
A microfluidic platform utilizing anchored water-in-oil-in-water double emulsions to create a niche for analyzing single non-adherent cells†
Bo
Cai
a,
Tian-Tian
Ji
a,
Ning
Wang
a,
Xin-Bo
Li
a,
Rong-Xiang
He
c,
Wei
Liu
b,
Guobin
Wang
d,
Xing-Zhong
Zhao
 *b,
Lin
Wang
*ae and
Zheng
Wang
*ad
*b,
Lin
Wang
*ae and
Zheng
Wang
*ad
aResearch Center for Tissue Engineering and Regenerative Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China. E-mail: zhengwang@hust.edu.cn; lin_wang@hust.edu.cn
bSchool of Physics and Technology, Wuhan University, Wuhan 430072, China. E-mail: xzzhao@whu.edu.cn
cInstitute for Interdisciplinary Research & Key Laboratory of Optoelectronic Chemical Materials, and Devices of Ministry of Education, Jianghan University, Wuhan 430056, China
dDepartment of Gastrointestinal Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China
eDepartment of Clinical Laboratory, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430022, China
First published on 14th December 2018
Abstract
Non-adherent cells play key roles in various biological processes. Studies on this type of cell, especially at single-cell resolution, help reveal molecular mechanisms underlying many biological and pathological processes. The emerging microfluidics technology has developed effective methods for analyzing cells. However, it remains challenging to treat and monitor single live non-adherent cells in an in situ, long-term, and real-time manner. Herein, a microfluidic platform was set up to generate and anchor cell-laden water-in-oil-in-water (W/O/W) double emulsions (DEs) to investigate these cells. Within the device, W/O/W DEs encapsulating non-adherent cells were generated through two adjacent flow-focusing structures and subsequently anchored in an array of microchambers. These droplets maintained the W/O/W structure and the anchorage status in the continuous perfusion fluid for at least one week. The mass transfer of different molecules with suitable molecular weights and partition coefficients between the interior and exterior of W/O/W DEs could be regulated by perfusion fluid flow rates. These features endow this platform with potential to continuously supply encapsulated non-adherent cells with nutrients or small-molecule stimuli/drugs through fluid perfusion. Meanwhile, the confinement of cells in the anchored DEs favored long-term monitoring of cellular dynamic behaviors and responses. As a proof of concept, fluorescein diacetate (FDA) was employed to visualize the cellular uptake and biochemical metabolism of TF-1 human erythroleukemia cells. We believe that this W/O/W DE anchorage and perfusion platform would benefit single-cell-level studies as well as small-molecule drug discovery requiring live non-adherent cells.
Introduction
Non-adherent cells play significant functional roles in biological and pathological processes in the human body. For instance, erythrocytes undertake the task of oxygen transportation; leukocytes are critical to immunity; pathologically, tumor cells in the anchorage-independent state in the bloodstream can disseminate to distant sites resulting in metastasis.1 Analyzing non-adherent cells helps to reveal various physiological and pathological mechanisms in human beings. Given the ability to precisely manipulate a trace of liquid and sensitively detect chemicals at a scale similar to cell size, microfluidics has emerged as a powerful platform for cell biology study over the last decade.2,3 Fast development of microfluidic single-cell techniques4–8 permits a thorough investigation of the biological mystery of non-adherent cells.To analyze cellular spatiotemporal behaviors (i.e. cell fate, cell–cell communications and cellular responses to drugs/stimuli) of non-adherent cells, various microfluidic techniques have been developed. Single emulsion droplets are one of the representative samples.9,10 In this setting, cells are encapsulated into individual droplets for investigation. For example, a microfluidic platform named “Dropspots” was developed to study the enzyme kinetics of yeast cells.11 Similar devices were utilized in serial studies on single non-adherent cells (mainly T cells), such as immune activation,12 IL-10 cytokine secretion,13 and T cell–dendritic cell/cancer cell interaction.14,15 Methods using microchambers with fluid supply channels are also employed to monitor non-adherent cell behaviors.16–18 Cells together with culture medium are loaded into microchambers, followed by introducing oil to seal each chamber to form individual compartmentalization for single-cell study. Nevertheless, the inert property of oil makes the droplets and microchambers closed compartments. Thus, these two methods are unable to regulate the environment where single non-adherent cells are located and to detect cellular communications within the environment. Although hydrogels can be utilized in droplet microfluidics for long-term culture and single-cell analysis through the exchange of culture medium and the doping of chemicals,19–22 this regime is inherently not suitable for non-adherent cells since the microenvironment provided by the gel is drastically different from the aqueous surroundings where non-adherent cells are freely suspended. Other microdevices with microstructures, such as filters or pillars, are also commonly employed to immobilize non-adherent cells for investigating their spatiotemporal behaviors.23–25 However, inside these devices, cells are placed in the same bulk chamber for treatment without physically separating them from one another, which presumably cannot achieve bona fide single-cell analysis. Moreover, the immobilization of cells using hydraulic force is unstable and readily influenced by fluid flow fluctuation. Also, the hydraulic force that cells directly endure can be detrimental. Thus, various active manipulation techniques have been integrated to improve this type of microstructured device.26,27 For example, pneumatic valves were utilized to achieve compartmentalization and regulate fluid flow.28–30 Nevertheless, the added actuators and their peripheral equipment would inevitably increase the complexity and difficulty of experimental manipulation.
Herein, we have developed a microfluidic platform utilizing anchored water-in-oil-in-water (W/O/W) double emulsions (DEs) to realize in situ, real-time treatment and monitoring of non-adherent cells at single-cell resolution (Fig. 1). W/O/W DEs allowed the mass transfer of small molecules, ions, and gas between their interior and exterior.22,31,32 Thus, the reagents for stimulating cells or analyzing cellular behaviors could be transported into DEs, making real-time cell treatment possible. In our work, cell-laden W/O/W DEs could be generated at various sizes and anchored inside the device, mainly through surface modification of microchannels and elaborate design for hydraulic resistance modulation. By balancing osmotic pressure, the anchored DEs in the device achieved good stability lasting for over a week. By regulating fluid flow rates, we could control the mass transfer of certain molecules between the interior and exterior of DEs, which was fundamentally important to achieve on-chip treatment of non-adherent cells within the anchored DEs. As a proof of concept, fluorescein diacetate (FDA) molecules were employed to monitor cellular uptake and subsequent FDA intracellular metabolism in TF-1 human erythroleukemia cells. The perfusion of FDA over anchored W/O/W DEs allowed FDA molecules to enter DEs, and the viability of encapsulated TF-1 cells was monitored by different cellular metabolisms of FDA to fluorescein. We believe that this microfluidic platform based on anchored W/O/W DEs is applicable to a wide range of single-cell studies on non-adherent cells, such as monitoring responses of these cells to various small-molecule drugs towards drug discovery and more.
