
High-throughput, non-equilibrium studies of single biomolecules using glass-made nanofluidic devices†
Mattia
Fontana‡
 a,
Carel
Fijen‡
a,
Serge G.
Lemay
b,
Klaus
Mathwig
*bc and
Johannes
Hohlbein
*ad
a,
Carel
Fijen‡
a,
Serge G.
Lemay
b,
Klaus
Mathwig
*bc and
Johannes
Hohlbein
*ad
aLaboratory of Biophysics, Wageningen University and Research, Stippeneng 4, Wageningen, 6708 WE, The Netherlands
bMESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, Enschede, 7500 AE, The Netherlands
cGroningen Research Institute of Pharmacy, Pharmaceutical Analysis, University of Groningen, P.O. Box 196, Groningen, 9700 AD, The Netherlands. E-mail: k.h.mathwig@rug.nl
dMicrospectroscopy Research Facility, Wageningen University and Research, Stippeneng 4, Wageningen, 6708 WE, The Netherlands. E-mail: johannes.hohlbein@wur.nl
First published on 20th November 2018
Abstract
Single-molecule detection schemes offer powerful means to overcome static and dynamic heterogeneity inherent to complex samples. However, probing biomolecular interactions and reactions with high throughput and time resolution remains challenging, often requiring surface-immobilized entities. Here, we introduce glass-made nanofluidic devices for the high-throughput detection of freely-diffusing single biomolecules by camera-based fluorescence microscopy. Nanochannels of 200 nm height and a width of several micrometers confine the movement of biomolecules. Using pressure-driven flow through an array of parallel nanochannels and by tracking the movement of fluorescently labelled DNA oligonucleotides, we observe conformational changes with high throughput. In a device geometry featuring a T-shaped junction of nanochannels, we drive steady-state non-equilibrium conditions by continuously mixing reactants and triggering chemical reactions. We use the device to probe the conformational equilibrium of a DNA hairpin as well as to continuously observe DNA synthesis in real time. Our platform offers a straightforward and robust method for studying reaction kinetics at the single-molecule level.
Introduction
Single-molecule techniques are playing an increasingly important role in identifying and analyzing static or dynamic interactions of (bio-)molecules with high spatiotemporal resolution.1–3 Among the large number of available frameworks,4 single-molecule fluorescence detection (SMFD) schemes, such as confocal or camera-based total-internal-reflection fluorescence (TIRF) microcopy, are frequently chosen due to their simplicity, robustness and ease of use.5 However, users often have to choose between high throughput or high time resolution,6–8 many experiments require surface immobilization techniques to extend the observation time, and it is difficult to access non-equilibrium conditions or follow biochemical reactions.Many creative solutions have been proposed to overcome these hurdles using fluidic platforms.9,10 These include mixers for studying single-molecule kinetics,11,12 titration devices,13 devices to confine molecules in polymer nanochannels,14 electroosmotic molecular traps,15 or microfluidic droplets containing individual enzyme analytes.16 These fluidic platforms are, despite their clear benefits, not yet widely used due to complex fabrication procedures, limited reusability and configurability or restrictions in integration into microscopy platforms.
Here, we introduce a fluidic platform for SMFD consisting of nano-/microchannel devices fabricated entirely in glass thereby overcoming limitations imposed by previous designs utilizing polydimethylsiloxane (PDMS) such as the susceptibility to swelling by some organic solvents and the limited modifiability of the surface to prevent non-specific adsorption. The design goals of our platform were (1) to achieve single-molecule detection at high time resolution by geometrically confining the flow through nanochannels (200 nm channel height and micro-scale width and length), (2) to use camera-based detection to monitor many molecules in parallel, (3) to enable straightforward device integration, and (4) to observe chemical reactions at the single-molecule level and in real time using mixing in a T-shaped nanochannel.
We utilize the new nanofluidic devices for high-throughput sensing of short DNA oligonucleotides in an array of parallel nanochannels by detecting single-molecule Förster resonance energy transfer (smFRET) between a donor and an acceptor fluorophore attached to the DNA. Using an updated excitation scheme, several hundred thousand events are combined to gather reliable single-molecule data within minutes.
We probe the conformational equilibrium of a DNA hairpin in nanochannel devices with a mixing geometry. By imaging the T-junction, we probe conformations before, during and after mixing with a buffer containing a high salt concentration that stabilizes the closed hairpin conformation.
Finally, we continuously monitor reaction kinetics at the single-molecule level by observing DNA synthesis upon mixing DNA oligonucleotides, DNA polymerases and nucleotides. Reactions are observed by a change in FRET efficiency downstream in the outlet nanochannel allowing us to translate a spatial position into a corresponding moment in the time evolution of a reaction. Thus, our platform demonstrates the ability for a wide range of applications in probing complex kinetics.
Materials and methods
Device fabrication and surface passivation
Nano-/microfluidic devices were fabricated by Micronit with a 45 mm × 15 mm footprint to be compatible with a commercially available chip holder Fluidic Connect PRO (Micronit Microtechnologies B.V., The Netherlands). In brief, the devices consist of two thermally bonded borosilicate glass layers with photolithographically defined and wet-etched nano- and microchannels. Microchannels and ports to connect to tubing are structured in the upper 1 mm thick glass, while nanochannels are etched into the bottom layer. Here, coverslip type D263 borosilicate glass (Schott) with a thickness of 175 μm was used enabling high-resolution fluorescence imaging with inverted oil-immersion microscope objectives with shortest working distances whilst minimizing undesirable autofluorescence.To prevent non-specific adsorption in experiments with proteins, channels were passivated with PEG using a variation of a method described previously.17 The fluidic devices were first flushed and incubated (3 × 5 min) with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (vol/vol) Vectabond:acetone solution. All subsequent washing and passivation steps were performed by flushing the channels with ∼100 μL of the respective solutions. PEGylated channels were filled with PBS and stored in a humid chamber at 4 °C.
:50 (vol/vol) Vectabond:acetone solution. All subsequent washing and passivation steps were performed by flushing the channels with ∼100 μL of the respective solutions. PEGylated channels were filled with PBS and stored in a humid chamber at 4 °C.
