrane with a thickness of 0.1–0.2 μm that is functionalized with a ligand that binds the cell secreted products of interest. While the microwells are pressed down onto the membrane the microwells with the cells are placed in an incubator under optimal cell culturing conditions. The cell secreted products diffuse through the pore in the bottom of the microwell towards the PVDF membrane (Fig. 1). The small distance between bottom of the microwells and the capturing membrane enables the capturing of antibodies in the immediate vicinity of the pore of the microwells, resulting in printed spots with a diameter of approximately 10–70 μm. To obtain nicely defined spots there should be no flow from the microwells towards the membrane and the printing should be based on diffusion only. For the used cell lines in the article, overnight incubation was sufficient to collect sufficient molecules on the capturing membrane to perform an ELISA analysis of the 6400 potential spots.Quantifying antibodies on a PVDF membrane
The feasibility of this approach was determined by using the microwells to print Herceptin antibody on protein A coated PVDF membranes using the spotting mechanism illustrated in Fig. 1. The microwells were filled with a buffer containing different concentrations of the Herceptin antibody (25, 50, 100 and 200 μg mL−1). After overnight incubation the membranes were detached from the microwell array and incubated with anti-human IgG-FITC. Fluorescence microscopy was used to acquire fluorescence images of the printed arrays. Fig. 2 presents a representative image of ±100 of the 6400 printed antibody spots. A well-defined and regular pattern of the antibody spots is obtained. The fluorescence intensity level in each spot in the printed microarray increased with increasing Herceptin antibody concentrations. To determine the variations in amount of printed antibody on the membrane, spots intensity and spot size were measured for each concentration of added Herceptin antibody. Fig. S2† shows a linear relation between added and captured amount of Herceptin (R2 = 0.93). | ||
| Fig. 2 Images of ±100 spots of the 6400 wells illustrating the printing of 25 (a), 50 (b), 100 (c) and 200 (d) μg mL−1 of Herceptin. The microwells were prefilled with Herceptin and brought into contact with a PVDF membrane coated with protein A. After detachment the membrane was stained with anti-human IgG FITC and images of the membrane were acquired by fluorescence microscopy. The images illustrate the printed antibody spots on the PVDF membrane. | ||
To determine the relation between the printed antibody concentration in pg μm−2 and the detected fluorescence intensity, the EpCAM and Herceptin antibodies were pipetted at different concentrations in drops of 1 μl on a protein A coated PVDF membrane, resulting in spots with an average diameter of 42 μm. After labeling the spots with anti-human IgG–FITC, the summed total fluorescence intensity level across every spot was determined. Panel A in Fig. 3 illustrates the obtained anti-human IgG–FITC fluorescence spots for EpCAM and Herceptin concentrations ranging from 0.78–200 μg ml−1. Panel B shows the calibration curve depicting the summed total average fluorescence FITC intensity level as detected on the microscope set-up as a function of the concentration of EpCAM and Herceptin in pg μm−2. These measurements were used to determine the amount of antibody produced by the individual cells.
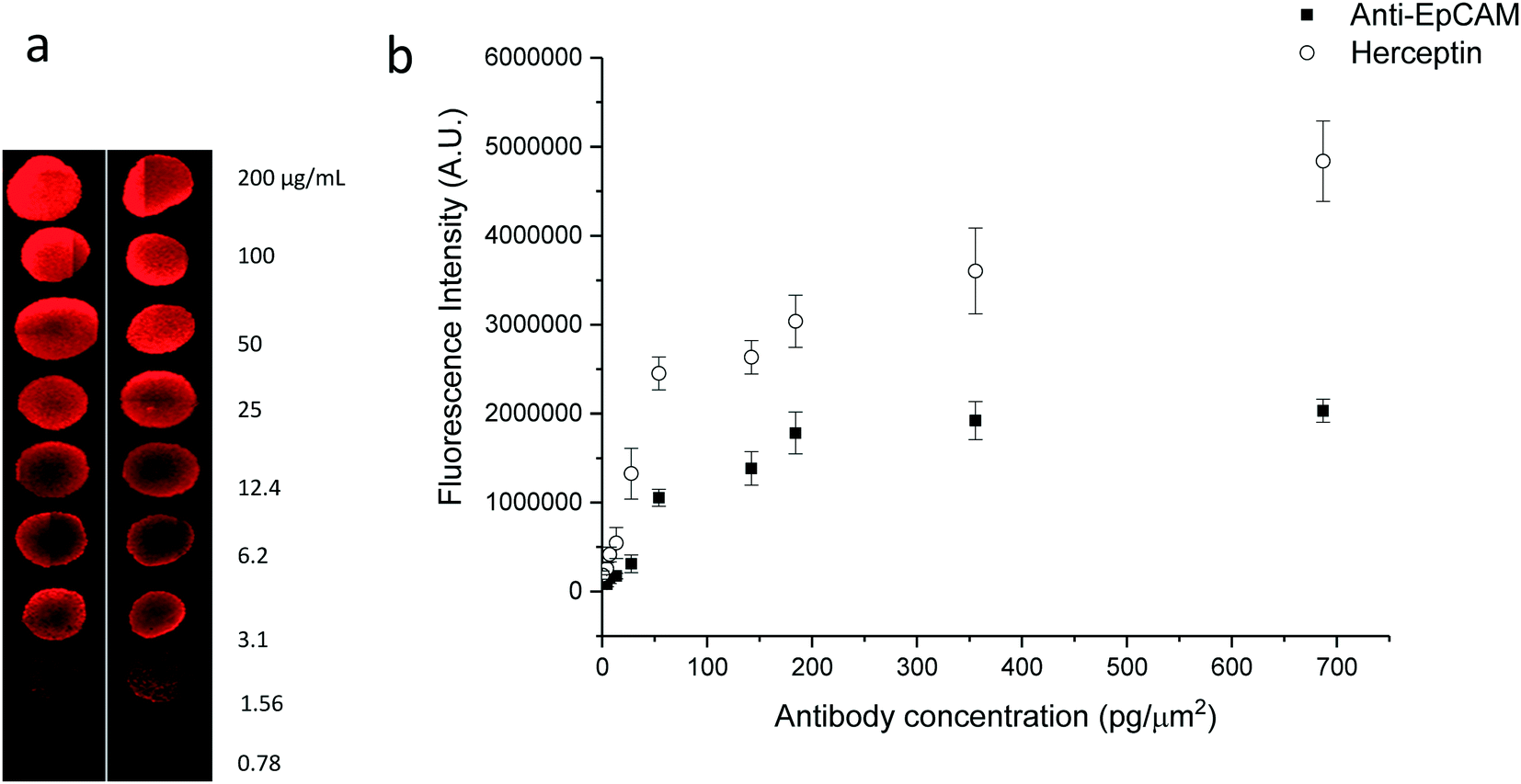 | ||
| Fig. 3 Titration of anti-EpCAM and Herceptin antibodies printed onto PVDF membranes. Panel (a). Fluorescence microscopy images of 9 concentration (in duplo) of a solution containing EpCAM antibody placed on a PVDF membrane and stained with anti-IgG-PE. Panel (b). Relation between the EpCAM (n = 5) and Herceptin (n = 5) antibody concentration in pg μm−2 and fluorescence intensity. | ||