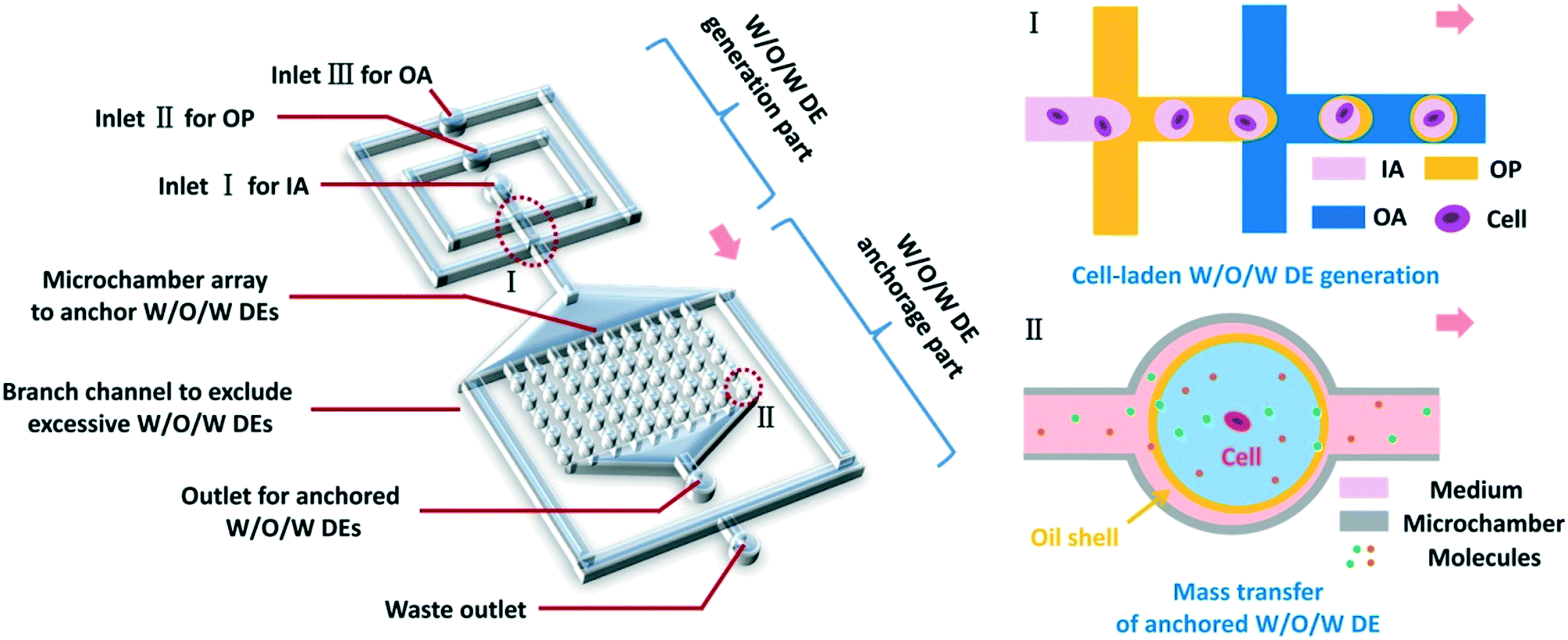 | ||
| Fig. 1 Schematics of the microfluidic platform utilizing anchored W/O/W DEs to treat and monitor non-adherent cells. This platform is composed of a DE generation part using two adjacent flow-focusing orifices and an array of microchambers to anchor DEs. The anchored W/O/W DEs enable continuous mass transfer of nutrients, small-molecular stimuli, or drugs between the interior and exterior of the DEs for encapsulated cells through fluid perfusion, and favor long-term monitoring of corresponding cellular behaviors and responses. (IA: inner aqueous phase; OP: oil phase; OA: outer aqueous phase). Pink arrows indicate the flow direction. | ||
Experimental
Chip design, fabrication and microchannel surface modification
Our microfluidic device is shown in Fig. 2(a–c). Two adjacent flow-focusing structures constituted the W/O/W DE generation part. All the microchannels of both flow-focusing orifices were 100 μm wide as well as the downstream connection microchannel. A 25 × 20 array of cylinder microchambers whose diameters were 100 μm formed the part to anchor W/O/W DEs on-chip. The microchambers in every row were connected by a rectangular microchannel (4200 μm-long and 50 μm-wide), and all the 25 microchannels finally led to an outlet for droplet collection. Two branch channels with 200 μm width were placed on each side of the microchamber array, respectively, and they led to a waste outlet. The hydraulic resistance of the branch channels and every anchorage microchannel was designed to be the same to ensure W/O/W DE loading and anchoring in the microchambers, and excessive DEs flowed into branch channels to the waste outlet (see Part S1 of the ESI†).11,33 The height of all the microchannels was 120 μm.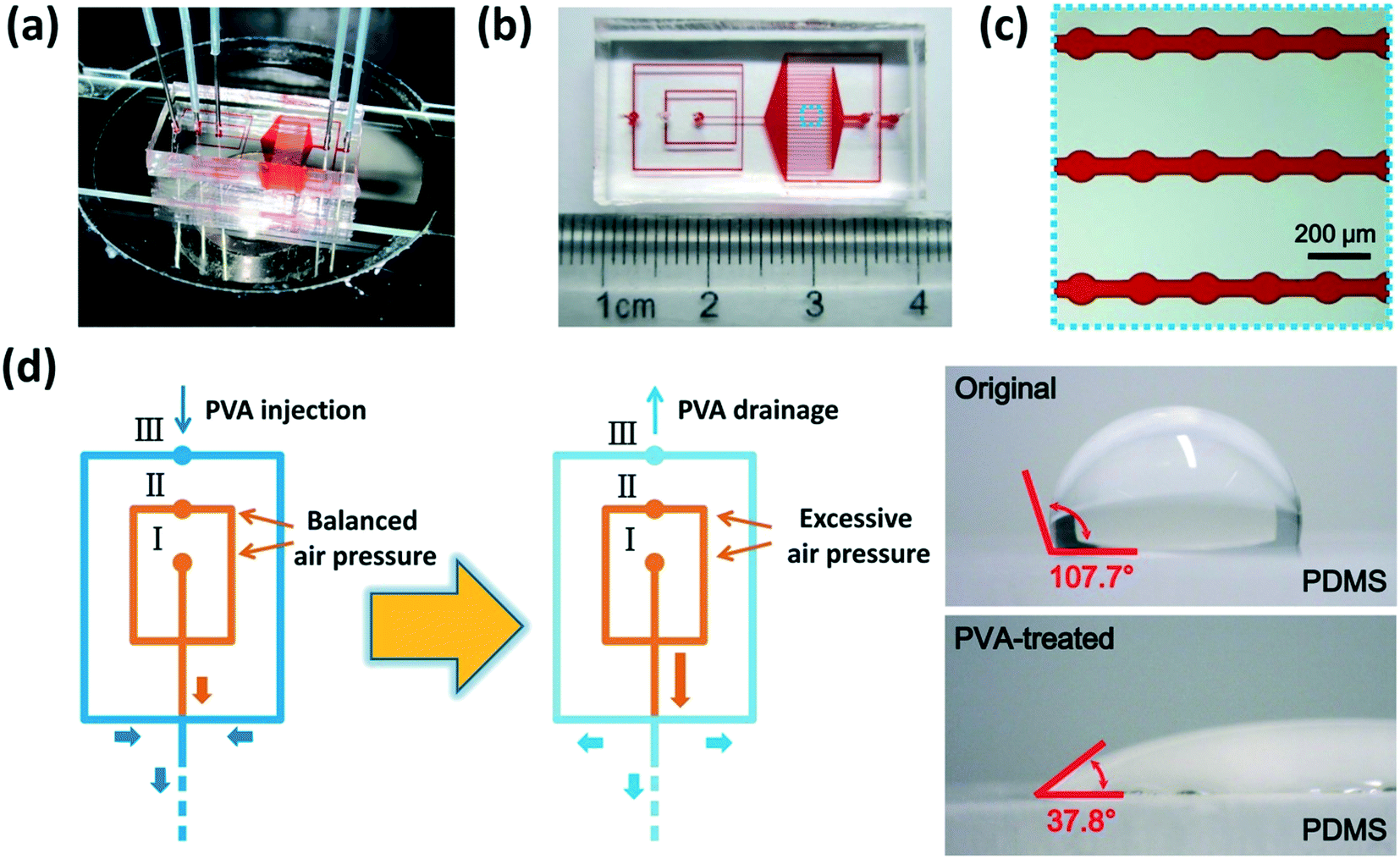 | ||
| Fig. 2 Device characterization and microchannel surface modification. (a) The microfluidic device mounted on an inverted microscope. (b) Photograph of the microfluidic device. (c) Microscopic magnification of the blue boxed area of the microchamber array in (b). (d) Hydrophilic modification of chosen microchannels by PVA coating. The illustration shows the modification cascade, and two photographs demonstrate the contact angle variation of the PDMS surface before and after PVA treatment. | ||
To fabricate the microfluidic device, at first, a mold on a Si wafer was manufactured according to standard photolithography using an SU-8 2100 photoresist (MicroChem, USA). Then a PDMS (polydimethylsiloxane, precursor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :crosslinker = 10:1, Sylgard 184, Dow Corning, USA) layer of the chip was obtained based on a soft lithography process. The glass slide used as the substrate was spin-coated with uncured PDMS (precursor:crosslinker = 10:1) at 1200 rpm and then baked at 80 °C for PDMS solidification. Finally, the hole-punched PDMS layer and PDMS-coated glass substrate were plasma-treated (Harrick, USA) to irreversibly bond to each other. The packaged device was baked at 110 °C for 30 min to regain hydrophobicity.
