
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDigestion-on-a-chip: a continuous-flow modular microsystem recreating enzymatic digestion in the gastrointestinal tract†
Pim
de Haan
 ab,
Margaryta A.
Ianovska
ab,
Klaus
Mathwig
a,
Glenn A. A.
van Lieshout
c,
Vassilis
Triantis
c,
Hans
Bouwmeester
de and
Elisabeth
Verpoorte
*a
ab,
Margaryta A.
Ianovska
ab,
Klaus
Mathwig
a,
Glenn A. A.
van Lieshout
c,
Vassilis
Triantis
c,
Hans
Bouwmeester
de and
Elisabeth
Verpoorte
*a
aUniversity of Groningen, Groningen Research Institute of Pharmacy, Pharmaceutical Analysis, P.O. Box 196, XB20, 9700 AD Groningen, The Netherlands. E-mail: e.m.j.verpoorte@rug.nl
bTI-COAST, Amsterdam, The Netherlands
cFrieslandCampina, Amersfoort, The Netherlands
dRIKILT – Wageningen UR, Wageningen, The Netherlands
eWageningen University, Division of Toxicology, Wageningen, The Netherlands
First published on 5th April 2019
Abstract
In vitro digestions are essential for determining the bioavailability of compounds, such as nutrients. We have developed a cell-free, miniaturized enzymatic digestive system, employing three micromixers connected in series to mimic the digestive functions of the mouth, stomach and small intestine. This system continuously processes samples, e.g. containing nutrients, to provide a constant flow of digested materials which may be presented to a subsequent gut-on-a-chip absorption module, containing living human intestinal cells. Our system incorporates three-compartment enzymatic digestion, one of the key functions of the gastrointestinal tract. In each of these compartments, we modify the chemical environment, including pH, buffer, and mineral composition, to closely mimic the local physiological environment and create optimal conditions for digestive processes to take place. It will therefore provide an excellent addition to existing gut-on-a-chip systems, providing the next step in determining the bio-availability of orally administered compounds in a fast and continuous-flow ex vivo system. In this paper, we demonstrate enzymatic digestion in each separate compartment using compounds, starch and casein, as model nutrients. The use of transparent, microfluidic micromixers based on chaotic advection, which can be probed directly with a microscope, enabled enzyme kinetics to be monitored from the very start of a reaction. Furthermore, we have digested lactoferrin in our system, demonstrating complete digestion of this milk protein in much shorter times than achievable with standard in vitro digestions using batch reactors.
Introduction
Digestion by means of enzymatic reactions is one of several key functions of the gastrointestinal (GI) tract. The process of digestion starts immediately upon ingestion, and continues until the terminal part of the colon. In the oral phase, ingested materials are first subject to mastication and simultaneous mixing with saliva. Starch digestion by amylase, an enzyme in saliva, starts in this phase at a pH of 6–7. Upon reaching the stomach, the pH of the ingested material is reduced to values as low as 1.0,1 causing the denaturation of proteins and providing optimal conditions for the enzyme, pepsin, to work. The gastric mixture is then transferred in small portions into the duodenum, the first part of the small intestine. The pH of this mixture is neutralized to 7–8 by a bicarbonate-rich juice flowing from the pancreas, which also contains high concentrations of proteases, lipases and other enzymes aiding the breakdown of nutrients into smaller molecules. Bile, secreted by the liver and stored in the gall bladder, helps to emulsify fats in the mixture, which in this phase is termed chyme. Smaller molecules (e.g. amino acids, monosaccharides) may be transported into the body by active or passive means by passage through the intestinal epithelium into the blood or lymph. Any ingested compound (e.g. nutrients, medicinal drugs or toxicants) is subject to the same digestive processes upon oral administration. This process may have an effect on the amount reaching and eventually absorbed by the intestinal epithelium, affecting the dose that reaches the bloodstream, or is bio-available. In order to reach the systemic circulation, a compound must dissolve from its matrix, withstand metabolism by digestive juices (enzymatic action, or acid-catalyzed reactions), and make its way into the epithelial cell layer. There it must withstand possible intracellular metabolism to move out of the epithelium into the blood or lymph, and finally pass through the portal vein system in the liver without being metabolized or excreted into the bile. The term bio-availability, F, is defined as the overall fraction of a compound which finally reaches the systemic bloodstream:2| F = FF·FG·FH | (1) |
In eqn (1), bio-availability is determined by FF, FG and FH. Bio-accessibility (or pharmaceutical availability3), FF, denotes the fraction of the dose which is liberated from the matrix, e.g. a tablet containing the compound of interest, or a toxicological sample, and is not degraded by digestive juices.4FG is the fraction which then is able to pass through the gut wall into intestinal blood vessels, by either actively being taken up by transporting proteins in the epithelial cells (intracellular transport), or by passive diffusion through the cell layer (or paracellular transport). After absorption, compounds will be transported to the liver through the vena portae, allowing the liver to process incoming materials before they reach the systemic circulation. The fraction that passes through the liver without being metabolized is FH. For both medicinal drugs and toxicants, it is of great importance to understand what happens to a compound during its entire trajectory through the GI tract, as this strongly influences the bio-availability of these compounds.
Most in vitro models of the GI tract to date have focused on better understanding and quantifying the absorption of compounds from the intestine over the intestinal wall into the circulatory system. The sample containing the compounds of interest is introduced to a barrier model of the human intestine. An intestinal barrier model can be any system containing two compartments separated by a layer representing the intestinal epithelium. The most commonly used cell-based system, having the lowest degree of complexity, consists of epithelial cells grown on a Transwell insert in well plates.5 These Transwell-based systems usually contain only one cell type (e.g. Caco-2 cells, a human intestinal cancer cell line), and static medium on either side of the cell layer. Caco-2 cells can develop an apical and a basolateral side, showing a polarization pattern analogous to the human intestine. The apical side is in direct contact with the intestinal lumen, whereas cells attach to a solid support, e.g. a basal membrane, to form the basolateral side. Samples may be added to the apical compartment, after which the concentration of the compound and its metabolites can be determined in the basolateral compartment, providing information on FG. Several different approaches have been developed to miniaturize and thus improve this type of barrier model of the human intestine, leading to so-called gut-on-a-chip devices. Various modifications have been made to more accurately represent the in vivo situation in gut-on-a-chip models, such as medium flow,6 three-dimensional scaffolds,7,8 peristaltic motions and bacteria,9–12 elements of the immune system,13 combinations with different organs-on-chips,14–16 and some elements of the digestive functions.17,18 Also noteworthy are specific devices to study the pharmacokinetic behavior of medicinal drugs in the intestine and liver.19–22 However, none of these gut-on-a-chip systems fully recapitulates enzymatic digestion, a key function of the gastrointestinal tract which ultimately determines the bio-availability of ingested compounds.
Common procedures to study the behavior of compounds in the GI tract involve in vitro digestions, in which the compound of interest is exposed to artificial digestive juices of varying complexities in a temperature-controlled and stirred container, having volumes from ten to hundreds of milliliters.23–26 These in vitro digestions are a tool to study the bio-accessibility of the compounds, but the methodology is relatively laborious. Even if performed in a fermenter-like container, with automated pH control and time-controlled addition of digestive juices, systems are not autonomous, and a skilled technician is still required to operate the system and collect the samples.25,27–29 Miniaturization of such a system would allow for automation, as well as a reduced need for valuable chemicals (such as enzymes) and solvents.