DNA and buffers
Fluorescently labelled oligonucleotides were ordered from IBA, Germany. To construct the DNA hairpin, a 30-mer primer sequence (biotin-5′-CCT CAT TCT TCG TCC CAT TAC CAT ACA TCC-3′) was annealed to a 75-mer hairpin sequence (5′-TGG ATT AAA AAA AAA AAA AAA AAA AAA AAA AAA AAA AAA TCC ATT GGA TGT ATG GTA ATG GGA CGA AGA ATG AGG-3′). The primer was internally labelled with ATTO647N at the −12 position; the hairpin was labelled with Cy3B at the 5′ end. Gapped DNA, used to study the binding of DNA polymerase I (Klenow fragment, KF), was constructed using the same primer sequence, annealed to a template strand (5′-CCA CGA AGC AGG CTC TAC TCT CTA AGG ATG TAT GGT AAT GGG ACG AAG AAT GAG G-3′) and a downstream complementary strand (5′-TAG AGA GTA GAG CCT GCT TCG TGG-3′). The template strand was labelled with Cy3B at the +12 position. The DNA sensor used for the polymerization experiment consists of the same primer and template sequences, but with ATTO647N on the −7 position and Cy3B on the +25 position, respectively. dNTPs were ordered from Sigma-Aldrich/Merck, Germany.DNA constructs (as well as KF, if specified) were diluted in an imaging buffer containing 50 mM Tris HCl (pH 7.5), 100 μg mL−1 BSA, 10 mM MgCl2, 5% glycerol, 1 mM DTT, 1 mM Trolox, 1% glucose oxidase/catalase and 1% glucose. Trolox is a triplet-state quencher and prevents fluorophore blinking. Glucose, glucose oxidase and catalase was used as an oxygen scavenger system to prevent premature photobleaching of fluorophores.18,19 The concentration of gapped DNA was 1 nM and, if used, the concentration of KF was 10 nM. DNA hairpin concentrations were 500 pM in parallel channels and 1 nM in the mixing channel (DNA hairpins were diluted in a similar imaging buffer without magnesium, but with additional NaCl as specified). Prior to mixing, the concentration of DNA polymerization sensors was 1 nM; the concentration of KF was 5 nM, and the concentration of dNTPs was 200 μM each. For this polymerization experiment, we added neutravidin directly to the imaging buffer in a concentration of 0.6 μg mL−1 to block 5′ end of the biotinylated DNA primer. We found this prevents the formation of a low E* state, the cause of which is probably binding of multiple KF polymerases to the same DNA molecule. The imaging buffer was applied to a 100 μL syringe (ILS, Germany), which was then connected to the nanofluidic device using ethylene tetrafluoroethylene (ETFE) tubing (1/16′′ outer diameter, 0.010′′ inner diameter; Micronit Microtechnologies B.V., The Netherlands).
Single molecule detection
We used a home built TIRF microscope and a fiber-coupled laser engine (Omicron, Germany) equipped with lasers of four different wavelengths (405 nm, 473 nm, 561 nm, and 638 nm). Laser intensities were independently controlled by a home-written LabVIEW program. Divergent light from the fiber output was collimated (f = 30 mm, Thorlabs, Germany) and focused by a second lens (f = 200 mm, Thorlabs, Germany) into the backfocal plane of a 100× NA 1.49 objective (Nikon, Japan). A polychroic filter and a multi-bandpass filter (Chroma, USA) prevented laser light from entering the emission path. A tube lens focused the emission on an aperture, which reduced the effective field of view to a rectangle. Next, the light was spectrally split into a blue, a green and a red emission channel. The three beams were focused on an Ixon Ultra 897 emCCD (Andor, UK) with 512 by 512 pixels (maximum acquisition rate: 56 Hz at full frame and 100 Hz after cropping the frame to 343 by 256 pixels). In our configuration, one pixel on the camera corresponded to a length and width of 112 nm in the sample plane. We used a rapid automated modular microscope (RAMM) system as a stage holder (ASI, USA), combined with motorized x, y-scanning stage and a z-piezo for focusing.Molecules were excited with a laser power of 130 mW (measured after the fiber output, 561 nm and 638 nm laser) for the fluidic experiments and with 30 mW (561 nm) and 15 mW (638 nm) for the surface immobilized experiments. Excitation in semi-total-internal reflection mode ensured the highest possible signal-to-noise ratio. A stroboscopic alternating-laser excitation (sALEX)20 scheme was used to reduce motion blur of diffusing molecules. Laser pulse widths were 1.5 ms (fluidic devices) and 3 ms (surface immobilized experiments) in a frame time of 10 ms. Green and red pulses were aligned back-to-back, so that particle movement between a green and a red frame was minimal. Shorter laser pulses, and the necessary higher laser powers, were found to cause rapid bleaching.
Extracting intensities and positions from single molecules
Particles were localized and tracked with a home-written variation on GaussStorm (Matlab).21 Time traces, histograms and binned maps were generated with custom-built software packages. We applied a bandpass filter to find local intensity maxima before fitting the local maxima with elliptical 2D Gaussian functions from which we obtained the photon count as well as the position with sub-pixel accuracy.21 After filtering of localizations based on their intensities, our algorithm links corresponding particles in the green and red channels and calculates FRET efficiency E* and stoichiometry ratio S for each molecule.Tracking of single molecules and determination of flow speeds and diffusion coefficients
A tracking algorithm was applied to track particles from frame to frame.22 The distribution of the displacements of single particles was used to calculate mean flow speed and mean diffusion coefficient in the entire field of view or in selected regions. In the case of one-dimensional diffusion along the direction of the flow, the probability distribution for the position of the particle (ρ) is distributed as a Gaussian with variance proportional to the diffusivity D of the particle (σ2 = 2Dt) and mean proportional to the flow speed v (μ = vt). For an example of displacement distribution see ESI† Fig. S5.FRET and alternating laser excitation (ALEX)
The apparent FRET efficiency E* is calculated from the emission intensities of donor and acceptor after donor excitation (denoted DD and DA, respectively) for each molecule in each camera frame according to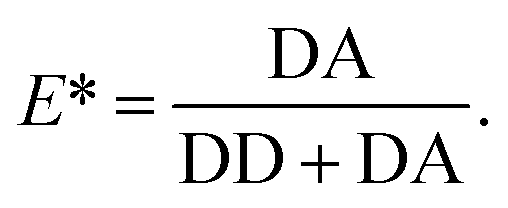 | (1) |
To verify the presence of an acceptor fluorophore on the DNA we applied alternating laser excitation (ALEX) in which every frame of donor excitation is followed by a direct excitation of the acceptor fluorophore using a second laser resulting in a third photon stream (AA) for each molecule.23–25 The detection of AA in addition to DD and DA allows for calculating the stoichiometry ratio S, defined as
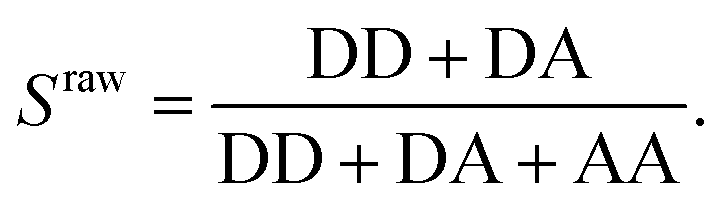 | (2) |
S can be used to filter molecules: molecules with a stoichiometry close to 0 have no photoactive donor (e.g., because of donor bleaching), and molecules with a stoichiometry close to 1 have no photoactive acceptor. Depending on the ratio of the laser intensities for direct donor and acceptor excitation, a stoichiometry around 0.5 represents molecules having both a photoactive donor and acceptor that can undergo FRET. In this work, we show FRET data based on molecules that have stoichiometry values 0.3 ≤ S ≤ 0.8 (nanochannel data) or 0.5 ≤ S ≤ 0.9 (immobilized hairpins).