:crosslinker = 10:1, Sylgard 184, Dow Corning, USA) layer of the chip was obtained based on a soft lithography process. The glass slide used as the substrate was spin-coated with uncured PDMS (precursor:crosslinker = 10:1) at 1200 rpm and then baked at 80 °C for PDMS solidification. Finally, the hole-punched PDMS layer and PDMS-coated glass substrate were plasma-treated (Harrick, USA) to irreversibly bond to each other. The packaged device was baked at 110 °C for 30 min to regain hydrophobicity.
Immediately after baking, parts of the microchannels were treated with PVA (polyvinyl alcohol, 87–90% hydrolyzed, molecular weight 30000–70000, Sigma-Aldrich, USA) to obtain hydrophilicity,34 as shown in Fig. 2(d). Briefly, a newly-prepared 4% (w/v) PVA solution was injected into the chip from inlet III, while compressed air was injected from inlets I and II to keep the PVA solution away from the microchannels of the inner and middle phases which needed to remain hydrophobic. The rest of the microchannels were filled with PVA and incubated for 15 min. Subsequently, the PVA solution was driven out from the microchannels by excessive compressed air injected from inlets I and II, and then the chip was baked at 100 °C for 5 min. This PVA incubation and baking process was repeated 3 times to form reliable PVA coating on the microchannel surface. All the PVA-treated chips were placed statically overnight before use. We measured the contact angle of the original and PVA-treated PDMS surfaces using a homemade image analysis system to validate the variation of surface wettability.
DE generation, loading and anchoring
In the prepared microfluidic chips, DEs could be generated through the two adjacent flow-focusing structures, and loaded into and anchored in the microchambers. The oil phase (OP) of all the experiments in this work utilized PMX-200 silicone oil (50 cSt, Xiameter, Dow Corning, USA) mixed with PDMS (precursor, Sylgard 184, Dow Corning, USA) at a ratio of 7:3 (v/v). Different aqueous solutions were prepared for different experiments, and 0.5% (w/v) PVA was added into the aqueous solutions as the surfactant.35 To balance the osmotic pressure of the interior and exterior of DEs,36 the main liquid ingredients were the same for both inner aqueous (IA) and outer aqueous (OA) solutions. For OA solutions, glycerol (Sigma-Aldrich, USA) was added at a 30% volume ratio to increase the liquid viscosity.37
The IA, OP and OA phases were injected into the chip from the corresponding inlets using syringe pumps (TS-1A, Longer, China). W/O/W DEs were generated with different sizes at different fluid flow rates. When their size was appropriate, these DEs could be loaded into the microchambers hydrodynamically. After all the microchambers were occupied, the DE generation process was stopped and a wash buffer (different in different experiments) was injected to remove excessive DEs or reagents. Different solutions such as culture medium, drugs, small molecules, etc. could be injected from inlets I and II and perfuse the microchamber array to incubate those DEs.
Mass transfer investigation
To investigate the mass transfer between the interior and exterior of the anchored DEs under perfusion conditions, Rhodamine B, Rhodamine 6G and fluorescein isothiocyanate-dextran (FITC-dextran, MW = 3000–5000, 10000 and 20000, respectively) (Sigma-Aldrich, USA) were prepared in deionized (DI) water (purified using a Millipore Q-3 system, Millipore, USA) at a concentration of 1 mM, 1 mM and 50 μM, respectively. These solutions were all doped with 0.5% PVA (w/v) to be used as IA phases. Glycerol and DI water were mixed at a volume ratio of 7:3 and also doped with 0.5% (w/v) PVA to be the OA phase. After the DEs were loaded and anchored, 0.5% (w/v) PVA solution was injected into the chip as the wash buffer to remove other solutions and as the perfusion solution to test the mass transfer ability of the DEs. The perfusion flow rate was set at different values to investigate the influence of fluid flow on the molecular transport. Images of different DEs were acquired under the same conditions every 10 min, and were analyzed by Image Pro Plus software (Media Cybernetics, USA) to obtain the optical or fluorescence intensity variation. We also analyzed the optical images of Rhodamine B at different concentrations to obtain the relationship between the optical intensity and Rhodamine concentration. The experiments were repeated 3 times to obtain the average values as well as the standard deviations.
FDA perfusion to detect cellular metabolism
TF-1 human erythroleukemia cells were provided by Zhongnan Hospital, Wuhan University. All the tools and solutions used for cell assays were sterilized by autoclaving or 0.22 μm filtration.To encapsulate cells in DEs, culture medium RPMI-1640 (Gibco™, Thermo-Fisher, USA) was used. After harvesting, cells were resuspended in RPMI-1640 doped with 0.5% (w/v) PVA as the IA phase. The OA phase was prepared using RPMI-1640 mixed with glycerol at a volume ratio of 7:3, and also doped with 0.5% PVA (w/v). After cell-laden W/O/W DEs were anchored in microchambers, another RPMI-1640 medium doped with 0.5% (w/v) PVA solution was injected from inlets I and II as the wash buffer to remove excessive DEs. This RPMI perfusion was kept for 7 days to validate the stability of the anchored W/O/W DEs in the microchamber array.
To monitor the perfusion of small molecules and their interaction with cells encapsulated inside DEs, RPMI-1640 doped with 0.5% (w/v) PVA and fluorescein diacetate (FDA, 1.6 μg mL−1, Sigma-Aldrich, USA) was injected into the chip at a flow rate of 100 μL h−1 to immerse the anchored cell-laden W/O/W DEs for 10 min. Subsequently, RPMI-1640 doped with 0.5% (w/v) PVA solution was again injected to remove the excess FDA dye and perfusion was maintained at a flow rate of 50 μL h−1. An IX71 fluorescence inverted microscope was employed to observe the interaction of FDA molecules and TF-1 cells.