In this report, we describe the design and development of a miniaturized digestive system, which can be coupled in series to a gut-on-a-chip to provide a continuous flow of digested materials (Fig. 1). In this system, samples are continuously mixed with saliva in the first compartment (‘mouth’). The output of the mouth is transferred to the next compartment (‘stomach’), where gastric juice is added. Finally, the gastric mixture flows to the third compartment (‘intestine’), where duodenal juice and bile are added. The result is a solution mixture comparable to chyme, the product of our digestive system from which compounds are absorbed in the intestinal tract. Our system will provide information on FF, and when combined with an absorption model of the intestine, the combination of FF and FG. Contrary to the commonly used batchwise in vitro digestions, our miniaturized system can be operated in continuous-flow mode, for both short- and long-term studies. A continuous flow has several advantages over a batch process, allowing processing of samples either continuously or in a pulsatile multi-dose regimen. At the same time, however, we need to very precisely control the conditions in the three separate compartments. Hence, solutions entering a compartment must be rapidly mixed with the relevant digestive juice. Therefore, each “organ” or compartment of the system consists of a 1.38 μL chaotic micromixer, based on a microchannel with an array of herringbone-shaped grooves, formed in the channel ceiling. These grooves perturb laminar flow profiles, and create two counter-rotating vortical flows in the channel, thereby folding the liquids into one another.30 Samples entering each micromixer are continuously mixed with artificial digestive juices containing enzymes in physiologically relevant ratios. Samples flow through three consecutive compartments, representing three digestive processes, to produce a flow of chyme of 25 μL min−1. This flow rate was chosen for two reasons: 1) the inlet flow rates used (ranging from 1–12 μL min−1) are low enough to save valuable artificial digestive juices and samples with respect to larger-scale in vitro digestions, but high enough to maintain steady-state flow conditions by using syringe pumps; and 2) we found in earlier work that mixing is very fast and efficient at these low flow rates, occurring within the first section of the micromixer channels (vide infra for a description of these micromixer channels containing cycles of herringbone-shaped grooves).31 The ratios of the inlet flow rates (1 + 4 + 8 + 12 μL min−1) in our system were chosen to approximate the volumetric ratios of digestive juices in vivo (Fig. 1).27 One of the key strengths of our system from a chemical point of view is that the conditions in each of the compartments are very different (i.e. mineral composition, buffer and pH, enzymes), but enzymatic reactions are taking place in all of the compartments.
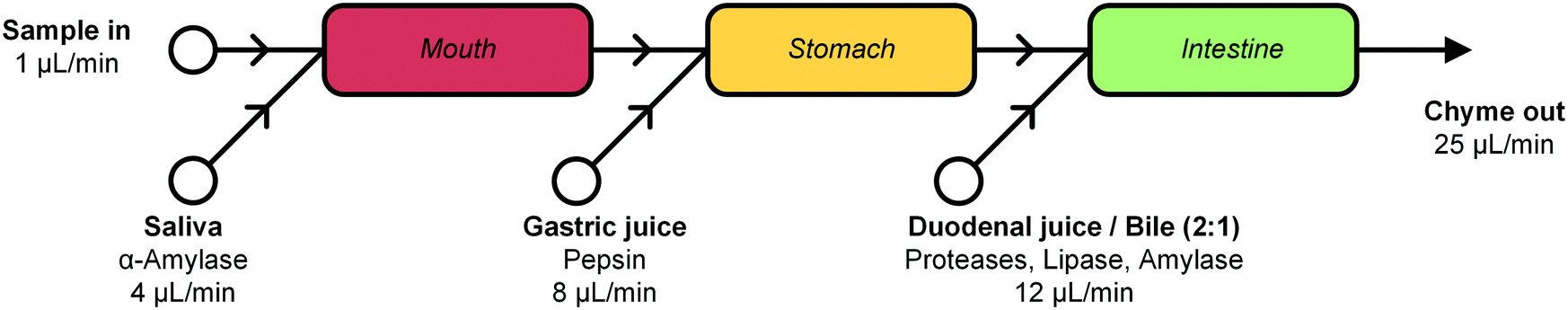 | ||
| Fig. 1 Schematic representation of the cell-free miniaturized digestive system. Samples are introduced at the top left, and are continuously mixed with artificial digestive juices, in physiologically relevant ratios (as shown by the different flow rates). The digested material, or chyme, is produced continuously and could be transferred by flow to a subsequent gut-on-a-chip module to study intestinal absorption. Each white circle represents an inlet pump set to the indicated flow rate. | ||
We have tested the individual compartments of our miniaturized digestive system with model compounds, to obtain enzyme kinetic plots to demonstrate enzyme activity in each stage of our digestion model. This was to make sure that the enzymes were fully active under the flow transport conditions inside the microchannels, where they are subjected to shear stress. Finally, the digestion of a sample containing lactoferrin, an iron-containing nutritional protein which is naturally present in milk,32–34 is shown and characterized.
Experimental section
Artificial digestive juices
Artificial saliva, gastric juice, duodenal juice and bile compositions are given in Table 1. The juices were freshly prepared on the day of the experiments, according to a modified literature procedure published by Walczak et al.,27 after optimization of the pH of each juice by changing the concentrations of HCl or NaOH. All chemicals (excluding enzymes) were dissolved in ultrapure water in volumetric flasks. HCl or NaOH was then added to adjust the pH of the buffer systems used for the mouth, stomach and intestine. The pH was verified with a calibrated pH meter (Metrohm, Herisau, Switzerland) at room temperature, and the enzyme content was added just before adding ultrapure water to the final volume. This sequence is essential, since the proteins may denature if added to a solution with a too high or too low pH value. The juices were warmed to 37 °C in an incubator least 1 h before the experiment, as described in other studies.25,27 All chemicals were obtained from Sigma-Aldrich/Merck (Zwijndrecht, the Netherlands), except sodium dihydrogen phosphate monohydrate and hydrochloric acid (Acros, Geel, Belgium), and potassium chloride and sodium chloride (Duchefa, Haarlem, the Netherlands).| Compound | Saliva (mg L−1) | Gastric juice (mg L−1) | Duodenal juice (mg L−1) | Bile (mg L−1) |
|---|---|---|---|---|
| CaCl2 | 302 | 151 | 167.5 | |
| Glucosamine HCl | 330 | |||
| Glucose | 650 | |||
| Glucuronic acid | 20 | |||
| KCl | 896 | 824 | 564 | 376 |
| KH2PO4 | 80 | |||
| KSCN | 200 | |||
| MgCl2·6H2O | 50 | |||
| Na2SO4 | 570 | |||
| NaCl | 298 | 2752 | 7012 | 5259 |
| NaH2PO4·H2O | 1021 | 306 | ||
| NaHCO3 | 3388 | 5785 | ||
| NH4Cl | 306 | |||
| Urea | 200 | 85 | 100 | 250 |
| Uric acid | 15 | |||
| HCl | 4.16 mM | 5.57 mM | 6.17 mM | |
| NaOH | 2.9 mM | |||
| α-Amylase (Bacillus sp.) | 145 | |||
| Bile (bovine) | 6000 | |||
| Lipase (porcine pancreas) | 500 | |||
| Pancreatin (porcine pancreas) | 3000 | |||
| Pepsin (porcine gastric mucosa) | 1000 |
Fabrication of devices