Results
Single-molecule fluorescence detection of nanometer sized, non-immobilized entities is challenging due to the limited photon budget of the fluorophores. In the case of surface-immobilized molecules, the integration of photon emission from a diffraction limited spot quickly leads to an acceptable signal-to-noise ratio, but moving particles distribute the emitted photons over a larger area on the camera, compromising the signal-to-noise ratio. For this reason, fluorophore excitation must be limited to an interval as short as possible. In the following, we describe the fluidic design and flow control enabling conditions in which the induced flow is slow enough as not to cause additional broadening of the point spread function, and enables the observation of DNA–protein interactions, salt-dependent conformational equilibria of DNA, and enzymatic driven DNA synthesis.Achieving low nanochannel flow speeds via parallel flow control
The first device consists of parallel nanochannels wet-etched into a first glass wafer and connected via wafer-bonding with feeding microchannel etched into a second glass wafer (Fig. 1, see also Material and methods). We use the principle of parallel flow control (PFC)26,27 in which liquid driven by a syringe pump is divided into nanochannels and a parallel bypassing microchannel. Due to the high ratio of hydraulic resistance between the nanochannels and the microchannel, the flow speed is reduced by orders of magnitude in the nanochannel, and the dead volume in feeding channels and tubing is replaced quickly (see also ESI† Fig. S2 and Note 1). The resulting typical flow rates in the nanochannels are in the range of 0.2 to 20 pL min−1 per channel, corresponding to average flow velocities of 5–500 nm ms−1. We validated our implementation of PFC by measuring the flow speeds (see Material and methods) and computing velocity profiles at different pump rates (ESI† Fig. S3). The shape and magnitude of the velocity profile show an excellent agreement with the theoretical model for Poiseuille flow in rectangular nanochannels at the pump rate tested experimentally (ESI† Fig. S4, ESI Note 2).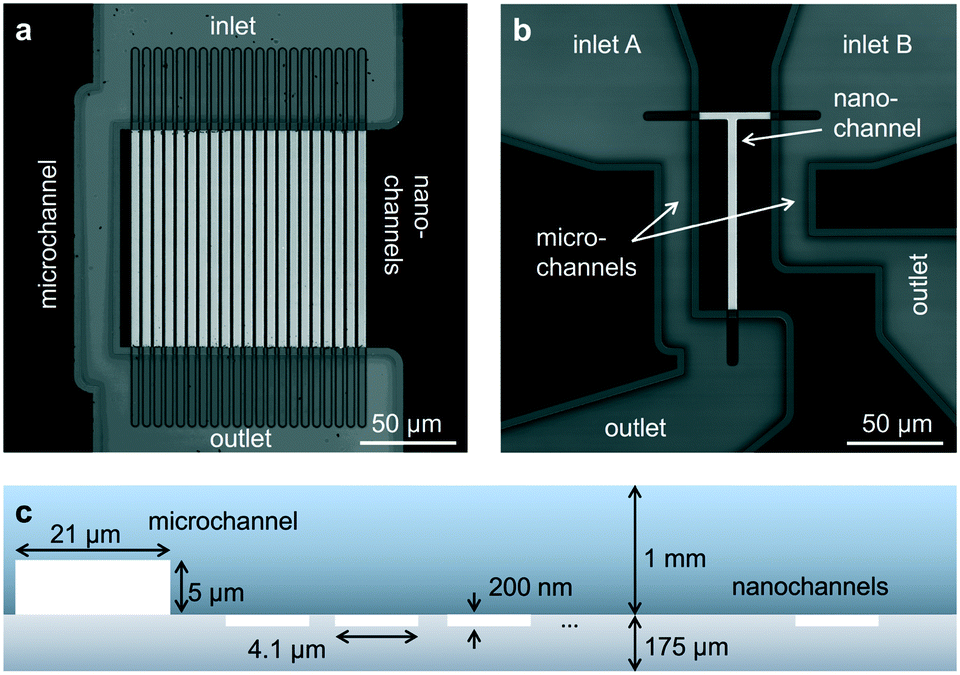 | ||
| Fig. 1 Design of glass nanofluidic chips. (a and b) High-resolution confocal scans based on reflection of light. The parallel channel design (a) contains 21 straight nanochannels (l × w × h: 120 by 4.1 by 0.2 μm) and a microchannel (w × h: 21 by 5 μm). The design of the mixing device (b) contains a single T-shaped nanochannel (horizontal part: l × w × h: 40 by 3.8 by 0.2 μm, vertical part l × w × h: 100 by 4.7 by 0.2 μm) in between two microchannels (l × w × h: 95 by 21 by 5 μm). Fluidic chips fit in a Micronit chip holder for easy connection to tubes and pumps (see also ESI† Fig. S1). (c) Schematic cross-section of the parallel channel array, showing the dimensions of the microchannel and the nanochannels (not to scale). | ||
The second device applies, for the first time, the PFC approach to a mixing geometry. Here, two syringe pumps deliver flow to two microchannels and two respective parallel feeding nanochannel inlets which merge to a single nanochannel at a T-junction (ESI† Fig. S2). The mass transfer in such device is summarized by the Péclet number
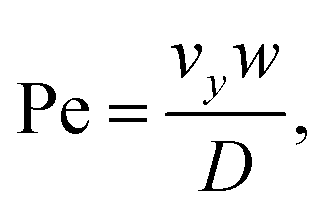 | (3) |
Detecting fluorescence of moving particles inside nanochannels
