
Extracellular vesicles as cancer liquid biopsies: from discovery, validation, to clinical application
 *bd
*bd
Abstract
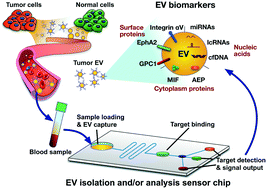
Substantial research has been devoted to elucidate the roles that extracellular vesicles (EVs) play in the regulation of both normal and pathological processes, and multiple studies have demonstrated their potential as a source of cancer biomarkers. However, several factors have slowed the development of liquid biopsy EV biomarkers for cancer diagnosis, including logistical and technical difficulties associated with reproducibly obtaining highly purified EVs suitable for diagnostic analysis. Significant effort has focused on addressing these problems, and multiple groups have now reported EV analysis methods using liquid biopsies that have the potential for clinical translation. However, there are still important issues that must be addressed if these discoveries and technical advances are to be used for clinical translation of EV cancer biomarkers from liquid biopsies. To address these issues, this review focuses on the potential application of EV biomarkers for diagnosis of major cancer types, discussing approaches for EV biomarker discovery and verification, EV clinical assay development, analytical and clinical validation, clinical trials, regulatory submission, and end user utilization for the intended clinical application. This review also discusses key difficulties related to these steps, and recommendations for how to best accomplish steps in order to translate EV-based biomarkers into clinical settings.
- This article is part of the themed collections: Personalised Medicine: Liquid Biopsy and Lab on a Chip Recent Review Articles
 Please wait while we load your content...
Please wait while we load your content...