Micromixer devices were fabricated by micromolding poly(dimethylsiloxane) (PDMS, Sylgard 184, Dow Corning, Midland, MI, USA) on SU-8 molds, according to a procedure we have described in earlier work.31 The 300-μm-wide, 51.5-mm-long channels contained 16 arrays of cycles of 12 herringbone-shaped grooves each, which greatly enhance mixing in this laminar-flow regime through chaotic advection.30,31 The channels were 60 μm deep, and 50 μm deeper in the groove regions for optimal mixing characteristics. Channels were sealed off with a glass microscope slide to yield a hybrid PDMS/glass device. Plateaus (areas with no grooves before and after each cycle) were incorporated to allow for a 300 × 100 μm measurement site, to monitor mixing and reaction progress (Fig. 2). Each compartment consisted of a micromixer; and compartments were connected with one another in series using poly(tetrafluoroethylene) (PTFE) tubing (0.8/1.6 mm inner/outer diameter, Polyfluor Plastics, Breda, the Netherlands). Blunt Fine-Ject 21G needles (HenkeSassWolf, Tuttlingen, Germany) were used to connect the tubing to syringes.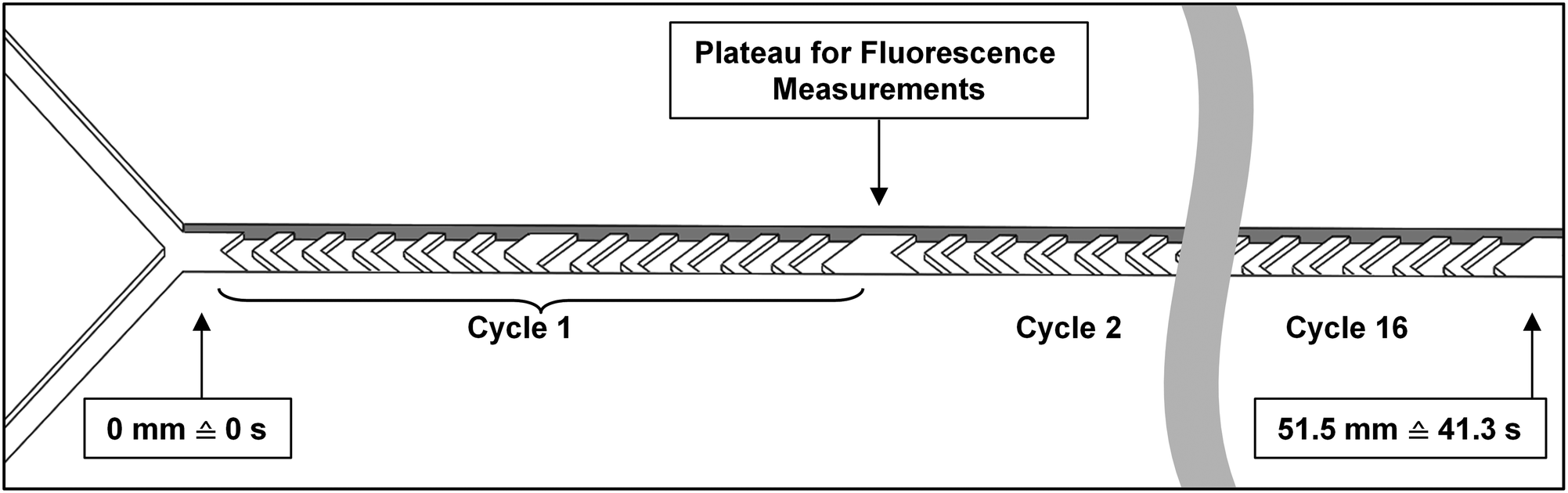 | ||
| Fig. 2 Diagram of a micromixer channel, containing herringbone-shaped grooves in its surface. Two inlet channels are visible at the left. The channel is just over 50 mm long, and contains 16 cycles of 12 grooves each. Between cycles, a plateau is incorporated to allow for a 300 × 100 μm measurement region. At a total flow rate of 2.0 μL min−1, as used in the enzyme kinetics experiments, 51.5 mm channel length corresponds to a residence time of 41.3 s. | ||
Enzyme kinetics
The overall function of the enzymes present in the artificial digestive juices (Fig. 1) is the digestion of food to release nutrients. Most biological macromolecules cannot be absorbed from the intestinal tract in intact form, and have to be hydrolyzed to smaller units (monomers or oligomers) before uptake through the intestinal wall. Polysaccharides such as starch are cleaved to smaller sugars by amylases in saliva and pancreatic juice. Proteins are broken down to smaller peptides and single amino acids by proteases in the stomach and intestine. Fats (triglycerides) are hydrolyzed to monoglycerides and free fatty acids by lipases in the intestine, assisted by bile salts to emulsify fatty content to achieve better enzyme accessibility. Enzymatic catalysis greatly enhances the processing rate of food. For these enzymes, the reaction rate, V, depends on the substrate concentration, [S], and enzyme parameters Vmax and Km, according to Michaelis–Menten kinetics:35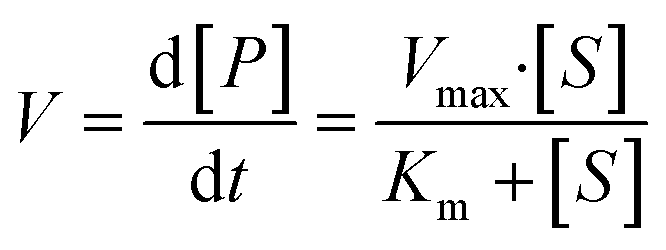 | (2) |
In this equation, the reaction rate, V, is equal to the formation of product, P, over time. Vmax is the maximum turnover rate of this enzyme under specified conditions, and Km denotes the concentration of substrate where V = ½ Vmax. This Michaelis–Menten equation predicts the typical kinetics plot (V vs. [S]), in which the V curve monotonically increases from zero and saturates or plateaus at Vmax.
Enzymatic assays – general
Each compartment was decoupled from the rest of the system and studied individually to study the enzyme kinetics of that compartment. Therefore, devices containing a single micromixer (two inlets, one outlet, as described by Ianovska et al.31) were used to demonstrate enzymatic function in the separate compartments. Two syringe pumps (Harvard Apparatus, Pump 11 Pico Plus Elite, Holliston, MA, USA) were used with glass syringes (250 μL, Kloehn, Whittier, CA, USA) or plastic syringes (1 mL, Braun, Melsungen, Germany), containing either the digestive juice or the solution containing the compound to be digested. Each assay consists of a labeled model nutrient, which when digested in our system causes an increase in fluorescence. Even though the single compartments were decoupled from the total system, we ensured proper functioning of each device by infusing the digestive juice in one inlet (e.g. gastric juice, in the stomach), and the simulated output of the preceding compartment in the other inlet (e.g. ‘mouth’ output, a mixture of water and saliva, at the appropriate ratios, to study the kinetics in the stomach). The chemical environment inside these decoupled compartments is therefore the same as in the assembled system. The total flow rate (Qjuice + Qsubstrate) was 2.0 μL min−1 in each separate stage of digestion in these tests. This lower flow rate was chosen to better be able to study enzyme kinetics in our system: at 2.0 μL min−1, the longer residence time of 41.3 s enables us to measure a detectable increase of fluorescence and model the enzyme kinetics on-chip.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixing ratio of saliva with samples, as indicated in Fig. 1. First, a syringe containing no substrate was connected, as a no-substrate control, and after flow stabilization for at least 1 min, fluorescence micrographs were taken at the Y-junction and after each cycle of grooves. Since the internal volume of the channel is 1.38 μL, the two solutions to be mixed have 41.3 s to mix at an overall flow rate of 2.0 μL min−1. Fluorescence micrographs (1.0 s exposure time per micrograph) were taken along the 51.5-mm-long channel, thus monitoring the reaction from the exact start up to 41.3 s. For measurements at each new substrate concentration, the syringe containing the substrate solution was exchanged, and steps were repeated to obtain new fluorescence micrographs after each cycle of grooves. The fluorescence intensity was measured using ImageJ software (NIH, Bethesda, MD, USA),36 calculating the 8-bit average gray value in a 300 × 100 μm rectangle drawn across the channel in each photo. The fluorescence observed in the channel increased after each consecutive cycle. The corresponding rate of product formation, dP/dt, was calculated by linear regression through the initial three data points (see ESI† Fig. S5) for curves obtained at each [S]. These initial reaction rates were then plotted versus the substrate concentration. Note that this method only allows for calculation of dP/dt in terms of fluorescence intensity. The constant required to convert measured fluorescence to actual moles of product per time per unit of enzyme remains unknown, but was not required for this initial proof-of-concept study.