As probes, we opted for doubly labelled DNA molecules providing a FRET efficiency E* that is calculated using photon numbers after donor excitation emitted by the donor (DD) and the acceptor (DA), respectively (see Material and methods). To verify the presence of the acceptor, we applied alternating-laser excitation (ALEX) in which every camera frame of donor excitation is followed by a frame of direct acceptor excitation creating photon numbers AA (Material and methods).23,24 Since motion blur severely affects the ability to determine emission intensities of individual molecules inside the nanochannels we opted for stroboscopic ALEX (sALEX)20 in which the molecules are only excited for a fraction of the camera exposure time. We further aligned the green and red laser pulses back-to-back to facilitate successive localizations in the acceptor channel allowing to link donor and acceptor signals (Fig. 2a, top). The stroboscopic excitation pulses (1.5 ms) lead to spots which are barely affected by motion blur and have an excellent signal-to-noise ratio allowing us to fit 2D Gaussians to obtain photon numbers and positions with sub-pixel localization accuracies for individual DNA molecules (Fig. 2a, bottom).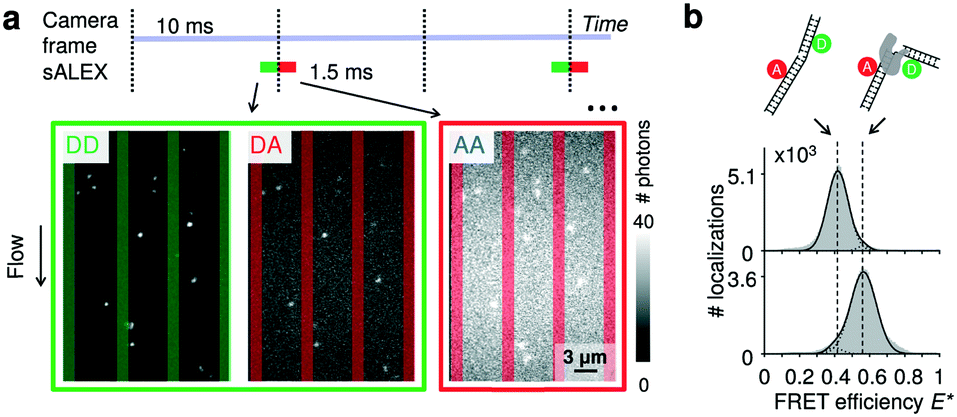 | ||
| Fig. 2 Detection of single molecules and DNA–protein interactions inside parallel nanochannels. (a) Top: Schematic of stroboscopic alternating laser excitation (sALEX); the excitation time is considerably shorter than the required acquisition time of a camera frame, reducing motion blur. The laser pulses are placed back to back to facilitate the linking of the AA signal to the correct DD/DA couple of signals. Bottom: Example of raw movie frames showing the gapped DNA construct flowing through the parallel nanochannels at a flow speed of 218(1) nm ms−1 (mean value and 95% interval of confidence). Excitation colors are indicated by the surrounding boxes. For each molecule, photon counts of DD and DA after donor excitation are determined simultaneously. The photon counts of AA after acceptor excitation are collected during the next camera frame. Nanochannel boundaries are indicated with green bars (green detection channel) or red bars (red detection channel). (b) Top: Schematic of a gapped DNA construct in its free and bound conformation. The dsDNA is labelled with a donor and acceptor dyes located on opposite sides of the one-nucleotide gap. Bottom: E* histograms (100 bins) of the gapped DNA construct, measured in the parallel channels for 5 minutes. Top: 1 nM DNA. Bottom: 1 nM DNA in presence of 10 nM DNA polymerase I (KF), a DNA polymerase known to change the conformation of gapped DNA upon binding. Dashed lines are added for visual guidance. For full E*/S histograms see ESI† Fig. S7a. | ||
Parallel nanochannels: visualizing DNA–protein interactions
To prove that, after passivating the glass surface using PEG (see Material and methods), our parallel device can be used to study protein binding without immobilization, we measured a sample containing 1 nM gapped dsDNA in the absence and presence of 10 nM of DNA polymerase I (Klenow fragment, KF). The nanochannels were imaged for 5 minutes obtaining ∼105 FRET efficiency readouts from single molecules flowing through the field of view; this allowed us to compute high quality FRET efficiency histograms (Fig. 2b).The mean FRET efficiency for the DNA molecules in the absence of KF shows a single species centered around E* = 0.4. The addition of KF causes an increase of FRET efficiency to 0.6 which reflects the expected shortened donor–acceptor distance upon binding and DNA bending.31 We note that using freely diffusing molecules, we obtained a second independent readout for the binding in the form of the decreased apparent diffusion coefficient along the channel upon KF addition and complex formation from DDNA = 35.3 ± 0.3 μm2 s−1 to DDNA/KF = 30.6 ± 0.3 μm2 s−1.