Measurement of antibodies secreted by single cells
CHO cell line producing Herceptin and VU1D9 hybridoma cell line producing anti-EpCAM antibodies were distributed across the microwells array. The cells were labelled with CellTracker Orange to be able to visualize the cells after filling of the microwells. Fluorescence images of the cells contained in the 6400 microwells are presented in Fig. 4 panel A. In this example 3350 of the 6400 microwells (52%) contained a single CHO cell. Panel C shows a higher magnification of these microwells which clearly illustrates the location of each cell in the microwell. After an overnight incubation the PVDF membrane was detached from the microwell array, stained with anti-human IgG FITC and imaged on a fluorescent microscope, using the same fluorescence imaging setting as used for obtaining the graph of Fig. 3b. The image of the captured Herceptin antibody secreted by the cells from panel A, is presented in panel B. Panel D shows a larger magnification of the Herceptin production of the cells from panel C and panel E shows the overlay of panels C and D. | ||
| Fig. 4 (a) Fluorescence images of cells stained with CellTracker Orange from the CHO cell line producing Herceptin, distributed into the self-sorting microwells. (b) The corresponding fluorescence image of the membrane captured Herceptin labelled with anti-human IgG FITC. (c) A section of the wells from panel (a). (d) The corresponding image of Panel (b) and (e) the overlay of Panels (c) and (d). | ||
Quantification of antibody secreted by cells
The amount of captured antibodies secreted by single cells was determined by measuring the fluorescence intensities of the printed spots and quantify this with the calibration curve of Fig. 3. For these experiments a number of 2000 cells were distributed into microwells. Cells seeded directly on a membrane (without microwell) was used as a positive control for antibody secretion. The fluorescence intensity in the captured antibody spots was automatically determined using the open source image analysis software ACCEPT (open-source ACCEPT software (http://github.com/LeonieZ/ACCEPT)).21 Images of arrayed cells and their corresponding microarrays were aligned manually and ACCEPT software was used to measure the total summed fluorescence intensity and diameter of the printed spot. Microwells without cells (n = 300) were used as an internal control to determine the background level and to set the detection limit. The calibration curve in Fig. 3 was used to convert the total fluorescence intensity to captured antibody in μg μm−2. Multiplying this number times the area of the printed spot results in the amount of captured antibody produced by a single cell in pg per cell per day.The production of EpCAM by VU1D9 hybridomas and Herceptin by CHO cells was measured in a triplicate experiment on separate days. After single cell distribution, the microwells were connected to the membrane and incubated overnight. The fluorescence intensity was analyzed and converted to captured antibody in pg per cell per day. The results of these three experiments conducted with the VU1D9 and CHO cells is shown in Fig. 5a and b respectively. Each spot in the graph represents the amount of captured antibody produced by a single cell during the 24 hours of incubation. The horizontal solid lines indicate the average collected antibody production of microwells that do contain a single cell and the dashed blue line presents the average from the wells without a cell. In all three experiments we could reproducibly detect single cell antibody secretion.