:1 mixing ratio of saliva with samples, as indicated in Fig. 1. First, a syringe containing no substrate was connected, as a no-substrate control, and after flow stabilization for at least 1 min, fluorescence micrographs were taken at the Y-junction and after each cycle of grooves. Since the internal volume of the channel is 1.38 μL, the two solutions to be mixed have 41.3 s to mix at an overall flow rate of 2.0 μL min−1. Fluorescence micrographs (1.0 s exposure time per micrograph) were taken along the 51.5-mm-long channel, thus monitoring the reaction from the exact start up to 41.3 s. For measurements at each new substrate concentration, the syringe containing the substrate solution was exchanged, and steps were repeated to obtain new fluorescence micrographs after each cycle of grooves. The fluorescence intensity was measured using ImageJ software (NIH, Bethesda, MD, USA),36 calculating the 8-bit average gray value in a 300 × 100 μm rectangle drawn across the channel in each photo. The fluorescence observed in the channel increased after each consecutive cycle. The corresponding rate of product formation, dP/dt, was calculated by linear regression through the initial three data points (see ESI† Fig. S5) for curves obtained at each [S]. These initial reaction rates were then plotted versus the substrate concentration. Note that this method only allows for calculation of dP/dt in terms of fluorescence intensity. The constant required to convert measured fluorescence to actual moles of product per time per unit of enzyme remains unknown, but was not required for this initial proof-of-concept study.
:1), to represent the dilution due to mixing in the first stage. Artificial gastric juice, containing 0.7 FIP-U mL−1 pepsin, was prepared fresh, and all solutions were heated to 37 °C before the experiments. As for the mouth, the set-up, containing all pumps, tubing and the micromixer, was placed on the stage of an inverted fluorescence microscope, located in a custom-built incubator kept at 37 °C. To maintain a 5:8 mixing ratio of mouth output:gastric juice at a total flow rate of 2.0 μL min−1, artificial gastric juice was infused at a rate of 1.23 μL min−1 (i.e. 8/13 × 2.0 μL min−1), and the various substrate solutions were infused at a rate of 0.77 μL min−1 (i.e. 5/13 × 2.0 μL min−1), yielding a total flow rate of 2.0 μL min−1. By using these ratios (5:8) between oral output (containing the substrate) and gastric juice, we ensure that the final gastric mixture has the same mineral composition as in our final digestion model. The same procedure as described for the first stage was used to obtain fluorescence micrographs along the channel and prepare kinetics plots. Initial rates were calculated from the first five data points.
:4:8) of water, saliva and gastric juice, omitting all enzymatic content, to represent the output of the second compartment. To maintain a 13:12 mixing ratio of stomach output:duodenal juices at a total flow rate of 2.0 μL min−1, the stomach mixture was infused at a rate of 1.04 μL min−1 (i.e. 13/25 × 2.0 μL min−1), and a freshly prepared mixture of 8:4 duodenal juice and bile containing 0.7 FIP-U mL−1 proteases was infused at a rate of 0.96 μL min−1 (i.e. 12/25 × 2.0 μL min−1). The same procedure as described for the first and second stages was used to obtain fluorescence micrographs and prepare kinetics plots. Initial reaction rates were calculated from the first five data points.
Proof of concept: lactoferrin digestion
Three microreactors were connected in series, as shown in Fig. 3. For these experiments, syringe pumps were replaced with a flow control system based on Bronkhorst Coriolis flow sensors.37 Bovine lactoferrin (50 mg mL−1 in ultrapure water, Vivinal Lactoferrin, courtesy of FrieslandCampina, Wageningen, the Netherlands), an iron-containing protein naturally occurring in bovine milk, was continuously introduced to the mouth and mixed with artificial saliva; the resulting solution was introduced to the stomach and mixed with gastric juice; and the resulting mixture was then introduced to the intestine and mixed with duodenal juice and bile, at the flow rate ratios depicted in Fig. 1. Loops of connective tubing were used to incubate mixtures after each mixing stage: a 2 cm (2 min) piece of tubing after the oral phase; a 312 cm (120 min) piece of tubing after the gastric phase; and a 600 cm (120 min) long piece of tubing after the intestinal phase (Fig. 3). The incubation loops of the gastric and intestinal phases were pre-cut at places corresponding to 5, 10, 30, and 60 min of incubation time to facilitate the collection of samples at these time points (see ESI† Fig. S4). The system was operated for more than 4 h to establish stable flows throughout the system. Samples were then collected starting from the last time point of the intestinal phase, working upstream towards the mouth. The samples were immediately diluted to 1 mg mL−1 lactoferrin, mixed with loading buffer containing lithium dodecyl sulfate (NuPAGE LDS Sample Buffer 4×, Invitrogen, Carlsbad, CA, USA) and stored at −20 °C to stop all enzymatic reactions. A batch-wise in vitro digestion of lactoferrin was done as a control experiment: a test tube containing 0.5 mL of lactoferrin (50 mg mL−1 in ultrapure water) was mounted on a rotary mixer (rotating head-over-heels, 24 rpm, in a 37 °C incubator), and 2 mL of artificial saliva was added. After 2 min, 4 mL of gastric juice was added and the mixture was incubated for 120 min. This was followed by the addition of 4 mL of duodenal juice and 2 mL of bile, and the mixture was incubated for another 120 min. Samples were collected from this test tube at the same time points as the microfluidic digestion and were processed in a similar way. The proteins in these samples (from both the microfluidic device and the test tube) were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), separating proteins and fragments thereof by size (4–12% Bis-Tris gels, sample denaturation for 5 min at 90 °C, 5 μL sample per well, run at 200 V for 30 min in 1× MES buffer, stained with Expedeon Coomassie Instant Blue).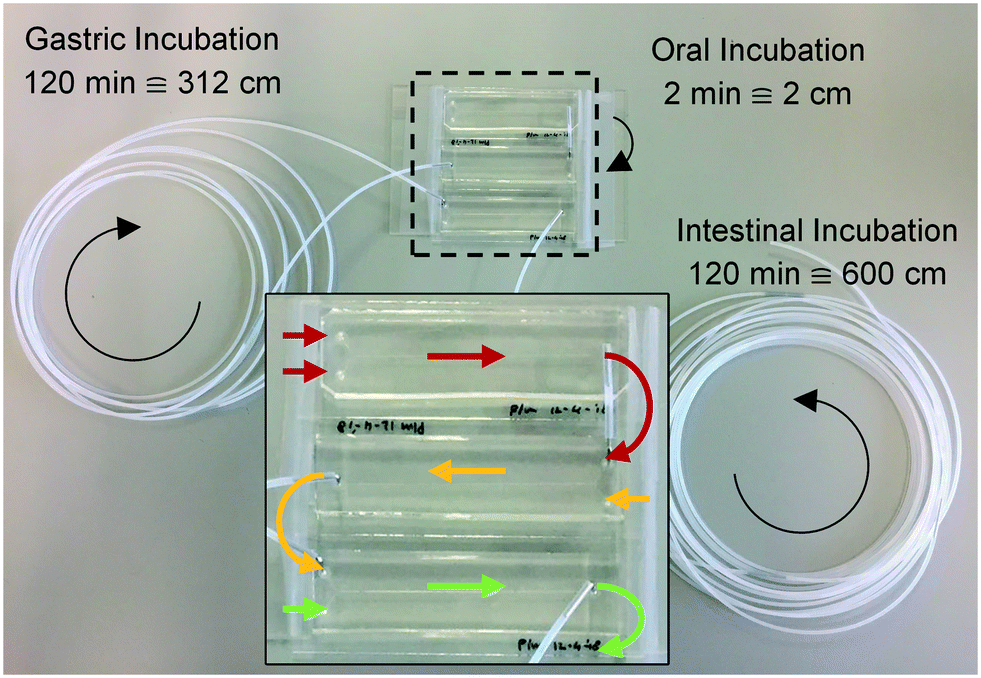 | ||