Parallel nanochannels: resolving salt-dependent conformational equilibria of DNA hairpins
To compare the accessible dynamic range of FRET measurements in nanochannels with those of immobilized entities, we utilized DNA hairpins that can interconvert between an open (low FRET) and closed (high FRET) conformations. The chosen DNA hairpin is mostly open at 0 M NaCl and mostly closed at 1 M NaCl, while showing fast opening and closing at an intermediate concentration of 0.5 M NaCl.20 Upon addition of salt, we further note a small shift of the FRET peak of the open conformation due to a salt-depended compaction of the single stranded DNA overhang.32 The FRET histograms of the freely flowing DNA hairpins are similar to the immobilized ones (Fig. 3a), indicating that we access a broad FRET range and resolve the individual conformational states of “open” and “closed”. Combining consecutive localizations from individual molecules allowed us to obtain time traces that show opening and closing of individual DNA hairpins in real time (Fig. 3b).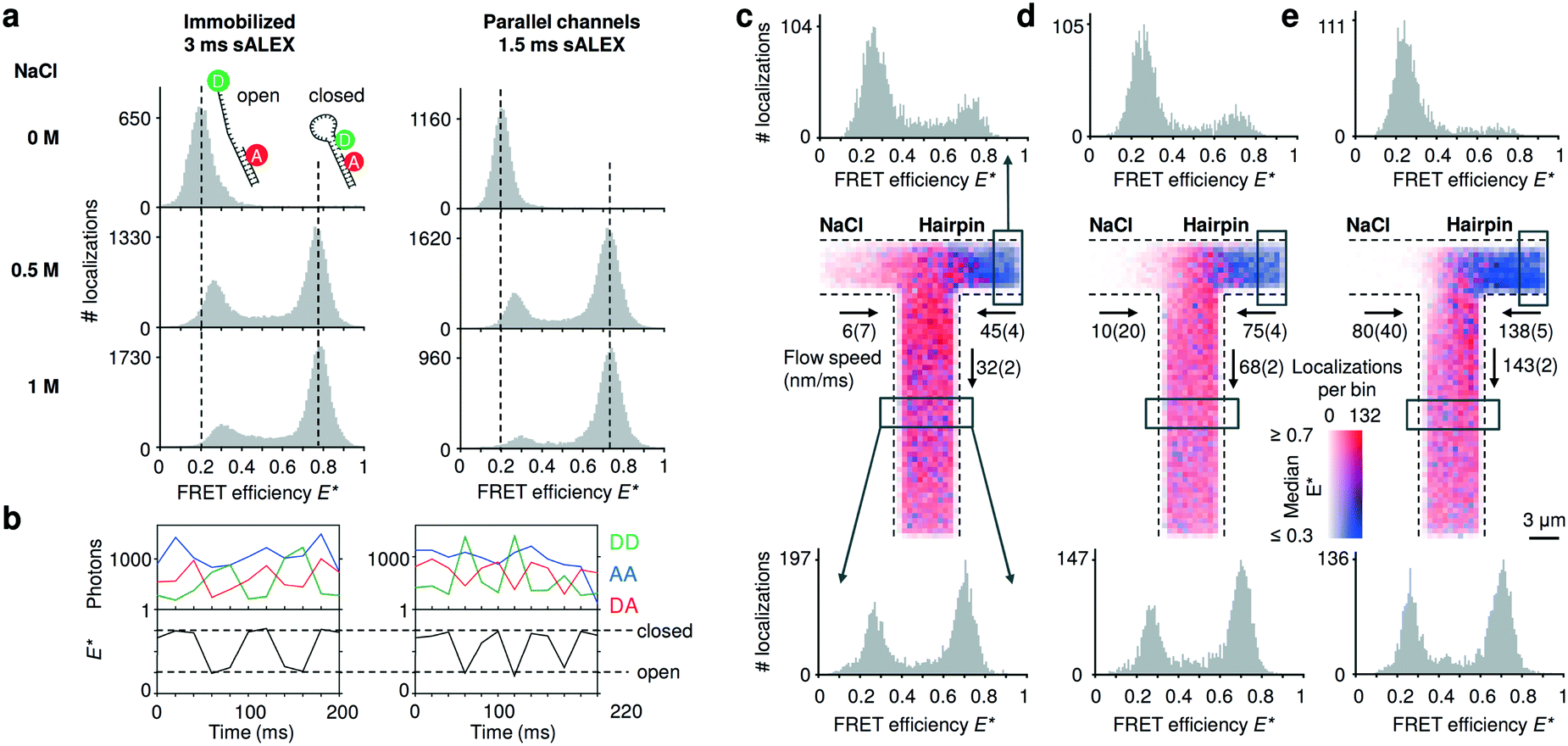 | ||
| Fig. 3 Detecting conformational equilibria of DNA hairpins in nanochannels. (a) Single-molecule detection of DNA molecules immobilized or non-immobilized within parallel nanochannels. The conformational equilibrium of DNA hairpins is salt dependent; DNA hairpins are mostly open (low FRET) at [NaCl] = 0 M; the conformational equilibrium shifts towards the closed state (high FRET) with increasing salt concentrations. The DNA hairpin shows similar behavior when imaged in parallel nanochannels compared to a standard immobilized sample. For full E*/S histograms see ESI† Fig. S7b. (b) Single-molecule time traces obtained from tracking individual molecules flowing through the channels at [NaCl] = 0.5 M. The AA signal (blue trace) remains largely constant. The clear anti-correlation of the DD (green trace) and the DA (red trace) signal and the resulting FRET time traces (black trace) indicate conformational changes in the millisecond timescale of the DNA hairpin during its passage. Dashed vertical lines are added for visual guidance indicating the expected FRET efficiencies of the open and closed DNA hairpin conformation, respectively. (c–e) Mixing of a high salt solution [NaCl] = 1 M with DNA hairpins [NaCl] = 0 M at increasing flow speeds. Center: binned maps (4 × 4 camera pixels per bin), in which color represents the median FRET efficiency and opacity indicates the number of localizations in a bin over the time span of the measurement. Given the low Péclet number, axial diffusion of the DNA hairpin in the salt inlet is visible at lower flow speeds (given with 95% interval of confidence) (c and d) but becomes negligible at the highest one (e). Top: FRET histograms reconstructed from the inlet (boxed region); the salt diffuses into the inlet channel of the hairpin stabilizing the closed conformation of the hairpin at the lowest flow rate; this effect decreases at higher flow rates. Bottom: FRET histograms reconstructed below the T-junction (boxed region); at higher flow speed, the concentration of the salt in the outlet decreases because the effect of axial diffusion becomes less prevalent originating a rise in the open conformation compared to the lower flow speeds. | ||
Nanofluidic mixing: spatially resolving shifts in the conformational equilibria of DNA hairpins
Next, we validated the mixing devices by shifting the conformational equilibrium of the DNA hairpin from mostly open to mostly closed. To this aim, we mixed a buffer containing 1 M NaCl with a buffer containing DNA hairpins at 0 M NaCl. The viscosity of the solution containing NaCl is expected to be approximatively 10% higher than the solution with zero salt;33 the pump rates were then adjusted by visual inspection.We constructed a map of spatially resolved, binned FRET efficiencies for various flow rates (Fig. 3c–e, center). For each bin, the color indicates the median FRET efficiency, but 1D FRET efficiency histograms can be reconstructed from any part of the field of view. We selected two regions, one from the right inlet and one below the mixing intersection (Fig. 3c–e, top and bottom histograms).
At the three pump settings tested, the mixing device operates at Pe < 20 for DNA (DDNA ≈ 35 μm2 s−1) and Pe < 0.4 (DNaCl ≈ 2000 μm2 s−1)28 for salt ions. Under these conditions, identification of a well-localized onset of mixing is not possible since axial diffusion becomes relevant: at the lowest flow speeds at which DNA molecules are transported with Pe ≈ 5 (Fig. 3c, center), many molecules diffuse back in the salt inlet whereas maintaining a convective displacement toward the junction. The effect of axial diffusion on DNA transport diminishes at higher flow rates in agreement with finite element Comsol simulations for these transport conditions (ESI† Fig. S6a, ESI Note 3). As the diffusion coefficient of salt is almost two orders of magnitude higher than the one of the DNA hairpin the simulations show a considerable amount of axial diffusion for this species; in particular, the concentration of the salt in the outlet is predicted to be between 400 mM for the highest flow rate up to 500 mM at the lowest flow rate. Experimentally, the salt concentration in a given region of a nanochannel can be estimated from the FRET efficiency profile reconstructed from that region. The histograms generated from the inlets (Fig. 3c and d, top) confirm that the salt diffuses to the other inlet indicated by a small “closed” population and its relative decrease with increasing flow speed. The histograms taken below the intersection show a relative increase of the “open” population for increasing flow speeds, consistent with the expected lower salt concentration.