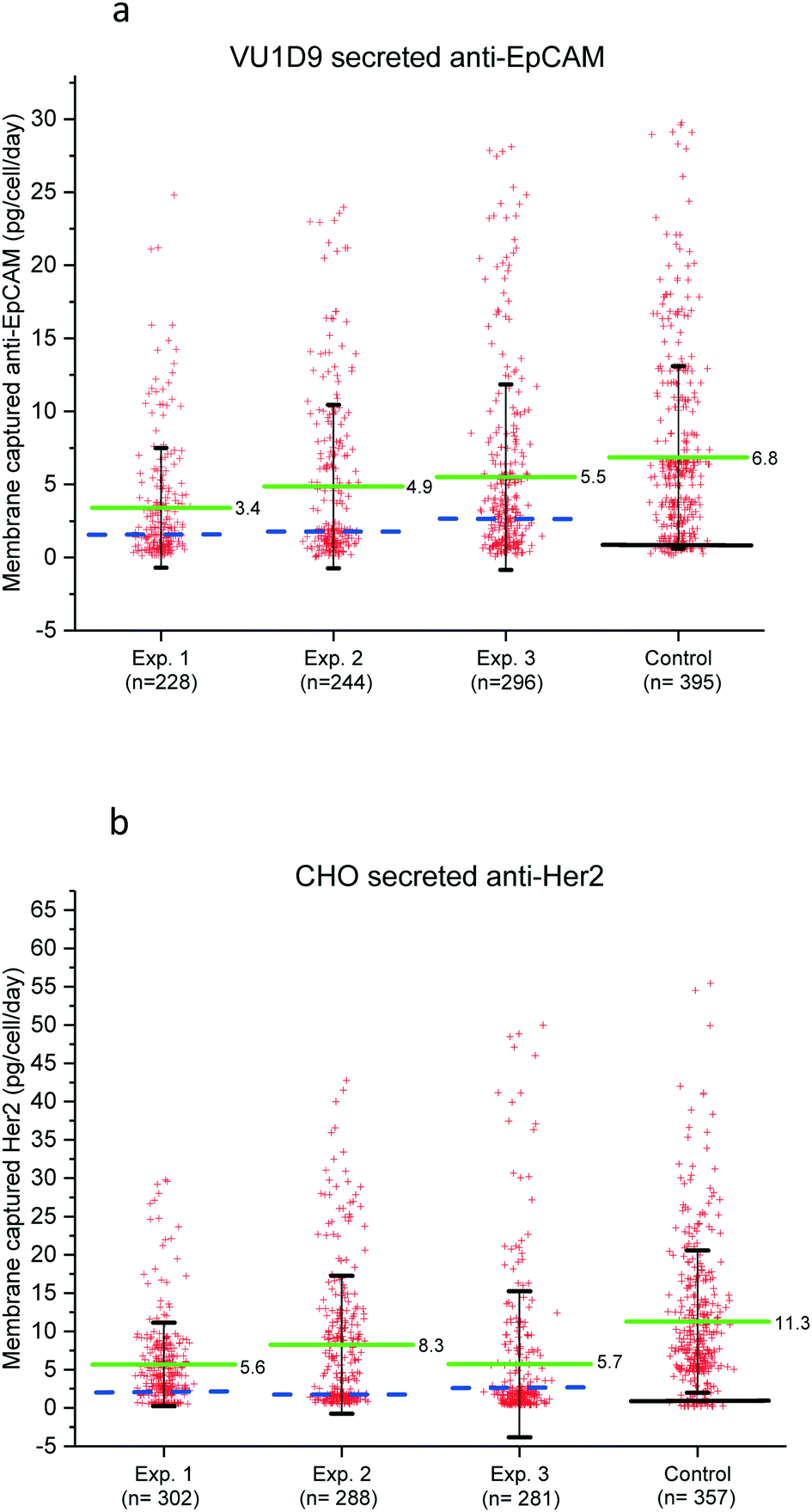 | ||
| Fig. 5 (a) Scatter plot showing the production of the anti-EpCAM antibody by the single VU1D9 cells. (b) Scatter plot showing the production of the Herceptin antibody by the single CHO cells. The scatter plots show the amount of membrane captured antibodies secreted by single cell in individual microwells for three experiments, as a control traditional FLUOROSPOT was performed. Each red cross represents the total amount of antibody secreted and produced by a single cell during 24 h of incubation. The green solid line indicates the average amount of captured antibodies with ±2SD in pg per cell per day. The dashed blue line indicates measured production rate of the empty microwells (background fluorescence). The black solid line indicates the background level of the PVDF without any fluorescence labeling. | ||
The data shows that most of the cells secrete low or non-detectable amounts of proteins and only a small fraction of the cells produce high amounts of antibodies. The high distribution in antibody secretion is also observed in the control group (traditional ELISPOT), which indicates that the observed heterogeneity in protein secretion is not caused by the microwells. The amount of membrane captured antibodies in the ELISPOT control group was on average 1,5 fold higher which is most probably due to the fact that not all secreted antibody is able to diffuse to the pore towards the capturing surface.