| Fig. 3 Photograph of the three micromixers, coupled in series, for the digestion of lactoferrin. The arrows represent the flow direction in each of the modules (red: mouth; yellow: stomach; green: intestine). Loops of tubing of 2, 312 and 600 cm were used for incubation after each stage, corresponding to 2, 120 and 120 min incubation time. All inlet tubings have been removed for clarity. | ||
Results and discussion
In order to be able to miniaturize an in vitro digestive system for ingested compounds, we first optimized the composition of artificial digestive juices. This ensured that only minimal precipitation and gas formation would occur inside our microfluidic channels. We then verified enzyme activity in all three compartments separately, and finally tested our continuous-flow microfluidic digestive system using lactoferrin, a milk protein, as a model compound.Optimization of juice composition
Artificial mixtures resembling saliva, gastric juice, duodenal juice and bile were initially prepared according to recipes reported in the literature by Walczak et al.27 It was found, however, in both off- and on-chip mixing experiments, that a precipitate was formed in the third (intestinal) stage upon mixing the output of the stomach (pH 1.38) with the solution containing duodenal juice and bile (pH 8.82 and 9.03) (Fig. 4). To understand the source of this precipitation, the pH of the solutions corresponding to the mouth, stomach and intestine was both calculated and determined experimentally (see ESI† Table S1–3). In Table 2, the main species that determine the pH are listed. The pH should be around 6.8 for saliva, around 1.3 for gastric juice, and around 8.1 and 8.2 for duodenal juice and bile, respectively.27 After mixing, this should yield a pH of 1.0–2.0 in the stomach phase and 5.5–6.5 in the intestinal phase. We calculated and measured correct values for the mixture in the stomach using these juices, but the mixture in the intestine was found to be too acidic, with both the calculated and measured values between 2.0–3.0. Physiologically, the pH in the small intestine varies, but is mostly reported as being between 7.0–8.0 after neutralization of gastric juice by bicarbonate in the duodenal (pancreatic) juice.1 A correct pH in each compartment will ensure optimal enzyme function in each compartment, as this strongly depends on pH. Moreover, protein solubilities may be lowered at non-optimal pH. To optimize the pH of the juices, the same concentrations of components were used, and only the concentration of species determining the pH were modified (see ESI† Table S3) to obtain pH values of 7.0, 3.0 and 7.0 after each successive phase. These pH values are based on a similar, standardized, in vitro digestion protocol named ‘Infogest’, reported by Minekus et al.25 A pH of 3.0 in the stomach was chosen in this study based on the same principle: the pH inside the stomach varies in vivo, at ∼2.0 when fasting but rising upon the ingestion of food, depending on the buffer capacity of the food; therefore the same static gastric pH of 3.0 was chosen for this study (as an estimated average of the varying gastric pH). Besides these changes in pH, two species which have no clear function in in vitro digestions, mucin and bovine serum albumin, were omitted from the juices. Adjusting the composition led to a slight modification of the pH in saliva, as displayed in the last column of Table 2. The concentration of hydrochloric acid in gastric juice was lowered from 100 mM to 4 mM to obtain a gastric mixture of pH 3.0. The concentration of bicarbonate and hydrochloric acid in duodenal juice and bile were modified to obtain a theoretical final pH of 7.0, experimentally determined to be around 7.3. After optimizing the composition of the juices, precipitation and gas formation were no longer apparent, and the system could be operated with no interference for extended periods of time (Fig. 5).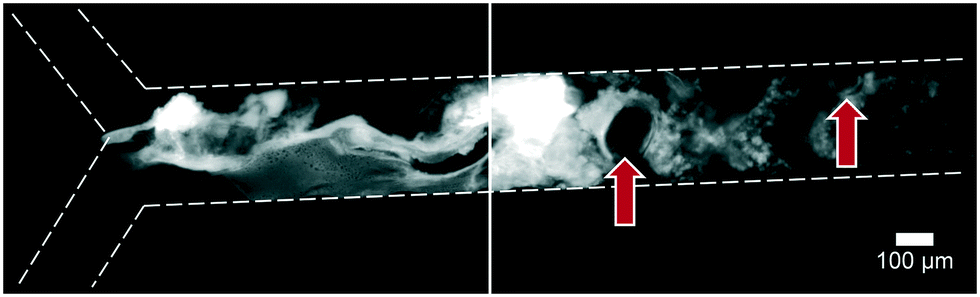 | ||
| Fig. 4 Precipitation and gas formation are visible at the start of the intestinal compartment when mixing non-optimized digestive juices (top inlet: 5 μM fluorescein, dissolved in a 1:4:8 mixture of water, saliva and gastric juice; bottom inlet: an 8:4 mixture of duodenal juice and bile). Red arrows indicate gas bubbles, most likely CO2. After optimization of juice compositions, no precipitation was visible (see Fig. 5). | ||
| Juice or mixture | Flow rate (μL min−1) | Main species determining pH | Physiological pH range38 | pH in original composition27 | pH in optimized composition |
|---|---|---|---|---|---|
| Measured (calculated) | Measured (calculated) | ||||
| Sample | 1 | Pure H2O | 6.96 (7.00) | 6.96 (7.00) | |
| Saliva | 4 | H2PO4−, OH− | 5.8–7.1 | 6.45 (6.71) | 6.75 (7.01) |
| Mixture in the mouth | 5 | All of the above | 6.47 (6.71) | 6.76 ( ) ) |
|
| Gastric juice | 8 | HCl, H2PO4− | 0.6–3.2 | 1.16 (1.02) | 2.57 (2.46) |
| Mixture in the stomach | 13 | All of the above | 1.38 (1.24) | 3.04 ( ) ) |
|
| Duodenal juice | 8 | HCO3−, HCl | 7.0–7.7 | 8.82 (7.51) | 8.32 (7.12) |
| Bile | 4 | HCO3−, HCl | 6.1–8.6 | 9.03 (7.76) | 8.81 (7.35) |
| Mixture in the intestine | 25 | All of the above | 2.53 (2.12) | 7.31 ( ) ) |
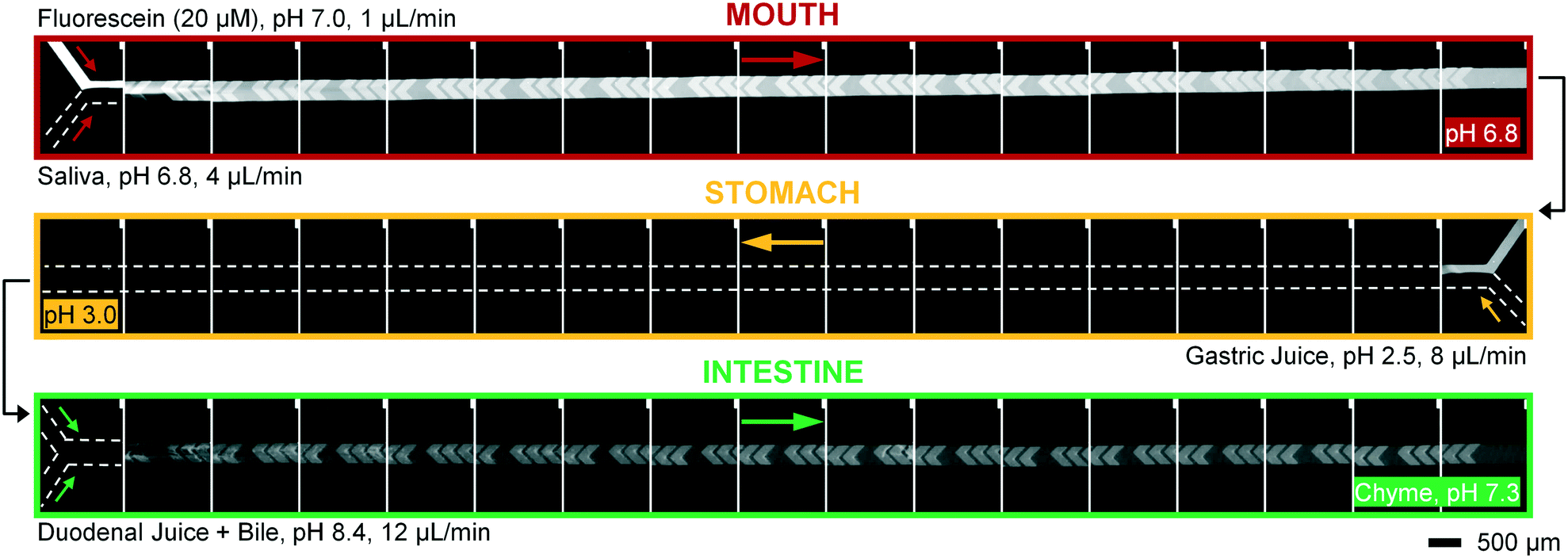 | ||