Nanofluidic mixing: triggering enzymatic catalysis of DNA synthesis
To establish our mixing device as a tool for non-equilibrium studies, we utilized a FRET-based DNA polymerization sensor. The sensor consists of a primer DNA, labelled with an acceptor fluorophore, that is annealed to a long template DNA in which the single stranded overhang (25 bases) is labelled with a donor fluorophore (see Material and methods). Since the single strand overhang is randomly coiled, the FRET efficiency is high (E* ∼ 0.6) before decreasing substantially upon addition of DNA polymerases and nucleotides (dNTPs: deoxyribonucleotide triphosphates) to E* ∼ 0.2. When the DNA construct is immobilized on a surface, we previously found that full polymerization adding 25 bases takes on average 1.6 s.34 Due to this long reaction time, we acquired movies using three different field of views and stacked them together (Fig. 4). Two different pump settings, which resulted in a flow speed of approximatively 20 μm s−1 and 10 μm s−1 in the common nanochannel and a residence time in a given field of view of 1.5 s and 3 s respectively.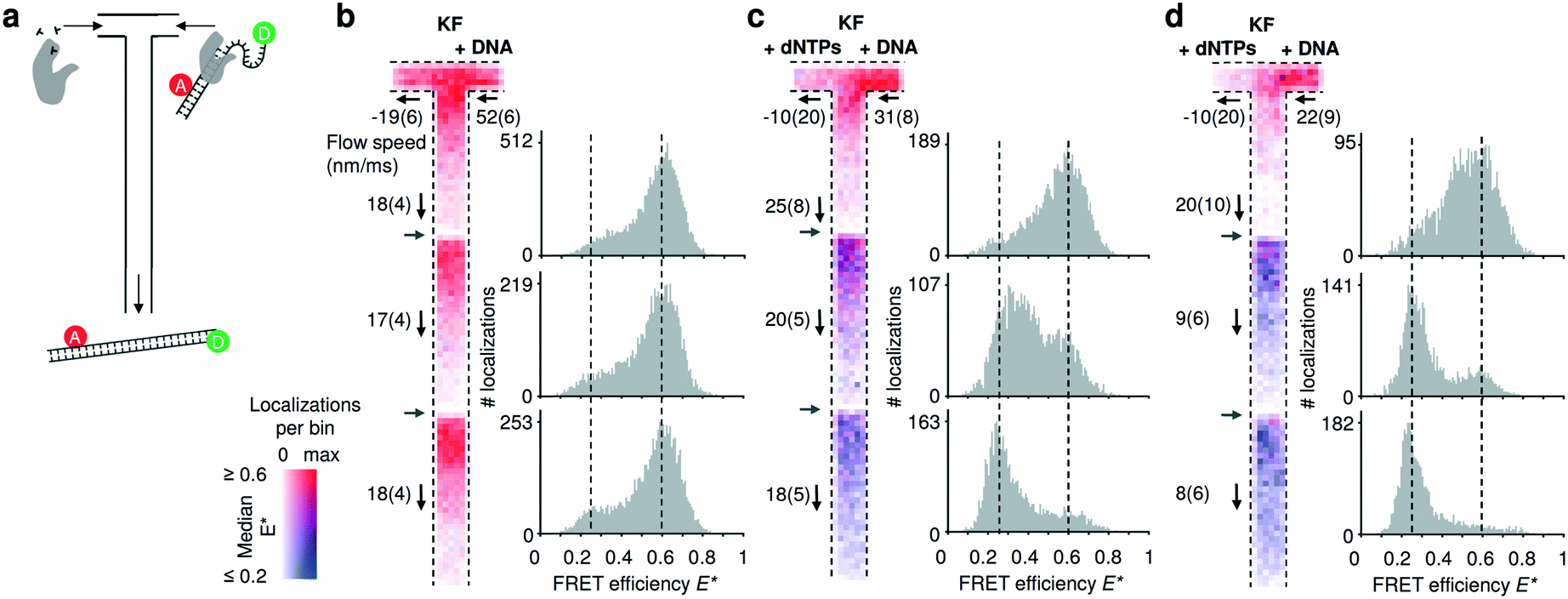 | ||
| Fig. 4 Monitoring DNA polymerization in mixing nanochannels. (a) Using the T-configuration, nucleotides and DNA polymerases (KF) are flowing from the left inlet encountering the doubly labelled DNA polymerization sensor and DNA polymerases from the right inlet. Successful DNA polymerization increases the distance between donor and acceptor fluorophore leading to a decrease in FRET efficiency. (b–d) Binned maps (8 × 8 camera pixels per bin) and corresponding FRET efficiency histograms containing data from three adjacent field of views (FOV); the borders of new FOVs are indicated with grey arrows. Color maps represent the median FRET efficiency; opacity indicates the number of localizations per bin, normalized for each map separately. Fewer molecules are localized in the lower parts of each FOV due to premature fluorophore photobleaching. Experimentally derived flow speeds are given with their mean value and 95% interval of confidence. (b) Control experiment in absence of nucleotides. The DNA sensors show no changes in the mapped FRET efficiencies (map max = 126 localizations per bin) and the FRET histograms (100 bins) establish E* = 0.6 as the center point of the high FRET species. (c) After addition of nucleotides via the left inlet, the mapped median FRET efficiencies of the FRET sensor decreases from ∼0.6 to 0.25 towards the outlet. Almost complete polymerization is achieved at the third FOV (map max = 66). (d) Decreasing the flow speed by half, shows that the polymerization is be almost completed in the second FOV, consistent with the doubling the residence time in the first FOV (map max = 58). For full E*/S histograms see ESI† Fig. S7c. | ||
In the control without dNTPs, the median FRET efficiencies binned and traced back to the field of view stays constant (Fig. 4b); upon addition of nucleotides in the left inlet (200 μM), the mapped median FRET efficiencies decrease from E* ∼ 0.6 to E* ∼ 0.2 (Fig. 4c; T-maps) as the underlying population of the fully polymerized sensor increases (Fig. 4b, 1D FRET histograms). Halving the flow speed doubles the residence time in a given field of view and the polymerization is almost complete already at the second field of view (Fig. 4d) confirming the expected polymerization time of 1–2 s. The single molecule displacement analysis revealed the presence of backflow in the dNTPs inlet; nevertheless, simulations of such conditions revealed a concentration of dNTP after the junction higher than 20 μM for the fast flow speed and higher than 40 μM for the slow flow speed (ESI† Fig. S6b). The effect of the axial diffusion of the dNTPs in the T-sensor allowed the polymerization to take place even in presence of backflow.