Results and discussion
W/O/W DE generation and their loading into microchambers
We utilized two adjacent flow-focusing orifices with different wettability to generate W/O/W DEs (Fig. 3(a)). At the first flow-focusing orifice, the intrinsic hydrophobicity of the channel surfaces due to PDMS enabled the generation of water-in-oil (W/O) droplets. Cells suspended in the IA phase were compartmentalized into these droplets. PVA coating on the surfaces of the second flow-focusing microchannels allowed the generation of oil-in-water (O/W) droplets owing to wettability switching from the hydrophobicity of the first flow-focusing orifice to the hydrophilicity of the second one (Fig. 2(d)). Therefore, when the cell-laden W/O droplets flowed from the first flow-focusing orifice to the second one, W/O/W DEs with cells inside were formed (Fig. 3(d), Movie S1 and S2†). The cell quantity in every DE was fitted with the Poisson distribution.38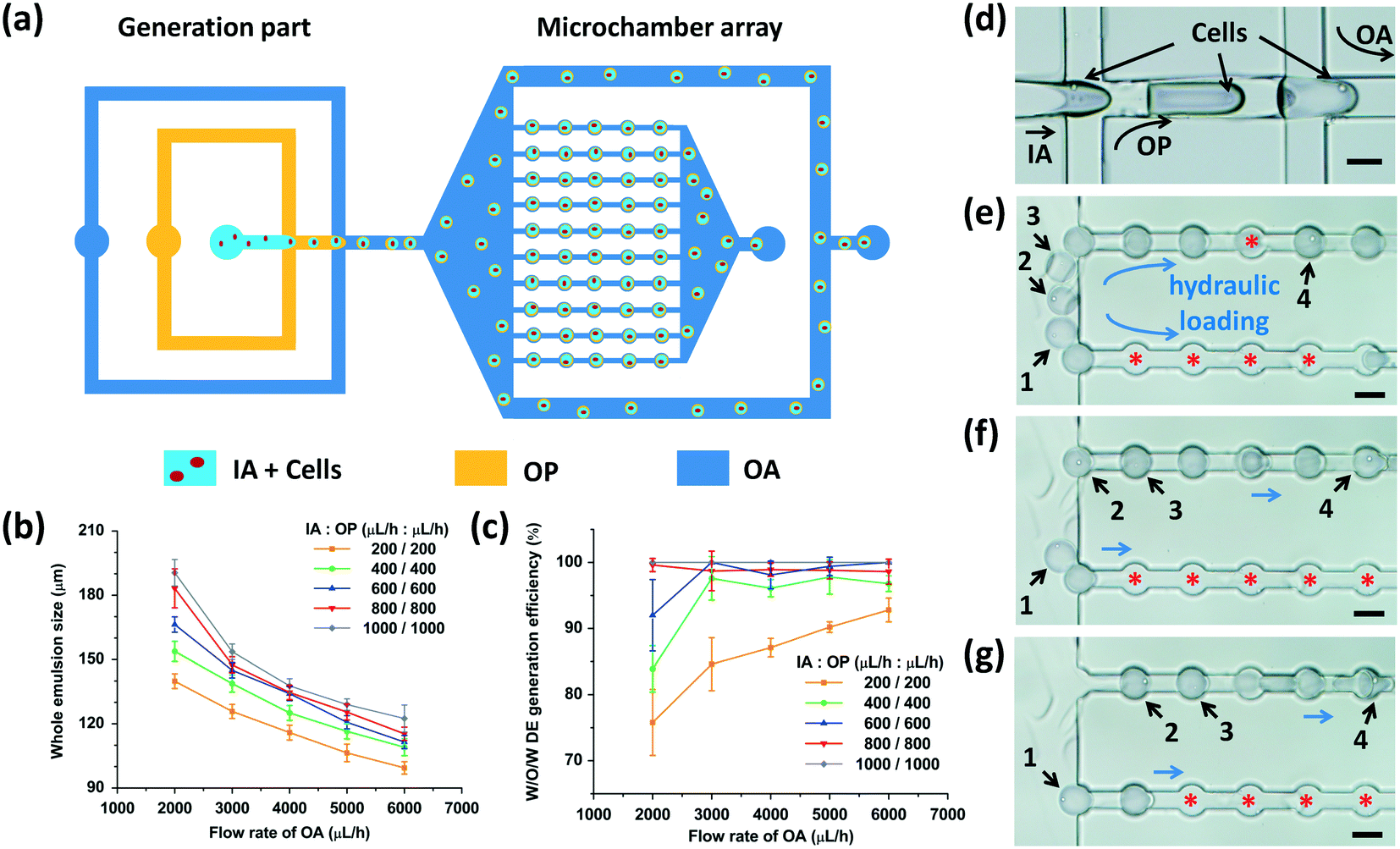 | ||
| Fig. 3 Cell-laden W/O/W DE generation and loading process. (a) Schematics showing that DEs are generated through two adjacent flow-focusing structures, and subsequently loaded into microchambers hydraulically. Excessive DEs are drained out through two branch channels. (b) Regulation of DE size by adjusting the flow rates of different fluid phases. (c) DE generation efficiency mediated by the flow rates of different fluid phases. (d) Microscopic image showing cell encapsulation into the interior of W/O/W DEs through the adjacent flow-focusing orifices. (e–g) Microscopic images showing the loading cascade of W/O/W DEs into microchambers. Four cell-laden W/O/W DEs are indicated by black arrows. Red asterisks mark the empty microchambers during the loading procedure. Scale bars, 100 μm. | ||
We next investigated the effects of fluid flow rates on the generation of W/O/W DEs. As shown in Fig. 3(b), through adjusting the fluid flow rates of the different phases, the DE size could be precisely modulated. When the flow rates of the IA and OP phases were fixed, the DE size decreased as the flow rate of the OA phase increased. In contrast, when the flow rate of the OA phase was fixed, the DE size increased as the flow rates of the IA and OP phases increased, consistent with a previous study reporting droplet size regulation by a flow-focusing regime.39
Due to the different viscosities of the fluid phases, varied fluid flow rates, and the function of surfactants, in our experiments, two different droplet generation regimes emerged in the two flow-focusing orifices: a dripping regime at the first orifice for W/O droplet generation and a jetting regime to generate O/W droplets at the second orifice. Given the poor monodispersity of the jetting regime to produce droplets compared to the dripping regime,40 the generation efficiency of DEs was affected by the fluid flow rates (Fig. 3(c)). Here, the generation efficiency was defined as the ratio of the DE quantity versus the quantity of all the generated emulsions. Some O/W droplets without a W/O droplet inside were produced at the second flow-focusing orifice due to unstable droplet generation resulting from the jetting regime (Fig. S2†). When the flow rates of all the fluid phases increased, faster droplet production was achieved.39 Thus, generation of W/O droplets and O/W droplets matched better to achieve a higher W/O/W generation efficiency. Given that the microchambers were 100 μm in diameter, flow rates of 200 to 400 μL h−1 for the IA and OP phases and 5000 to 6000 μL h−1 for the OA phase were chosen to obtain W/O/W DEs with appropriate sizes to be anchored. Of note, depending on the physical and chemical properties (such as viscosity and surface tension) of the chosen liquids, the fluid flow rates should be optimized accordingly.
After generation, the DEs entered the microchamber array (Fig. S2†). The hydraulic resistance of every row of microchambers was the same as that of the branch microchannels with a designed channel structure (see Part S1 of the ESI†).11,33 When the size of the DEs was adjusted to fall within 100 to 120 μm, they could be pressed into the microchambers one by one hydraulically, when the flow rate of the OA phase was sufficiently high (Movie S3†). Fig. 3(e–g) show the loading cascade of the W/O/W DEs into the microchambers. The hydraulic force of the OA phase drove the W/O/W DEs into the microchambers one after another. The latter DE that entered the fluid passage forced the former DE to move to the next microchamber and then occupied the empty microchamber that was left. This process was repeated until all the microchambers were occupied by the emulsions (double or single). The excessive DEs were drained out from the two branch microchannels. Our device had 500 microchambers in total to enable parallel analysis of cells encapsulated at the same time.