| Fig. 5 Micrographs of the three digestive modules, coupled in series. A microscope photograph was taken before and after each cycle of 12 grooves. Fluorescein, a fluorescent dye visible between pH 5–9, was used as a sample molecule (appearing white to grayish) and is not digested in this system. In the upper series (mouth), the sample (20 μM fluorescein in ultrapure water) is continuously mixed with artificial saliva in a 1:4 ratio. The resulting mixture is transferred to the middle module (stomach), where it is mixed with artificial gastric juice in a 5:8 ratio (flowing from right to left). This mixture, in turn, is transferred to the lowest module (intestine), where it is mixed in a 13:12 ratio with a flow of artificial duodenal juice and bile (pre-mixed in a 8:4 ratio). The final output is a flow of “chyme” at 25 μL min−1. The entire system, including all devices, syringe pumps and tubing, was placed in a 37 °C microscope-incubator. Note that fluorescein is not visible in the stomach compartment, since the local pH is too low; fluorescence re-appears in the intestinal compartment after neutralization of the gastric juice. The disappearance and reappearance of fluorescence is almost simultaneous when a pH change is effected in the system, indicating that mixing is highly efficient. Note that the fluorescence intensity in the intestine is lower, due to dilution. | ||
To operate our miniaturized digestive system in continuous flow mode, three micromixers were coupled in series and all juices were infused at the flow rates described in Fig. 1. The fluorescent dye, fluorescein (20 μM in ultrapure water), was used as a sample to visualize flows inside the channels (Fig. 5). Fluorescein is not susceptible to digestion or metabolism under these conditions. We observed a rapid mixing process in the mouth, which we have confirmed by characterizing the mixing process (see ESI† Fig. S3) of these liquids. This was done in the same fashion as the mixing process for water and several organic solvents in mixers with herringbone-shaped grooves in an earlier study.31 The output from the first (mouth) stage was transferred to the second (stomach) stage using a short (2 cm) piece of tubing. We observed that the fluorescence disappears within one cycle of grooves upon mixing with gastric juice. This is because gastric juice causes the pH to drop to 3.0, which is out of range for the occurrence of fluorescence in fluorescein (pH 5–9). After neutralizing with a mixture of duodenal juice and bile, in the third (intestinal) stage, a reappearance of fluorescence is observed. The resulting chyme has a pH of 7.3, and only minimal precipitation and gas formation were observed. Note that the intensity of the fluorescence is decreased when it reappears in the intestine, due to dilution.
Enzyme kinetics
To ascertain that the enzymes in the artificial digestive juices are also active as catalysts, we performed studies of enzyme kinetics to determine if we observed typical Michaelis–Menten behavior. For each separate compartment, a fluorescent substrate was dissolved in the appropriate matrix (i.e., water, or a mixture of water and digestive juice to mimic the output of preceding compartments) and was mixed on-chip with a flow of digestive juice containing the enzyme in question. Enzyme concentrations were fixed at their usual values (Table 1), and the concentration of substrate, [S], in the other inlet was increased in increments. For the first stage (mouth), quenched, labeled starch was digested by the α-amylase (11.5 U mL−1) present in artificial saliva (Fig. 6A), yielding an increase in fluorescence upon hydrolysis of the substrate. No increase in fluorescence was visible when artificial saliva without α-amylase was used (Fig. 6B, for all raw data see ESI† Fig. S5). The reaction rates were measured on-chip: in our micromixers, the absolute start of the reaction (t0) is continuously visible at the Y-junction, where enzyme and substrate meet. Each point downstream of the Y-junction corresponds to a fixed time point in the progressing reaction, up to 41.3 s at the end of the channel. In this way, we can transpose distance to time and continuously monitor a reaction from the very start, allowing us to study initial reaction rates without missing initial data points. This is in contrast to what happens when the same reaction is monitored in a conventional fluorimeter set-up, where there is a delay between the time to mix reactants and starting fluorescence measurements (see ESI† Fig. S8). We note that at the highest concentration of starch (Fig. 6A), the fluorescence intensity reached a maximum value after 4 cycles (∼10 s). This raised the question if the reaction had stopped because 1) all the substrate had been completely converted or 2) the enzyme had ceased to catalyze the reaction, due to e.g. shear flow-induced damage in our micromixers. A series of control experiments was undertaken to address this question. We performed the exact same experiments in a 96-well plate, monitoring reactions with a fluorimeter (ESI† Fig. S6). After 1 h, the reaction mixture was transferred to chips to measure the fluorescence on-chip, and we found similar endpoint fluorescence values (ESI† Fig. S7), indicating that the same endpoint is reached both on- and off-chip. Noteworthy is that the reaction on-chip seems faster than the reaction off-chip in wells (ESI† Fig. S8), possibly because of the highly efficient mixing processes in the channel. In a final control experiment, we transferred the output of one micromixer (saliva and labeled starch mixed 4:1) to another micromixer, to which we added fresh α-amylase (ESI† Fig. S9). If the reaction in the first micromixer had stopped because of enzyme damage, we would expect an increase of fluorescence in the second micromixer upon enzyme addition as remaining labeled starch was consumed. We did not observe an increase, however, which supports the hypothesis that the reaction is indeed complete after only ∼10 s in the first mixer; and the reaction does not stop because of enzyme inactivation, but rather as a result of substrate depletion.