Discussion
We introduced two nanofluidic device designs for single-molecule detection under equilibrium and non-equilibrium conditions: the first one with a series of parallel nanochannels for equilibrium studies, and the second one with a T-shaped nanochannel for non-equilibrium studies. Both designs combine camera-based single-molecule detection, which allows monitoring many fluorescent molecules in parallel, with constant replenishment of freely flowing molecules. An updated stroboscopic excitation scheme utilizing back-to-back illumination resulted in near-circular emission spots on the camera sensor. Furthermore, in both designs we employed the concept of parallel flow control allowing us to work with simple syringe pumps whilst obtaining extremely low flow speeds that are necessary for single-molecule detection using image-based data acquisition.Our design featuring the parallel array of nanochannels is especially suited for high-throughput measurements. Using 1 nM of fluorescently labelled DNA hairpins, we obtained a throughput of ∼104 FRET data points per minute of measurement, even though the current field of view on our setup (∼29 μm by 19 μm) covers only 3 out of 21 channels.
In single-focus, diffusion based confocal microscopy, fluorescence bursts of molecules passing the focus are collected with the time between individual bursts being kept long enough to avoid doubly occupancy in the focal volume.35 Assuming 500 ms required between obtaining two single FRET data points, a 1 minute measurements yields only around 100 FRET data points; more than two orders of magnitude less than in our nanochannels. The throughput of camera-based smFRET detection with immobilized samples can in principle be higher than in our devices,36 but requires elaborate protocols for surface immobilization restricting the range of potential applications and potentially introducing surface-induced artefacts. Furthermore, as experiments at high time resolution require high laser powers leading to premature photo-bleaching, the continuous replenishment of molecules in our method is advantageous.
Using our nanochannel mixing devices, we first accessed non-equilibrium conditions by mixing primarily open DNA hairpins from one inlet with a high-salt solution from a second inlet triggering hairpin closing. Additionally, we observed polymerization of 25 bases on a DNA template by a DNA polymerase, illustrating that complex biological reactions can be followed in real time and in a continuous fashion.
In our current mixing design, the distance from junction to outlet is 100 μm, corresponding to a maximum residence time in the nanochannel of around 10 s. To gain access to further time points after mixing, designs using meandering channels could be implemented as demonstrated for confocal microscopy37,38 or widefield microscopy.39 Furthermore, our current field of view is cropped by a factor of two do ensure data acquisition at 100 Hz. With the use of faster cameras (sCMOS) and by reducing the overall magnification of the optical system (e.g., by replacing the 100× TIRF objective with a 60× objective), more molecules could be simultaneously observed for a longer time over a larger area and with greater time resolution.36
Conclusions
The introduced devices enable single-molecule studies with high throughput and millisecond time resolution under both equilibrium and non-equilibrium conditions. We used our platform to determine conformational changes and kinetic DNA synthesis by mapping the timescale of single reaction as a position in the nanochannel. We believe that the flexibility of the underlying design principles and the glass-only fabrication will allow for many applications in the (bio-) chemical sciences.Author contributions
J. H. and K. M. conceived the project and designed the fluidic devices. M. F. and J. H. performed preliminary experiments. C. F. performed the shown experiments. M. F., C. F., and J. H. wrote and adapted software for data analysis and analysed the data. M. F. and K. M. performed fluidic simulations. All authors contributed in designing the research, participating in discussions and writing the manuscript.Conflicts of interest
J. H. and K. M. filed a patent application on the nanofluidic designs.Acknowledgements
We thank Timo Wenzel and Ebru Acun for initial characterizations of the fluidic devices. We thank Adrie Westphal for help with the confocal scans. J. H. acknowledges support from a Marie Curie Career Integration Grant (#630992). S. G. L. acknowledges financial support from The Netherlands Organization for Scientific Research (NWO) and the European Research Council (ERC) under Project 278801. M. F. acknowledges financial support from the Graduate School Experimental Plant Sciences (EPS) under Project EPS3 3b 092. M. F and C. F. contributed equally to this work.References
- R. D. Smiley and G. G. Hammes, Chem. Rev., 2006, 106, 3080–3094 CrossRef CAS PubMed.
- V. I. Claessen, H. Engelkamp, P. C. M. Christianen, J. C. Maan, R. J. M. Nolte, K. Blank and A. E. Rowan, Annu. Rev. Anal. Chem., 2010, 3, 319–340 CrossRef CAS PubMed.
- A. Küchler, M. Yoshimoto, S. Luginbühl, F. Mavelli and P. Walde, Nat. Nanotechnol., 2016, 11, 409–420 CrossRef PubMed.
- N. G. Walter, C.-Y. Huang, A. J. Manzo and M. A. Sobhy, Nat. Methods, 2008, 5, 475–489 CrossRef CAS PubMed.
- C. Joo, H. Balci, Y. Ishitsuka, C. Buranachai and T. Ha, Annu. Rev. Biochem., 2008, 77, 51–76 CrossRef CAS PubMed.
- J. Hohlbein, K. Gryte, M. Heilemann and A. N. Kapanidis, Phys. Biol., 2010, 7, 031001 CrossRef PubMed.
- Y. Santoso, C. M. Joyce, O. Potapova, L. Le Reste, J. Hohlbein, J. P. Torella, N. D. F. Grindley and A. N. Kapanidis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 715–720 CrossRef CAS PubMed.
- J. Hohlbein, L. Aigrain, T. D. Craggs, O. Bermek, O. Potapova, P. Shoolizadeh, N. D. F. Grindley, C. M. Joyce and A. N. Kapanidis, Nat. Commun., 2013, 4, 2131 CrossRef PubMed.
- K. Mathwig, Q. Chi, S. G. Lemay and L. Rassaei, ChemPhysChem, 2016, 17, 452–457 CrossRef CAS PubMed.
- N. Banterle and E. A. Lemke, Curr. Opin. Biotechnol., 2016, 39, 105–112 CrossRef CAS PubMed.
- B. Wunderlich, D. Nettels, S. Benke, J. Clark, S. Weidner, H. Hofmann, S. H. Pfeil and B. Schuler, Nat. Protoc., 2013, 8, 1459–1474 CrossRef CAS PubMed.