W/O/W DE anchoring and long-term structural stability within fluid perfusion in microchambers
As shown in Fig. 4(a), when the loading process was finished, the DEs occupied the microchambers. The anchorage was attributed to the low surface energy of the DEs (due to the surfactant).41 These DEs would not readily deform to flow through the narrow fluid passages unless the flow rate was considerably high to generate sufficiently strong hydraulic force. Therefore, different solutions could be employed to perfuse the microchambers at adjusted flow rates to maintain the anchorage of these W/O/W DEs in the microchambers (see Part S3 of the ESI†). Excessive DEs were directed to the waste outlet through the branch channels hydraulically by the perfusion fluid as well.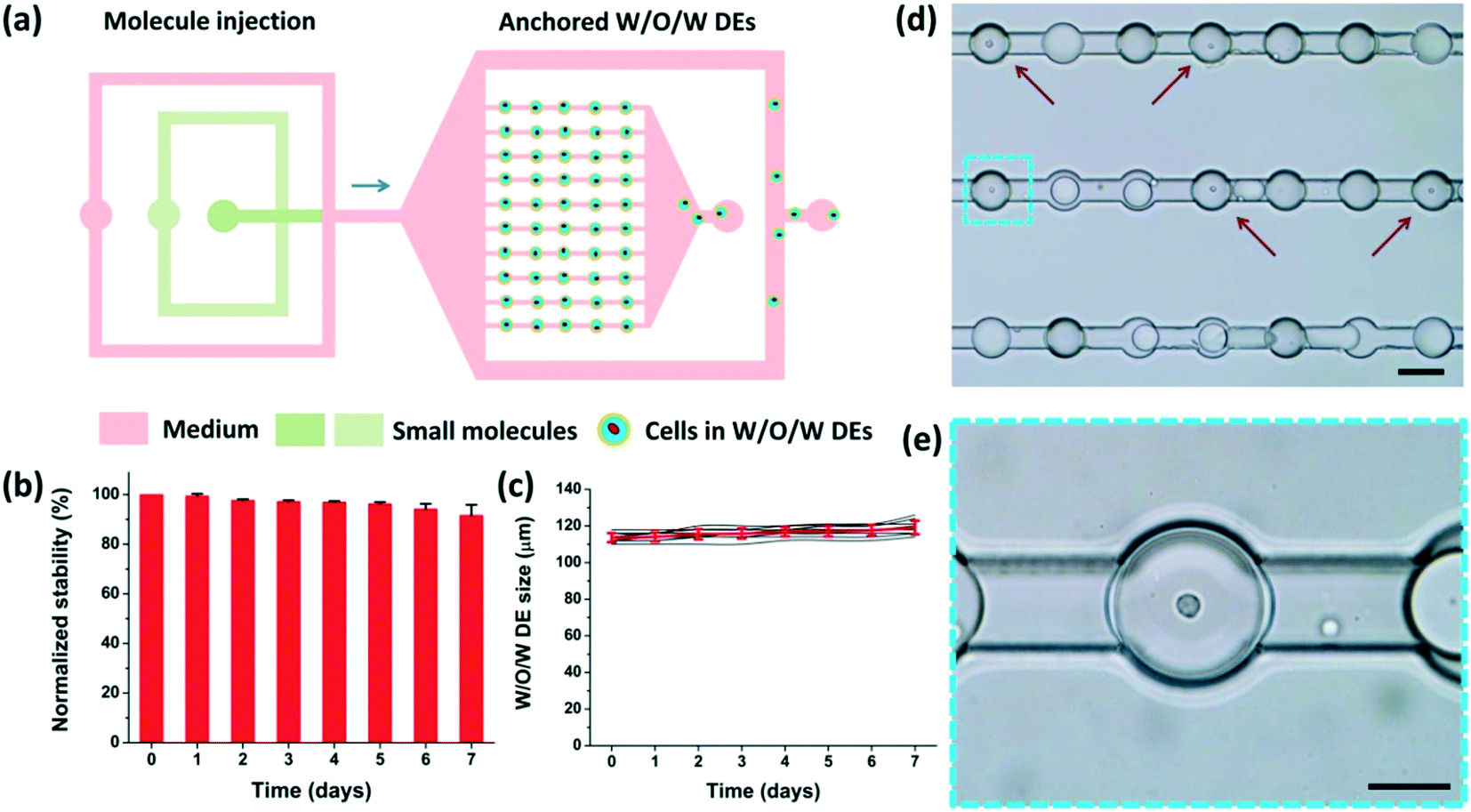 | ||
| Fig. 4 W/O/W DE anchoring and stability under continuous perfusion. (a) After the DEs are anchored in the microchambers, medium is injected into the device to drain out excessive DEs, demonstrating the potential to supply the encapsulated non-adherent cells with nutrients, small-molecule stimuli, or drugs continuously through fluid perfusion. (b) Normalized stability of anchored cell-laden W/O/W DEs during the perfusion process. The flow rate of the culture medium is set at 50 μL h−1. (c) The size variation of 10 typical anchored W/O/W DEs during the perfusion process. The red curve is the mean diameter and corresponding standard deviation of these 10 DEs. (d) Typical microscopic image of cell-laden W/O/W DEs (indicated by red arrows) anchored in the microchambers. Scale bars, 100 μm. (e) Magnification of a blue boxed region in (d) to show an anchored DE encapsulating a single cell. Scale bars, 50 μm. | ||
The stability of the anchored DEs in a perfusion flow, i.e. the maintenance of the W/O/W structure and size, was critical to implement long-term on-chip treatment/monitoring of encapsulated cells. Thus, after DE generation using RPMI-1640 culture medium (containing fetal bovine serum and penicillin–streptomycin) and their anchorage in the microchambers, we investigated the morphological stability of these DEs over 7 days. During these days, the same medium perfused the whole chip continuously from inlets I and II at a flow rate of 50 μL h−1 (Movie S4†). As shown in Fig. 4(b), over 90% anchored droplets maintained their W/O/W DE structure after 7 days. Fig. 4(c) shows that no obvious size variation (less than 5%) of the anchored W/O/W DEs was observed. These results indicate a good stability of these anchored DEs in the perfusion fluid in a confined environment. As the osmotic pressure of the perfusion fluid and the DE interior liquid was tightly associated with stability,36 the concentrations of the ingredients in the perfusion fluid and the DE interior liquid should be carefully selected and adjusted. Meanwhile, the confinement of living cells in anchored DEs offers a unique opportunity for long-term monitoring of cellular behaviors or responses (Fig. 4(d) and (e)).
Mass transfer between the interior and exterior of DEs
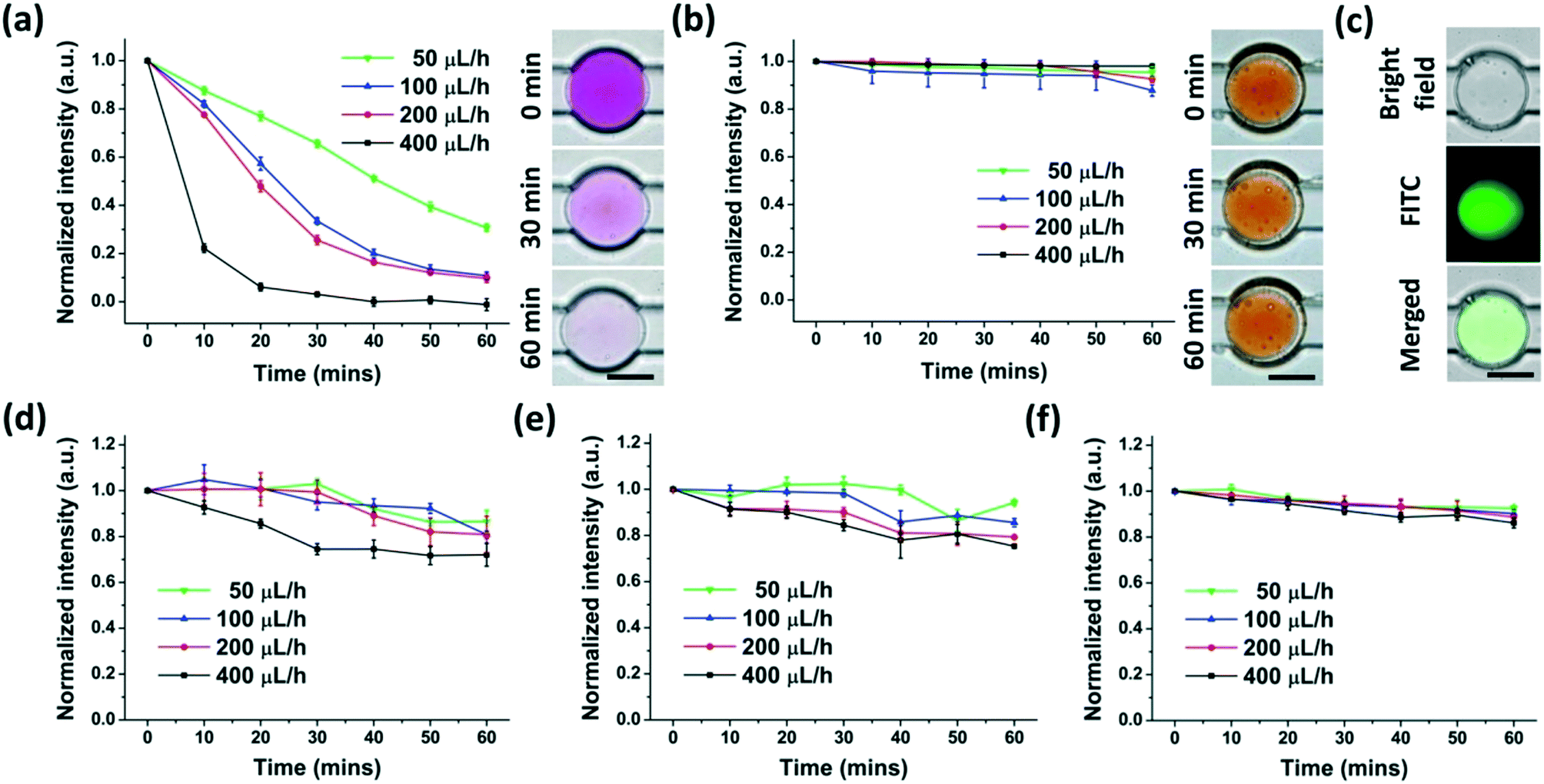
To supply DE-encapsulated cells with nutrients or drugs by the perfusion fluid, the anchored DEs should have the ability to transport molecules between the interior and exterior. It was reported that water and water-soluble materials (gas, ions, and small molecules) were able to shuttle through the oil film of some types of DEs. The mechanisms underlying this phenomenon were mainly due to (1) “swelling-breakdown” mechanisms, and (2) diffusion and/or permeation through oil membranes.31,37,42–44We then investigated the molecule transport of anchored W/O/W DEs in our experiments using molecules with different molecular weights and partition coefficients. A colorimetric method was set up to analyze the concentrations of chemicals in a solution by measuring the optical or fluorescence intensity as shown in Fig. S4.