The VU1D9 hybridoma cells show a significant lower amount of membrane captured anti-EpCAM antibody, which indicate a lower antibody secretion rate compared to CHO cells. The CHO produced 0.4–50 pg per cell per day with an average of 6.5 pg per cell per day which equals a membrane capture rate of 272 IgG molecules per minute. For the VU1D9 cells the amount captured ranged between 0.12–24.8 pg per cell per day, with an average of 4.6 pg per cell per day corresponding to a capture rate of 194 IgG molecules per minute. The data presented here for the VU1D9 shows to be comparable to previous studies in which VU1D9 IgG secretion was measured using SPRi.22,23 Recently we have shown that cells from different cancer cell lines can be isolated from the microwells and expanded.24 In Fig. S3a and b† we demonstrate that individual CHO cells can be individually recovered from the microwell and expanded. We are pursuing experiments to show if the high antibody producing cells indeed remain high producers. For these experiments we however need to assure that sterile conditioned can be maintained over a longer time period.
Dynamics of antibody secretion
After measuring the secretion from the two cell lines, we next investigated the antibody secretion dynamics of the same cells at different time periods. CHO cells were sorted in the microwell chip and the secreted antibodies were measured at 8 h, 16 h, 24 h, 32 h, 40 h and 48 h. For each measurement a new membrane was placed under the chip to spot the secreted proteins from the same cells. These time points provide the cells enough time to secrete detectable amounts of antibodies between the different assays. Fig. 6a shows a heatmap demonstrating the secretion profile of the individual CHO cells at different time points. The cells were grouped into low, average and high producer cells based on the total amount of secreted anti-Her2 during the assay (summation of the total amount of antibody at 8–48 h period). Fig. 6b shows the secretion of CHO cells which initially started as a single secretor but proliferated during assay (at 24 h). Also, the cells were grouped into low, average and high producer cells. The heatmap clearly indicates an increase in antibody secretion for divided cells when compared to single cells. However, on average anti-Her2 secretion for each single cell show no big differences. Fig. 6c and d show typical secretion dynamics for single and proliferated cells respectively. The data indicates that the secretion profile remained steady for each time interval during the assay. Interestingly, low producer cells remained low producer, while high producer cells remained high producer during the assay.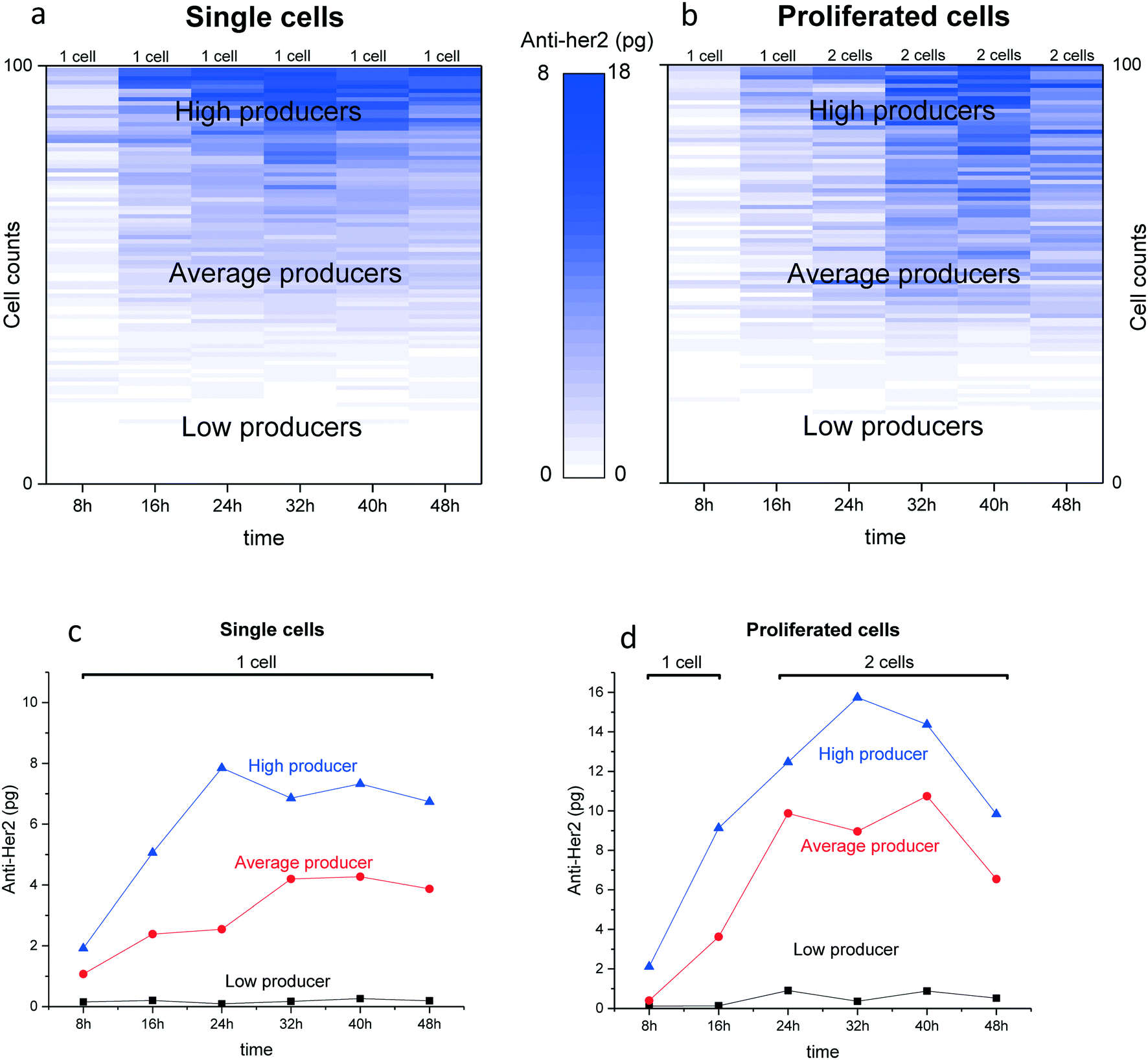 | ||
| Fig. 6 Antibody secretion profile. (a) The dynamics of single CHO cell-line secreting anti-Her2 is plotted in a heatmap, n = 100 cells for each time-point. The cells were sorted based on the total amount of Anti-Her2 antibody produced in 48 h. (b) The secretion profile of CHO cells that started as single cells in the microwells and at 24 h proliferated (2 cells, see red bar), n = 100 cells for each time point. The anti-Her2 secretion dynamics is plotted in a heatmap. The cells were sorted based on the total amount of membrane bound anti-Her2 antibody produced in 48 h. (c) A typical example illustrating the secretion profile of a high producing cell, an average producing and a low producing single CHO cell. (d) An example showing the secretion profile of a high producing, average producing and low producing CHO cells that started as a single cell in the well (1 cell) and proliferated at 24 h in two cells (2 cells) and remained as two cells throughout the experiment. | ||
Discussion
The detection of protein excretion on the single cell level has the potential to for example screen antibody secreting cells or to study cell-to-cell secretion heterogeneity in a wide variety of biological systems. The current available methods used to interrogate single cells on the single cell level are limited.16,17,25 The technology described in this paper is able to detect the secreted product of individual viable cells at different time points and if desired the cells of interest can be isolated for clonal expansion. The technology consists of hardware, software and protocols to analyze the cellular responses of single cells using a microwell chip that has 6400 microwells filled with a single cell. The sorting of single cells in individual microwells eliminates the effect of cell–cell contact with neighboring cells, and purely targets single cell secretion.To proof of concept was demonstrated by determining the antibody secretion of two model cell lines secreting antibodies against EpCAM and Her-2. Firstly, it was shown that the microwell chips can be used to print biomolecules on a capturing surface, resulting in a protein microarray. Secondly, we observed that single cells can be kept alive in the microarrays and Thirdly the molecules secreted by the cells can be captured at multiple time points on a surface and identified and quantified. The secreted molecules migrate through the small pore in the bottom of the individual microwells and are immobilized on the capturing surface. Using this approach different antibody secreting cell populations are identified with varying levels of protein secretion. The majority of the cells were identified as low or non-secretors (0–3 pg per cell per day) and only a small subpopulation of cells secreted high concentrations of antibodies (>5 pg per cell per day). Furthermore, our method has a sensitivity and specificity comparable with traditional ELISPOT but overcomes the limitation in collecting the cells of interest.