- Y. Gambin, V. VanDelinder, A. C. M. Ferreon, E. A. Lemke, A. Groisman and A. A. Deniz, Nat. Methods, 2011, 8, 239–241 CrossRef CAS PubMed.
- S. Kim, A. M. Streets, R. R. Lin, S. R. Quake, S. Weiss and D. S. Majumdar, Nat. Methods, 2011, 8, 242–245 CrossRef CAS PubMed.
- S. Tyagi, V. VanDelinder, N. Banterle, G. Fuertes, S. Milles, M. Agez and E. A. Lemke, Nat. Methods, 2014, 11, 297–300 CrossRef CAS PubMed.
- J. F. Lesoine, P. A. Venkataraman, P. C. Maloney, M. E. Dumont and L. Novotny, Nano Lett., 2012, 12, 3273–3278 CrossRef CAS PubMed.
- R. Arayanarakool, L. Shui, S. W. M. Kengen, A. van den Berg and J. C. T. Eijkel, Lab Chip, 2013, 13, 1955–1962 RSC.
- G. W. Evans, J. Hohlbein, T. Craggs, L. Aigrain and A. N. Kapanidis, Nucleic Acids Res., 2015, 43, 5998–6008 CrossRef CAS PubMed.
- I. Rasnik, S. A. McKinney and T. Ha, Nat. Methods, 2006, 3, 891–893 CrossRef CAS PubMed.
- T. Cordes, J. Vogelsang and P. Tinnefeld, J. Am. Chem. Soc., 2009, 131, 5018–5019 CrossRef CAS PubMed.
- S. Farooq and J. Hohlbein, Phys. Chem. Chem. Phys., 2015, 17, 27862–27872 RSC.
- S. J. Holden, S. Uphoff, J. Hohlbein, D. Yadin, L. Le Reste, O. J. Britton and A. N. Kapanidis, Biophys. J., 2010, 99, 3102–3111 CrossRef CAS PubMed.
- J. C. Crocker and D. G. Grier, J. Colloid Interface Sci., 1996, 179, 298–310 CrossRef CAS.
- A. N. Kapanidis, N. K. Lee, T. A. Laurence, S. Doose, E. Margeat and S. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8936–8941 CrossRef CAS PubMed.
- J. Hohlbein, T. D. Craggs and T. Cordes, Chem. Soc. Rev., 2014, 43, 1156–1171 RSC.
- B. Hellenkamp, S. Schmid, O. Doroshenko, O. Opanasyuk, R. Kühnemuth, S. R. Adariani, B. Ambrose, M. Aznauryan, A. Barth, V. Birkedal, M. E. Bowen, H. Chen, T. Cordes, T. Eilert, C. Fijen, C. Gebhardt, M. Götz, G. Gouridis, E. Gratton, T. Ha, P. Hao, C. A. Hanke, A. Hartmann, J. Hendrix, L. L. Hildebrandt, V. Hirschfeld, J. Hohlbein, B. Hua, C. G. Hübner, E. Kallis, A. N. Kapanidis, J.-Y. Kim, G. Krainer, D. C. Lamb, N. K. Lee, E. A. Lemke, B. Levesque, M. Levitus, J. J. McCann, N. Naredi-Rainer, D. Nettels, T. Ngo, R. Qiu, N. C. Robb, C. Röcker, H. Sanabria, M. Schlierf, T. Schröder, B. Schuler, H. Seidel, L. Streit, J. Thurn, P. Tinnefeld, S. Tyagi, N. Vandenberk, A. M. Vera, K. R. Weninger, B. Wünsch, I. S. Yanez-Orozco, J. Michaelis, C. A. M. Seidel, T. D. Craggs and T. Hugel, Nat. Methods, 2018, 15, 669–676 CrossRef CAS PubMed.
- H. Liang, W. J. Nam and S. J. Fonash, Proc. NSTI Nanotechnol. Conf. Trade Show, Boston MA USA, 2008, vol. 3, p. 281283 Search PubMed.
- K. Mathwig, D. Mampallil, S. Kang and S. G. Lemay, Phys. Rev. Lett., 2012, 109, 118302 CrossRef PubMed.
- T. M. Squires and S. R. Quake, Rev. Mod. Phys., 2005, 77, 977–1026 CrossRef CAS.
- H. Song, Y. Wang and K. Pant, Microfluid. Nanofluid., 2012, 12, 265–277 CrossRef CAS PubMed.
- A. Sadeghi, AIChE J., 2016, 62, 4119–4130 CrossRef CAS.
- T. D. Craggs, M. Sustarsic, A. Plochowietz, M. Mosayebi, H. Kaju, A. Cuthbert, J. Hohlbein, L. Domicevica, P. C. Biggin, J. P. K. Doye and A. N. Kapanidis, bioRxiv, 2018, p. 263038 Search PubMed.
- M. C. Murphy, I. Rasnik, W. Cheng, T. M. Lohman and T. Ha, Biophys. J., 2004, 86, 2530–2537 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, 86th Edition, ed. D. R. Lide, CRC Press, Boca Raton, 86th edn, 2005 Search PubMed.
- C. Fijen, A. M. Silva, A. Hochkoeppler and J. Hohlbein, Phys. Chem. Chem. Phys., 2017, 19, 4222–4230 RSC.
- Y. Santoso, C. M. Joyce, O. Potapova, L. Le Reste, J. Hohlbein, J. P. Torella, N. D. F. Grindley and A. N. Kapanidis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 715–720 CrossRef CAS PubMed.
- M. F. Juette, D. S. Terry, M. R. Wasserman, R. B. Altman, Z. Zhou, H. Zhao and S. C. Blanchard, Nat. Methods, 2016, 13, 341–344 CrossRef PubMed.
- B. Wunderlich, D. Nettels, S. Benke, J. Clark, S. Weidner, H. Hofmann, S. H. Pfeil and B. Schuler, Nat. Protoc., 2013, 8, 1459–1474 CrossRef CAS PubMed.
- F. Dingfelder, B. Wunderlich, S. Benke, F. Zosel, N. Zijlstra, D. Nettels and B. Schuler, J. Am. Chem. Soc., 2017, 139, 6062–6065 CrossRef CAS PubMed.
- C. Freitag, C. Noble, J. Fritzsche, F. Persson, M. Reiter-Schad, A. N. Nilsson, A. Granéli, T. Ambjörnsson, K. U. Mir and J. O. Tegenfeldt, Biomicrofluidics, 2015, 9, 044114 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01175c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |