
Multilayer microfluidic array for highly efficient sample loading and digital melt analysis of DNA methylation†

Abstract
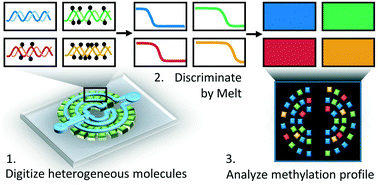
Liquid biopsies contain a treasure of genetic and epigenetic biomarkers that contain information for the detection and monitoring of human disease. DNA methylation is an epigenetic modification that is critical to determining cellular phenotype and often becomes altered in many disease states. In cancer, aberrant DNA methylation contributes to carcinogenesis and can profoundly affect tumor evolution, metastatic potential, and resistance to therapeutic intervention. However, current technologies are not well-suited for quantitative assessment of DNA methylation heterogeneity, especially in challenging samples such as liquid biopsies with low DNA input and high background. We present a multilayer microfluidic device for quantitative analysis of DNA methylation by digital PCR and high resolution melt (HRM). The multilayer design facilitates high-density array digitization aimed at maximizing sample loading efficiency. The platform achieves highly parallelized digital PCR-HRM-based discrimination of rare heterogeneous DNA methylation as low as 0.0001% methylated/unmethylated molecules of a classic tumor suppressor gene, CDKN2A (p14ARF).
- This article is part of the themed collection: Personalised Medicine: Liquid Biopsy
 Please wait while we load your content...
Please wait while we load your content...