† Three types of molecules (Rhodamine B, Rhodamine 6G, and FITC-dextran) with different molecular weights and partition coefficients were employed. While the molecular weights of Rhodamine B and Rhodamine 6G were the same (479.01), their partition coefficients were different.22 Three types of FITC-dextran had much higher molecular weights (3000–5000, 10000 and 20000, respectively) than Rhodamine B and Rhodamine 6G. These molecules were encapsulated in the interior of the anchored W/O/W DEs, and a 0.5% PVA solution continuously perfused these DEs at different flow rates. This perfusion fluid continuously carried off the molecules that were transported from the interior to the exterior. The mass transfer procedure was monitored by analyzing colorimetric or fluorescence intensity changes within the DE interior.
Rhodamine B with a high partition coefficient (water to silicone oil) rapidly moved from the interior to the exterior of the DEs, which was affected significantly by the flow rate of the perfusion fluid (Fig. 5(a)). Faster perfusion flow made mass transport faster, likely due to the rapid clearance of molecules around the DEs. This clearance maintained a high concentration disparity between the interior and the exterior of W/O/W DEs, resulting in swift molecule transport. Moreover, as the concentration of Rhodamine B decreased, the transport slowed down (Fig. S5†). However, the sensitivity of colorimetric image analysis limited the reliable detection of Rhodamine B at very low concentrations.
 | ||
| Fig. 5 Mass transfer between the interior and exterior of W/O/W DEs. (a) Rhodamine B transport influenced by the perfusion fluid, measured by the optical intensity. (b) Rhodamine 6G transport influenced by the perfusion fluid, measured by the optical intensity. (c) Typical fluorescence micrographs of FITC-dextran in the DE interior. (d–f) FITC–dextran transport influenced by the perfusion fluid, measured by the fluorescent intensity. The molecular weight of FITC-dextran used is 3000–5000 (d), 10000 (e) and 20000 (f), respectively. Scale bars, 50 μm. | ||
Rhodamine 6G with a low partition coefficient (water to silicone oil) but the same molecular weight as Rhodamine B almost could not be transported through the oil film, suggesting that the partition coefficient is also an important factor in the mass transfer of DEs. In this setting, the flow rate of the perfusion fluid did not affect the mass transport (Fig. 5(b)). These results indicate that the partition coefficient of the chemicals and the oil/aqueous phase should be carefully selected if this anchored W/O/W DE platform is adopted to modulate the interior environment through continuous external fluidic perfusion.
The molecular weight was another important factor affecting the mass transfer of DEs. As shown in Fig. 5(c–e), given possible fluorescence bleaching, FITC-dextran with a higher molecular weight than Rhodamine B was transported from the interior to the exterior of the DEs at a much slower speed. However, the flow rate of the perfusion fluid still had a considerable impact on the transport when the molecular weight was less than 10000 (Fig. 5(c) and (d)). However, for molecules with a molecular weight around 20000, their transport across the oil film of PMX-200 and the PDMS precursor was reduced and almost not influenced by the flow rate of the perfusion fluid (Fig. 5(e)). The experiments also showed that this oil film had poor performance for ion transport between the interior and exterior of the DEs (data not shown). Therefore, the oil phases and corresponding surfactants with the properties for fast mass transfer of large molecules (e.g. various cytokines with a molecular weight larger than 10 kDa) as well as ions are preferred for the generation of W/O/W DEs for potential bio-applications related to non-adherent cells, such as long-term cell culture.
Assessment of the cellular uptake and biochemical metabolism of encapsulated non-adherent cells
To demonstrate the potential of our device in treating and monitoring non-adherent cells, we attempted to dynamically visualize the cellular uptake activity and biochemical metabolism of human erythroleukemia cells (TF-1 cell line) using FDA, a molecule which can be taken up by viable cells that use intracellular enzymes to biochemically convert non-fluorescent FDA to green fluorescent metabolite fluorescein. TF-1 cells were encapsulated into DEs and anchored in the microchamber array (Fig. 6(a)). After a perfusion of the wash buffer to remove the excess emulsions (Fig. 6(b), original), FDA solution was injected into the chip and perfused continuously (Fig. 6(b), FDA perfusion) for 10 min. As aforementioned, W/O/W DEs enabled mass transfer between their interior and exterior. FDA molecules continuously entered the interior of the DEs. They were then taken in by live cells within the DEs. The cells enzymatically converted intracellular FDA molecules to a fluorescein metabolite that emitted green fluorescence (Fig. 6(b), washed). Notably, after another perfusion of the wash buffer, the excess FDA molecules were effectively removed from the microchannels as well as from the interior of the DEs, indicating an effective two-way mass transfer (Movie S5 and S6†). Moreover, cellular viability could be reflected by the intensity of fluorescence emitted from the cells (Fig. 6(c–e)). This is because cells with different viabilities had different uptake efficacies and converting metabolisms of FDA molecules, resulting in different amounts of fluorescein accumulated in the cells. Dead cells inside the DEs did not absorb FDA molecules at all (Fig. 6(d)), neither could they convert FDA into a fluorescein metabolite. This mass transfer feature of W/O/W DEs enables us to precisely regulate the environment within DEs through delivering different chemicals to encapsulated cells via perfusion, thus helping reveal the dynamic behaviors of these cells in a real-time manner.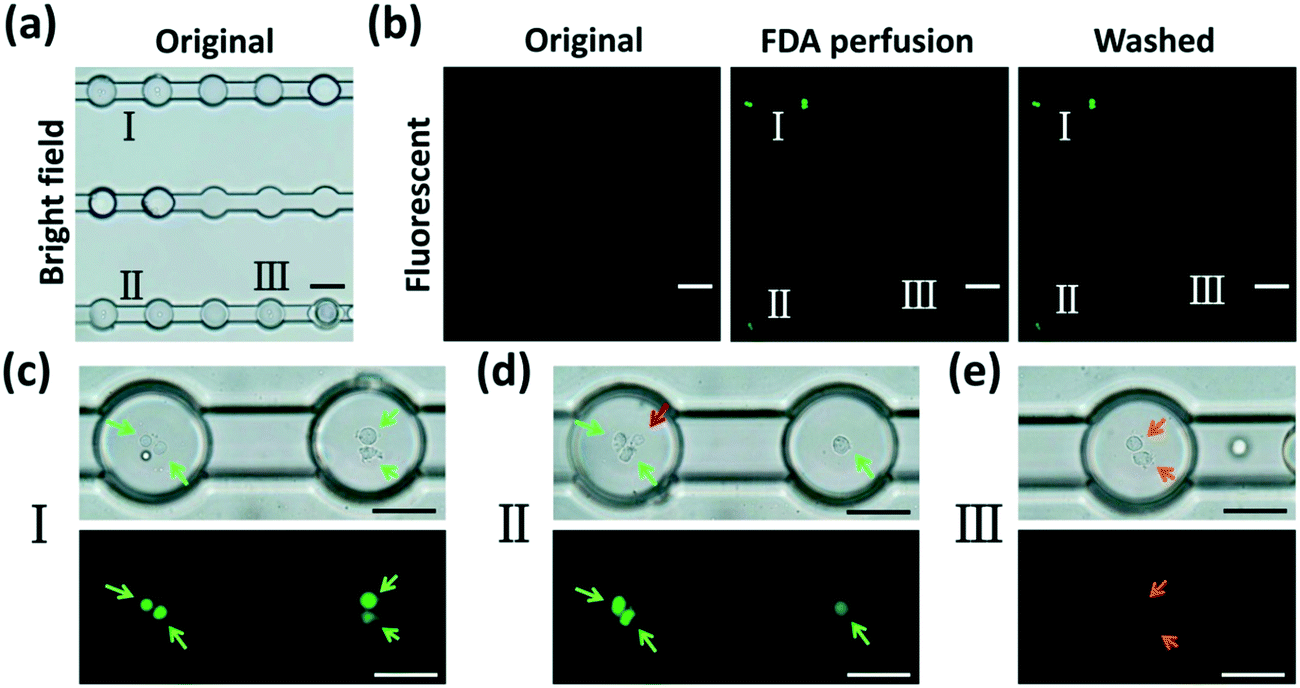 | ||
| Fig. 6 FDA perfusion and corresponding cellular uptake monitoring to show the different viabilities and FDA metabolism of human erythroleukemia cells (TF-1 cell line). (a) Cells are encapsulated in DEs and anchored in the microchambers. I, II and III label DEs encapsulating cells with different viabilities. (b) Fluorescence images showing that cellular metabolic dye FDA diffuses into the DEs shown in (a). No obvious fluorescence is observed before and during FDA perfusion. After FDA perfusion and wash-away processes, cells with different viabilities shown in (a) uptake FDA and show green fluorescence at different intensities. (c–e) Magnifications of cells with different viabilities encapsulated in DEs (I, II and III in (a), respectively) showing green fluorescence of different intensities. Green arrows in (c) and (d) indicate live cells that uptake non-fluorescent FDA molecules and convert them effectively to fluorescein emitting bright green fluorescence. The red arrow in (d) indicates a dead cell that cannot uptake FDA and thus does not show any fluorescence. Orange arrows in (e) indicate dying cells whose metabolism of converting FDA to green fluorescent fluorescein is weak. Scale bars, 100 μm ((a) and (b)) and 50 μm ((c–e)), respectively. | ||
Although the PVA surfactant in the aqueous solutions was proven not to impact cellular viability (see Part S6 of the ESI†), TF-1 cells encapsulated in anchored W/O/W DEs could only remain viable and continue to proliferate during 24 h of culture under continuous perfusion (see Part S7 of the ESI†). After optimizing the liquid used for improved mass transfer activity (mainly ions and molecules with a molecular weight larger than 5 kDa), we believe that our platform has the potential for long-term culture of encapsulated non-adherent cells in a single-cell manner by continuously supplying them with nutrients and gas, and carrying off wastes. Moreover, by using suitable aqueous and oil phases to form W/O/W DEs, small-molecule drugs could be effectively delivered to encapsulated cells of interest in a controlled fashion to achieve in situ, real-time, long-term monitoring of cellular responses, which would undoubtedly benefit pharmaceutical development towards non-adherent-cells involved in diseases, such as leukemia.
Conclusions
In summary, we developed a microfluidic platform utilizing anchored W/O/W DEs for in situ, real-time, long-term treatment and monitoring of non-adherent cells. By coating hydrophilic PVA on the surfaces of PDMS microchannels to adjust wettability, W/O/W DEs were generated inside two planar adjacent flow-focusing structures with sizes ranging from 99 μm to 190 μm and production efficiency varying from 75.8% to 100% at different flow rates. Given the elaborate design of hydraulic resistance inside the microchips, suitable W/O/W DEs could be anchored in an array of microchambers with a diameter of 100 μm. These DEs were utilized to encapsulate cells and the structure of the DEs could be stably maintained within the microchambers over 7 days in a continuous perfusion of culture medium. This good stability of the anchored W/O/W DEs provided ideal compartments for encapsulated cells. Moreover, W/O/W DEs were capable of mass transfer between their interior and exterior, which was regulated by perfusion flow rates, molecular weights and partition coefficients. Molecules with smaller molecular weights or higher partition coefficients (water to oil) could be exchanged more rapidly between the inner and the outer phases of the DEs, revealing the potential of using our platform to treat and monitor encapsulated non-adherent cells in a single-cell manner. As a proof of concept, TF-1 human erythroleukemia cells were encapsulated in W/O/W DEs that were anchored inside the device, and FDA molecules were employed to successfully visualize and assess cellular uptake and chemical compound metabolism in cells. Thus, our work demonstrates the potential value of this microfluidic platform to studies requiring in situ, long-term, and real-time treatment and monitoring of non-adherent cells at single-cell resolution.Author contributions
B. C., X. -Z. Z, L. W. and Z. W. conceived the idea of the study. B. C., T. -T. J., N. W., X. -B. L, and R. -X. H. performed the experiments, and collected and analyzed the data. B. C. and Z. W. wrote the draft of the paper. W. L, G. -B. W. and L. W. commented on the manuscript. All the authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate Mr. Zhu-Hao Wu, Mr. Zi-Xiang Wang, Miss Yue Sun, Mr. Ke-Ke Chen (Wuhan University, Wuhan, China), Mr. Lei Zhang and Mr. Jia-Hao Dai (Huazhong University of Science and Technology, Wuhan, China) for their assistance and discussion. This work was supported by the National Key R&D Program for Major Research Instruments (81527801), the National Natural Science Foundation of China (81741019, 81572866, 81773104, 81773263, and 81472740), the Natural Science Foundation of Hubei Province (2018CFB124 and 2015CFA049), and the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST.References
- C. L. Buchheit, K. J. Weigel and Z. T. Schafer, Nat. Rev. Cancer, 2014, 14, 632 CrossRef CAS PubMed.
- J. Sibbitts, K. A. Sellens, S. Jia, S. A. Klasner and C. T. Culbertson, Anal. Chem., 2018, 90, 65–85 CrossRef CAS PubMed.
- D. Huber, A. Oskooei, X. Casadevall i Solvas, A. deMello and G. V. Kaigala, Chem. Rev., 2018, 118, 2042–2079 CrossRef CAS PubMed.
- S. Taheri-Araghi, S. D. Brown, J. T. Sauls, D. B. McIntosh and S. Jun, Annu. Rev. Biophys., 2015, 44, 123–142 CrossRef CAS PubMed.
- T. Konry, S. Sarkar, P. Sabhachandani and N. Cohen, Annu. Rev. Biomed. Eng., 2016, 18, 259–284 CrossRef CAS PubMed.