We further used our platform to track the antibody secretion dynamics of cell in microwell chip. To track the dynamics from the same cells at different time intervals, secreted antibodies were monitored by printing onto different membranes every 8 h hours (8–48 h). This preliminary data indicates that the secretion of anti-Her2 form the single CHO cells used in this study is stable during the complete assay and changes only slowly. For cells that gave rise to division in the wells an increase in antibody production was found, when compared with single cells. However, on average per cell there was no difference between divided and non-divided cells. The elevated secretion from divided cells was maintained for at least 16 h. Furthermore, it was found that based on their total secretion, CHO cells can be grouped into low, average and high producer cells. Cells that were high producer remained a high secretor also after division, while low secretors remained low after divisions. These single cells assays can give more insight in cell behavior and provide valuable information when compared to bulk assay which only provide an average signal.
These findings demonstrate the power of the developed technology to screen a library of single cells for protein secretion, phenotyping, identification and isolation. This article is focuses on the identification of single cells that produce high amounts of a specific protein. The described method can be combined with the Puncher technology as described by Swennenhuis et al.,20 to transfer the cells from the microwells to a culture plate for clonal expansion. Therefore, it would be interesting to use this method to further determine if these antibody producing cells are truly high producers. Future single cell isolation studies should show if the cells of interest remain high producer cells after isolation and clonal expansion. Currently were are, experiments are being performed where single cell antibody is measured followed by recovery from the microwells for clonal expansion.
In this paper, we have demonstrated a method to measure antibody secretion from single VU1D9 hybridoma and CHO cell line over a period of 24 h. We also showed with some preliminary data that the microwell array can be used to track the secretion dynamics of cells. Nevertheless, the ability of this method to provide information on the secretion of proteins from single cells as well as the ability to assure monoclonality has potential to be used as a platform for the screening of cells for therapeutic antibody development. To this end, the method described here can also be adapted to screen other cells (e.g., T cells and cancer cells) secreting different molecules (e.g., cytokines).
Online Methods
Recombinant human EpCAM (rhEpCAM) and Protein A were purchased from Acrobiosystems (Newark, USA). Anti-mouse IgG-PE (Fab'2) and anti-human IgG-FITC (H+L) (Fab) were purchased from Sigma Aldrich (St Louis, USA).Preparation of the capture membranes
Low fluorescence polyvinylidene membrane (PVDF) with a pore size of 0.45 was purchased from Bio-rad laboratories (California, USA). PVDF were cut to 1 × 1 cm dimensions and wetted in 100% methanol (Fisher scientific, Leicestershire, UK) followed by 3 washes in sterile MilliQ. Next PVDF membranes were coated with ligands, rhEpCAM (25 μg mL−1) or Protein A (30 μg mL−1) overnight at 4 °C. Next membranes were washed in Phosphate buffered saline (PBS), (EMD Milliepore, Milleroca, USA) and blocked in blocking buffer consisting of 3% BSA (Sigma Aldrich, St Louis, USA) in PBS for 1 h at RT. Finally, the PVDF membranes were washed once in PBS and incubated in cell culture medium prior to use.Preparation of cells
Hybridoma cell line (VU1D9) secreting anti-EpCAM IgG were provided by Immunicon Corp., Huntingdon Valley, PA, USA. Cells were cultured in chemically defined CD Hybridoma medium (Invitrogen, Carlsbad, USA) supplemented with 4 mM L-glutamine, 100 U per mL penicillin/streptomycin (Sigma, St. Louis, MO). CHO cells producing Herceptin (anti-Her2) were kindly provided by Bioceros (Bioceros BV, Utrecht, The Netherlands). CHO cells were cultured in serum free ProCHO medium (Lonza, Verviers, Belgium) supplemented with 2% pluronic F68 (Invitrogen, Carlsbad, USA) and 4 mM L-glutamine. For passaging, suspension cultures were harvested by thorough pipetting and cells were pelleted by centrifugation and resuspended in fresh complete medium. Subsequently cell number and viability were determined by incubation of the cells with 4% tryphan blue (Invitrogen, Carlsbad, USA) and counted on a Luna-FL™ automated cell counter (Logos Biosystems, Westburg B.V., Leusden, The Netherlands). Cells were cultured in T25 non-treated culture flasks (VWR international B.V, Amsterdam, The Netherlands) suitable for suspension cultures at a density of 2 × 105 cells per mL in a humidified incubator at 37 °C under 5% CO2.Preparation of microwell array
Microwell arrays (VyCAP B.V, Deventer, the Netherlands) were sterilized in 70% ethanol for 60 minutes and washed in PBS. To remove any ethanol and air in the microwells, the array was placed into a desiccator and vacuum was applied to allow PBS to enter the wells. Subsequently, the microwell array was filled with complete culture medium and incubated for 30 min at 37 °C.Single-cell antibody printing
Cells were incubated with 1 mM of Calcein AM stain (Invitrogen, Carlsbad, USA) for 30 minutes or with CellTracker Orange (Invitrogen, Invitrogen, Carlsbad, USA) at a dilution of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5000 v/v for 60 minutes. Labeled cells were spun-down and washed twice in PBS and once in complete culture medium to remove any secreted antibodies present in the supernatant. Next, a cell suspension of containing around 6500 single cells suspended in complete medium were distributed into the microwells array by applying a small negative pressure of 5–10 mbar. The microwell arrays were imaged with an automated inverted epifluorescence microscope. The prepared PVDF capturing membrane was next sandwiched between the bottom of the microwell arrays and a PDMS slab using a clamping device. Finally, the device was incubated at 37°C and 5% CO2 for 24 h. For tracking cell secreting dynamics of the same cells at different time points the same protocol was used as described above, main difference was the chip filled with cells received a new membrane at each time interval to capture the secreted antibodies from the same cells and the cells were imaged to determine cell division during the assay.
:5000 v/v for 60 minutes. Labeled cells were spun-down and washed twice in PBS and once in complete culture medium to remove any secreted antibodies present in the supernatant. Next, a cell suspension of containing around 6500 single cells suspended in complete medium were distributed into the microwells array by applying a small negative pressure of 5–10 mbar. The microwell arrays were imaged with an automated inverted epifluorescence microscope. The prepared PVDF capturing membrane was next sandwiched between the bottom of the microwell arrays and a PDMS slab using a clamping device. Finally, the device was incubated at 37°C and 5% CO2 for 24 h. For tracking cell secreting dynamics of the same cells at different time points the same protocol was used as described above, main difference was the chip filled with cells received a new membrane at each time interval to capture the secreted antibodies from the same cells and the cells were imaged to determine cell division during the assay.
Detection of printed antibody arrays
For the detection of cell secreted proteins, the PVDF membranes were washed once in PBS with 0.05% Tween 20 and PBS (5 min) to remove cell debris. Hereafter membranes were blocked in blocking buffer for 1 h at RT and incubated with FITC-conjugated detection antibodies, anti-human IgG (1:1000 v/v) for 1 h at RT. Finally, the membranes were briefly washed twice in PBS (5 min) and once in MilliQ and then dried. The membranes were imaged using an automated inverted epifluorescence microscope.
Calibration curves
To convert the measured fluorescence intensity to concentrations of anti-EpCAM and anti-Her2 drops of 1 μl containing different concentrations of recombinant anti-EpCAM (0–400 μg mL−1) and anti-Her2 (0–400 μg mL−1) were spotted on the PVDF membrane. The recombinant antibodies were reconstituted in complete culture medium (CD hybridoma or CHO5 medium). The amount of antibody in each spot is known and dividing this by the area of the spot results the antibody concentration in pg μm−2.Image analysis and quantification of fluorescent signal
The images from the microwell array and from the PDVF membrane were stored as tiff files and were analyzed with the open source image analysis program ACCEPT.21 The ACCEPT toolbox, developed in the EU funded CANCER-ID & CTC-Trap programs, is an open source toolbox for cell analysis downloadable from https://github.com/LeonieZ/ACCEPT. To quantify the total number of cells present in each microwell array and their printed antibody spot, the fluorescent images were loaded in ACCEPT. The software analysis these fluorescent images and detects all events present in the images by an advanced multi-scale segmentation approach and extracts several intensity and shape measurements for every event found.21 In practice images of the microwell array containing the cells and the corresponding the microarray were aligned. Aligned images were used to extract a panel of parameter (e.g. total cell counts, antibody fluorescence signal and spot area) in ACCEPT. The extracted data was then used to estimate the total amount of antibody captured on the membrane.Conflicts of interest
Authors Arjan Tibbe and Joska Broekmaat are employed by VyCAP. Author Leon Terstappen is shareholder of VyCAP. Authors Fikri Abali, Richard Schasfoort and Leonie Zeune declare to have no potential conflict of interest.Acknowledgements
This research is part of the HTSM research programme and was partly funded by the NWO, The Netherlands Organization for Scientific Research (Utrecht, The Netherlands) and is part of Project McSPRinter under project number 15327. The authors would like to thank Bioceros BV for providing Herceptin and Herceptin secreting CHO cells.References
- J. R. Birch and A. J. Racher, Antibody production, Adv. Drug Delivery Rev., 2006, 58, 671–685 CrossRef CAS PubMed.
- M. Butler, Animal cell cultures: recent achievements and perspectives in the production of biopharmaceuticals, Appl. Microbiol. Biotechnol., 2005, 68, 283–291 CrossRef CAS PubMed.
- C. Michel and C. Lily, Development and Production of Commercial Therapeutic Monoclonal Antibodies in Mammalian Cell Expression Systems: An Overview of the Current Upstream Technologies, Curr. Pharm. Biotechnol., 2008, 9, 447–467 Search PubMed.
- S. K. Dessain, et al. High efficiency creation of human monoclonal antibody-producing hybridomas, J. Immunol. Methods, 2004, 291, 109–122 CrossRef CAS PubMed.
- H.-Y. Kim, A. Stojadinovic and M. J. Izadjoo, in Monoclonal Antibodies: Methods and Protocols, ed. V. Ossipow and N. Fischer, Humana Press, Totowa, NJ, 2014, pp. 33–45 Search PubMed.
- C. Zhang, in Antibody Methods and Protocols, ed. G. Proetzel and H. Ebersbach, Humana Press, Totowa, NJ, 2012, pp. 117–135 Search PubMed.
- S. M. Browne and M. Al-Rubeai, Selection methods for high-producing mammalian cell lines, Trends Biotechnol., 2007, 25, 425–432 CrossRef CAS PubMed.
- V. Fitzgerald and P. Leonard, Single cell screening approaches for antibody discovery, Methods, 2017, 116, 34–42 CrossRef CAS PubMed.
- C. C. Czerkinsky, L.-Å. Nilsson, H. Nygren, Ö. Ouchterlony and A. Tarkowski, A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells, J. Immunol. Methods, 1983, 65, 109–121 CrossRef CAS PubMed.
- C. Czerkinsky, et al., Reverse ELISPOT assay for clonal analysis of cytokine production I. Enumeration of gamma-interferon-secreting cells, J. Immunol. Methods, 1988, 110, 29–36 CrossRef CAS PubMed.
- P. Gille-Johnson, et al., Circulating Monocytes Are Not the Major Source of Plasma Cytokines in Patients With Sepsis, Shock, 2012, 38, 577–583 CrossRef CAS PubMed.
- M. Kouwenhoven, et al., Enzyme-Linked Immunospot Assays Provide a Sensitive Tool for Detection of Cytokine Secretion by Monocytes, Clin. Diagn. Lab. Immunol., 2001, 8, 1248–1257 CAS.
- P. Holmes and M. Al-Rubeai, Improved cell line development by a high throughput affinity capture surface display technique to select for high secretors, J. Immunol. Methods, 1999, 230, 141–147 CrossRef CAS PubMed.
- J. C. Weaver, P. McGrath and S. Adams, Gel microdrop technology for rapid isolation of rare and high producer cells, Nat. Med., 1997, 3, 583–585 CrossRef CAS PubMed.
- J. F. B. A. C. J. Mann, Rapid Isolation of Monoclonal Antibody Producing Cell Lines BioProcess International, 2006, pp. 48–51 Search PubMed.
- A. Jin, et al., A rapid and efficient single-cell manipulation method for screening antigen-specific antibody–secreting cells from human peripheral blood, Nat. Med., 2009, 15, 1088 CrossRef CAS PubMed.
- A. Jin, et al., Rapid isolation of antigen-specific antibody-secreting cells using a chip-based immunospot array, Nat. Protoc., 2011, 6, 668 CrossRef CAS PubMed.
- J. C. Love, J. L. Ronan, G. M. Grotenbreg, A. G. van der Veen and H. L. Ploegh, A microengraving method for rapid selection of single cells producing antigen-specific antibodies, Nat. Biotechnol., 2006, 24, 703–707 CrossRef CAS PubMed.
- A. O. Ogunniyi, C. M. Story, E. Papa, E. Guillen and J. C. Love, Screening individual hybridomas by microengraving to discover monoclonal antibodies, Nat. Protoc., 2009, 4, 767–782 CrossRef CAS PubMed.
- J. F. Swennenhuis, et al., Self-seeding microwell chip for the isolation and characterization of single cells, Lab Chip, 2015, 15, 3039–3046 RSC.
- L. Zeune, G. V. Dalum, L. W. M. M. Terstappen, S. A. V. Gils and C. Brune, Multiscale Segmentation via Bregman Distances and Nonlinear Spectral Analysis, SIAM J. Imaging Sci., 2017, 10, 111–146 CrossRef.
- I. Stojanović, W. Baumgartner, T. J. G. van der Velden, L. W. M. M. Terstappen and R. B. M. Schasfoort, Modeling single cell antibody excretion on a biosensor, Anal. Biochem., 2016, 504, 1–3 CrossRef PubMed.
- I. Stojanović, T. J. G. van der Velden, H. W. Mulder, R. B. M. Schasfoort and L. W. M. M. Terstappen, Quantification of antibody production of individual hybridoma cells by surface plasmon resonance imaging, Anal. Biochem., 2015, 485, 112–118 CrossRef PubMed.
- K. C. Andree, et al., Self-Seeding Microwells to Isolate and Assess the Viability of Single Circulating Tumor Cells, Int. J. Mol. Sci., 2019, 20, 477 CrossRef PubMed.
- J. C. Love, J. L. Ronan, G. M. Grotenbreg, A. G. van der Veen and H. L. Ploegh, A microengraving method for rapid selection of single cells producing antigen-specific antibodies, Nat. Biotechnol., 2006, 24, 703 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00100j |
| This journal is © The Royal Society of Chemistry 2019 |