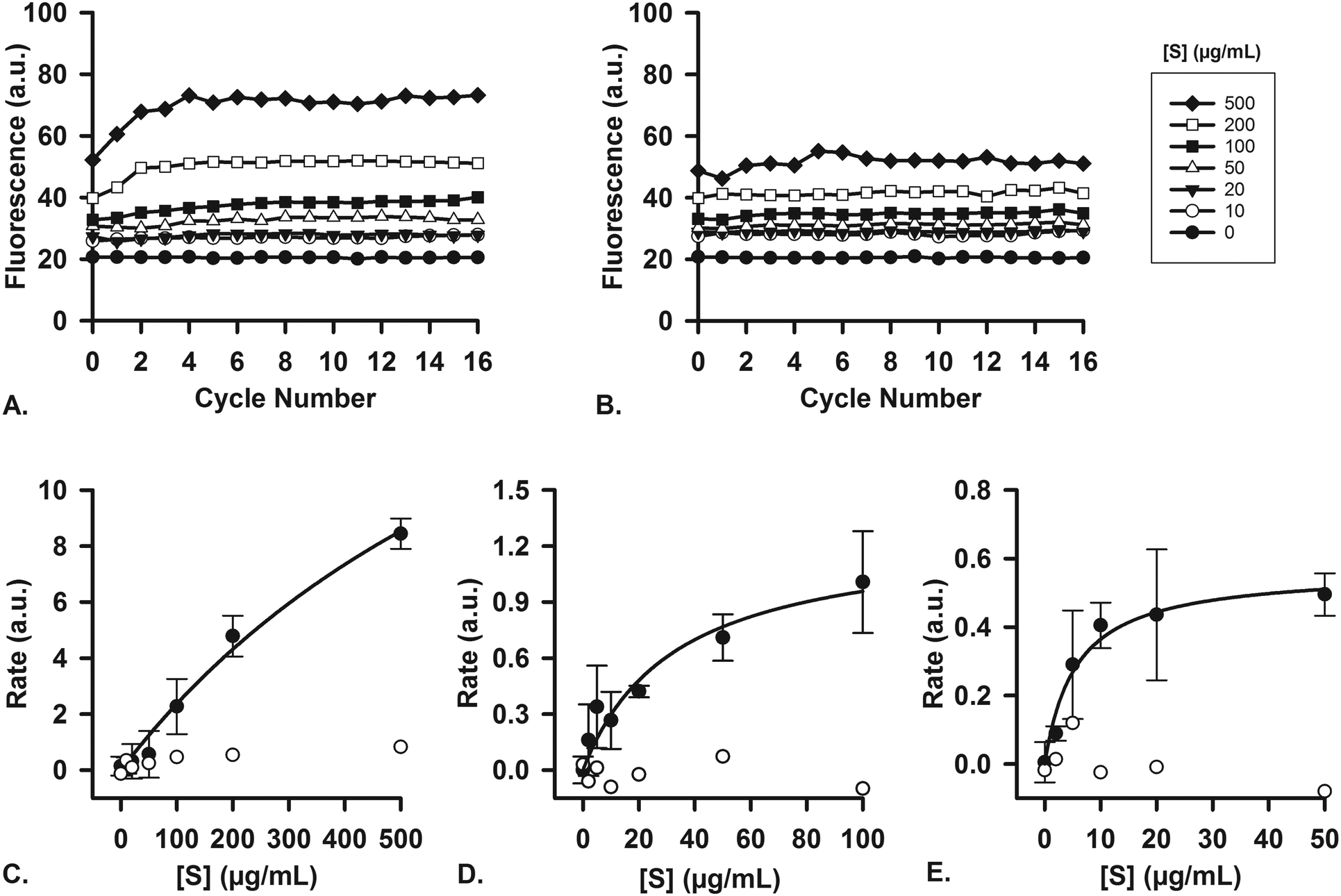 | ||
| Fig. 6 Enzyme kinetics studies were performed in single micromixers, to demonstrate the enzymatic function of each juice. For experiments with each separate stage, the digestive juice containing a fixed concentration of enzyme, [E], was introduced in one micromixer inlet, and a solution containing a varying concentration of substrate, [S], was introduced in the other inlet. Cycle numbers count the number of cycles passed from the Y-shaped confluence. A) Example of raw data (1 out of n = 3) showing the α-amylase-catalyzed digestion of different concentrations of labeled starch in a chip (oral compartment); B) no reaction takes place when artificial saliva without α-amylase is used under the same conditions (raw data). Initial reaction rates were measured (in graphs such as A and B) and plotted versus [S] (n = 3, average ± standard deviation). These Michaelis–Menten plots show the digestion of C) labeled starch in the oral compartment, D) labeled casein by pepsin in the gastric compartment, and E) labeled casein by proteases in the intestinal compartment. The displayed concentrations of substrates are those of the substrate solution (before mixing in the device). The white circles are control experiments, using the same juices but without any enzymatic content. | ||
Reaction rates were measured both with (n = 3) and without the enzyme, α-amylase, and a typical near-saturation Michaelis–Menten plot was obtained in the former case (Fig. 6C). No conversion factor is available to convert these arbitrary units to moles substrate reacted per time per unit of enzyme, but we can clearly observe Michaelis–Menten behavior. These results confirm enzyme-catalyzed reactions in our miniaturized digestive system.
For the second (stomach) and third (intestine) compartments, quenched, labeled casein was used as a substrate for proteases. Both the artificial gastric and duodenal juices contain proteases, and their presence and activity is demonstrated in this assay. In the same fashion as described for the first compartment, the enzyme and substrate were injected into the two separate inlets of the channel. An increase in fluorescence occurred upon hydrolysis of casein into smaller peptides, thereby indicating enzymatic activity. For gastric juice (Fig. 6D, 0.7 FIP-U mL−1 pepsin), a typical Michaelis–Menten plot was obtained, showing near-saturation at high [S]. In the third compartment (Fig. 6E, 0.7 FIP-U mL−1 protease in the 8:4 mixture of duodenal juice and bile), a similar kinetics plot was recorded, with clear saturation at high [S] values (for raw data, see ESI† Fig. S10 and S11). The Michaelis–Menten behavior is evident, and we have ascertained the activity of these enzymes in our system by digesting fluorescent model nutrients on-chip.