- L. Valihrach, P. Androvic and M. Kubista, Int. J. Mol. Sci., 2018, 19, 807 CrossRef PubMed.
- Y. F. S. Seah, H. Hu and C. A. Merten, Mol. Aspects Med., 2018, 59, 47–61 CrossRef CAS PubMed.
- V. C. Shukla, T.-r. Kuang, A. Senthilvelan, N. Higuita-Castro, S. Duarte-Sanmiguel, S. N. Ghadiali and D. Gallego-Perez, Trends Biotechnol., 2018, 36, 549–561 CrossRef CAS PubMed.
- L. Shang, Y. Cheng and Y. Zhao, Chem. Rev., 2017, 117, 7964–8040 CrossRef CAS PubMed.
- Z. Zhu and C. J. Yang, Acc. Chem. Res., 2017, 50, 22–31 CrossRef CAS PubMed.
- C. H. J. Schmitz, A. C. Rowat, S. Köster and D. A. Weitz, Lab Chip, 2009, 9, 44–49 RSC.
- S. Sarkar, V. Motwani, P. Sabhachandani, N. Cohen and T. Konry, J. Clin. Cell. Immunol., 2015, 6, 334 Search PubMed.
- T. Konry, M. Dominguez-Villar, C. Baecher-Allan, D. A. Hafler and M. L. Yarmush, Biosens. Bioelectron., 2011, 26, 2707–2710 CrossRef CAS PubMed.
- T. Konry, A. Golberg and M. Yarmush, Sci. Rep., 2013, 3, 3179 CrossRef PubMed.
- S. Sarkar, P. Sabhachandani, D. Stroopinsky, K. Palmer, N. Cohen, J. Rosenblatt, D. Avigan and T. Konry, Biomicrofluidics, 2016, 10, 054115 CrossRef CAS PubMed.
- A. Dewan, J. Kim, H. McLean Rebecca, A. Vanapalli Siva and N. Karim Muhammad, Biotechnol. Bioeng., 2012, 109, 2987–2996 CrossRef CAS PubMed.
- J. Shemesh, T. Ben Arye, J. Avesar, J. H. Kang, A. Fine, M. Super, A. Meller, D. E. Ingber and S. Levenberg, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11293–11298 CrossRef CAS PubMed.
- G. Amselem, C. Guermonprez, B. Drogue, S. Michelin and C. N. Baroud, Lab Chip, 2016, 16, 4200–4211 RSC.
- L. Zhang, K. Chen, H. Zhang, B. Pang, C.-H. Choi, S. Mao Angelo, H. Liao, S. Utech, J. Mooney David, H. Wang and A. Weitz David, Small, 2018, 14, 1702955 CrossRef PubMed.
- T. Kamperman, S. Henke, W. Visser Claas, M. Karperien and J. Leijten, Small, 2017, 13, 1603711 CrossRef PubMed.
- S. Sart, R. F. X. Tomasi, G. Amselem and C. N. Baroud, Nat. Commun., 2017, 8, 469 CrossRef PubMed.
- H. F. Chan, Y. Zhang, Y.-P. Ho, Y.-L. Chiu, Y. Jung and K. W. Leong, Sci. Rep., 2013, 3, 3462 CrossRef PubMed.
- N. Moore, D. Doty, M. Zielstorff, I. Kariv, L. Y. Moy, A. Gimbel, J. R. Chevillet, N. Lowry, J. Santos, V. Mott, L. Kratchman, T. Lau, G. Addona, H. Chen and J. T. Borenstein, Lab Chip, 2018, 18, 1844–1858 RSC.
- L. Liang, Y. X. Jin, X. Q. Zhu, F. L. Zhou and Y. Yang, Lab Chip, 2018, 18, 1422–1429 RSC.
- B. Dura, M. M. Servos, R. M. Barry, H. L. Ploegh, S. K. Dougan and J. Voldman, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E3599–E3608 CrossRef CAS PubMed.
- L.-Y. Ke, Z.-K. Kuo, Y.-S. Chen, T.-Y. Yeh, M. Dong, H.-W. Tseng and C.-H. Liu, Lab Chip, 2018, 18, 106–114 RSC.
- F. Guo, P. Li, J. B. French, Z. Mao, H. Zhao, S. Li, N. Nama, J. R. Fick, S. J. Benkovic and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2014, 112, 43–48 CrossRef PubMed.
- M. Junkin, A. J. Kaestli, Z. Cheng, C. Jordi, C. Albayrak, A. Hoffmann and S. Tay, Cell Rep., 2016, 15, 411–422 CrossRef CAS PubMed.
- R. A. Kellogg, R. Gómez-Sjöberg, A. A. Leyrat and S. Tay, Nat. Protoc., 2014, 9, 1713 CrossRef CAS PubMed.
- C. Ma, R. Fan, H. Ahmad, Q. Shi, B. Comin-Anduix, T. Chodon, R. C. Koya, C.-C. Liu, G. A. Kwong, C. G. Radu, A. Ribas and J. R. Heath, Nat. Med., 2011, 17, 738–743 CrossRef CAS PubMed.
- J. Cheng, J.-F. Chen, M. Zhao, Q. Luo, L.-X. Wen and K. D. Papadopoulos, J. Colloid Interface Sci., 2007, 305, 175–182 CrossRef CAS PubMed.
- K. K. Y. Ho, L. M. Lee and A. P. Liu, Sci. Rep., 2016, 6, 32912 CrossRef CAS PubMed.
- K. W. Oh, K. Lee, B. Ahn and E. P. Furlani, Lab Chip, 2012, 12, 515–545 RSC.
- Q.-Q. Liao, S.-K. Zhao, B. Cai, R.-X. He, L. Rao, Y. Wu, S.-S. Guo, Q.-Y. Liu, W. Liu and X.-Z. Zhao, Sens. Actuators, A, 2018, 279, 313–320 CrossRef CAS.
- Y. Jia, Y. Ren, L. Hou, W. Liu, T. Jiang, X. Deng, Y. Tao and H. Jiang, Lab Chip, 2018, 18, 1121–1129 RSC.
- C.-X. Zhao, D. Chen, Y. Hui, A. Weitz David and P. J. Middelberg Anton, ChemPhysChem, 2017, 18, 1393–1399 CrossRef CAS PubMed.
- S. A. Nabavi, G. T. Vladisavljević, M. V. Bandulasena, O. Arjmandi-Tash and V. Manović, J. Colloid Interface Sci., 2017, 505, 315–324 CrossRef CAS PubMed.
- J. Clausell-Tormos, D. Lieber, J.-C. Baret, A. El-Harrak, O. J. Miller, L. Frenz, J. Blouwolff, K. J. Humphry, S. Köster, H. Duan, C. Holtze, D. A. Weitz, A. D. Griffiths and C. A. Merten, Chem. Biol., 2008, 15, 427–437 CrossRef CAS PubMed.
- C. N. Baroud, F. Gallaire and R. Dangla, Lab Chip, 2010, 10, 2032–2045 RSC.
- A. S. Utada, A. Fernandez-Nieves, H. A. Stone and D. A. Weitz, Phys. Rev. Lett., 2007, 99, 094502 CrossRef PubMed.
- J.-C. Baret, Lab Chip, 2012, 12, 422–433 RSC.
- K. Pays, J. Giermanska-Kahn, B. Pouligny, J. Bibette and F. Leal-Calderon, J. Controlled Release, 2002, 79, 193–205 CrossRef CAS PubMed.
- S. Lee, T. Y. Lee, E. Amstad and S.-H. Kim, Adv. Mater. Technol., 2018, 3, 1800006 CrossRef.
- Y. Lee, H.-R. Lee, K. Kim and S. Q. Choi, Anal. Chem., 2018, 90, 1660–1667 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01130c |
| This journal is © The Royal Society of Chemistry 2019 |