Lactoferrin digestion
A milk protein, lactoferrin, was dissolved in water (50 mg mL−1) and continuously digested in our three-compartment miniaturized digestive system as a model compound. A piece of tubing was connected to the outlet of each compartment to incubate the mixture of that compartment to allow for continued enzymatic action (Fig. 3). Samples were collected at different time points in these loops and analyzed by SDS-PAGE to assess digestion progress. Since SDS-PAGE separates proteins and peptides by size, it yields information about the digestion of lactoferrin, a 78 kDa protein, to smaller peptides (Fig. 7A). In the oral phase, no digestion of lactoferrin is visible. In the gastric phase, however, we see a rapid decrease in the intensity of the lactoferrin band (78 kDa), and bands corresponding to smaller peptides appear at the bottom of the gel. From 60 min in the gastric phase on, the lactoferrin band is no longer visible. In the intestinal phase, only a faint band around 50 kDa can be seen, which likely originates from lipase in the artificial duodenal juice. The same digestion was carried out in a test tube rotating head-over-heels, as a control experiment under the same conditions (Fig. 7B). There, we observe a slower digestion in the gastric phase, with the lactoferrin band (around 78 kDa) becoming less intense over time, but not disappearing. After transferring the mixture to the intestinal phase, the lactoferrin band disappeared within 5 min by rapid protease activity of the duodenal juice. The digestion of lactoferrin was therefore faster in our miniaturized system, as compared to a rotating test tube. We hypothesize that most of the digestive action takes place in the micromixer of the stomach, even though it only has a residence time of 6 s: the output of the oral phase and gastric juice are thoroughly mixed by the folding-like motions of the liquid in the micromixer, causing a fully mixed gastric mixture after only a few cycles. Digested lactoferrin fragments are visible at the bottom of the gel. The digestive enzymes, α-amylase and pepsin, will also be digested, but because of their low abundance, they are not visible in these gels. Even after 120 min in the gastric phase, the lactoferrin fragments are not fully digested. After transferring to the intestine, however, these bands <10 kDa quickly disappear, indicating complete digestion. The digestion of lactoferrin occurs partly in the stomach and partly in the intestine, and is therefore a good example to show multi-compartment digestion in our system.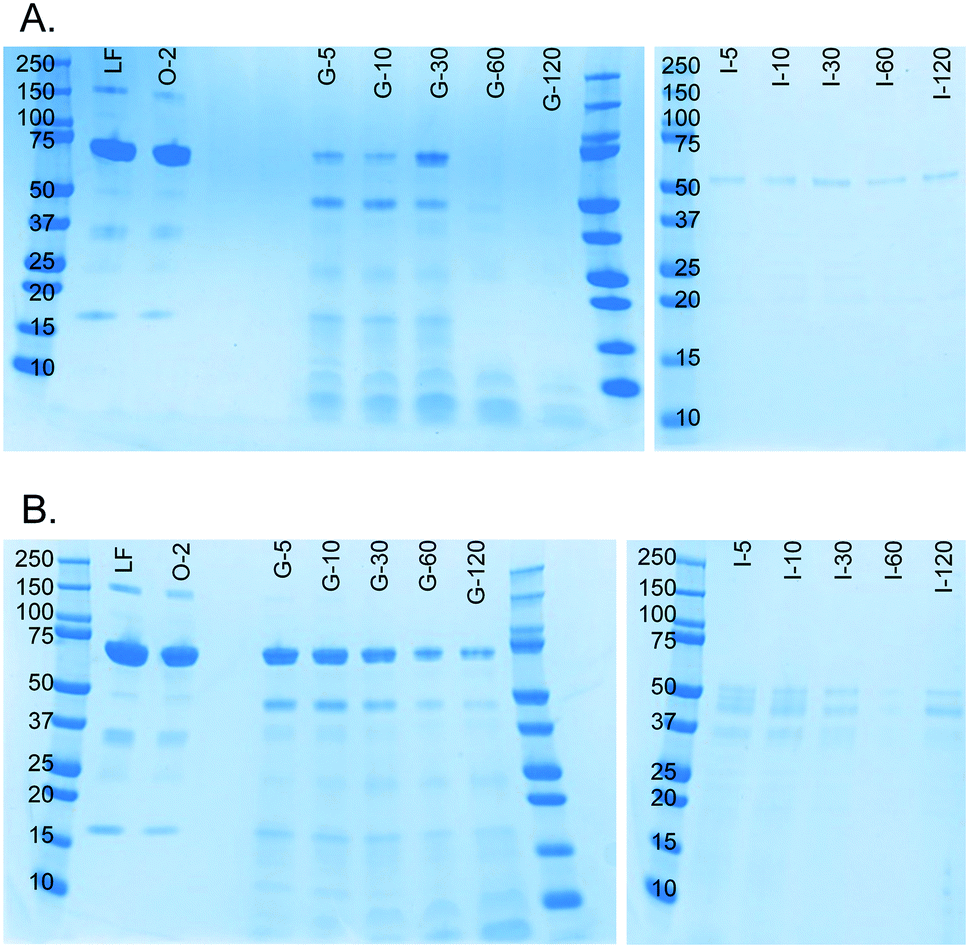 | ||
| Fig. 7 SDS-PAGE gels showing results of the digestion of lactoferrin (50 mg mL−1 in water) in A) our microfluidic digestive system and B) a test tube rotating head-over-heels. The same artificial digestive juices were used for both experiments, and both were carried out in incubators set at 37 °C. All samples were diluted to 1 mg mL−1 lactoferrin before analysis (marker protein size displayed in kDa). LF: lactoferrin control sample; O-2: oral phase sample after 2 min; G-5: gastric phase sample after 5 min, etc.; I-5: intestinal phase sample after 5 min, etc. | ||
Conclusion and future outlook
We have described the development of a three-compartment, microfluidic digestion system, in which we can vary the reaction conditions from one compartment to the next, allowing different reactions to take place. We have demonstrated enzymatic function by digesting fluorescent compounds and by digesting molecules which are of interest in the field of nutrition. As shown for the milk protein, lactoferrin, the digestion in our miniaturized system is considerably faster than batch-wise digestions in vitro, and will save time as well as chemicals (with a combined net consumption of 6 mL of digestive juices per 4 h digestion, at 25 μL min−1). We expect that our system will need a shorter timespan for digestion than in the physiological situation, due to fast and thorough mixing. Digestion times in our model system need to be compared to physiological conditions in order to further optimize incubation times. In the future, this system will be coupled to a gut-on-a-chip, a barrier model of the human intestine containing living epithelium. This would allow integrated testing of the terms FF and FG, related to compound bioavailability, for new drugs, toxicants and nutrients. When coupled in-line to a device mimicking the human liver (e.g. a liver-on-a-chip), the full bioavailability F (including FF, FG and FH) could be studied, by transferring the basolateral flow of the gut-on-a-chip via a ‘portal vein’ connection to a piece of liver tissue. We will be investigating the in-line conjugation to a gut-on-a-chip next, to study the uptake of medicinal drugs and toxicants after digestion.Author contributions
Conceptualization: H. B. and E. V.; Data curation: P. d. H.; Formal analysis: P. d. H.; Funding acquisition: H. B. and E. V.; Investigation: P. d. H.; Methodology: P. d. H., M. A. I. and E. V.; Project administration: P. d. H. and E. V.; Resources: M. A. I., K. M., G. A. A. v. L., V. T. and H. B.; Software: Not applicable; Supervision: E. V.; Validation: P. d. H., G. A. A. v. L., V. T. and H. B.; Visualization: P. d. H.; Writing – original draft: P. d. H. and E. V.; Writing – review & editing: P. d. H., M. A. I., K. M., G. A. A. v. L., V. T., H. B., and E. V.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research received funding from the Netherlands Organization for Scientific Research (NWO) in the framework of the Technology Area PTA-COAST3 (GUTTEST, Project #053.21.116) of the Fund New Chemical Innovations. We thank Margot Jeronimus-Stratingh (Interfaculty Mass Spectrometry Centre, University of Groningen) for assistance in protein analysis.References
- O. H. Petersen, Human Physiology, Blackwell Publishing, Malden, MA, USA, 5th edn, 2007, pp. 494–539 Search PubMed.
- M. Rowland and T. N. Tozer, Clinical Pharmacokinetics and Pharmacodynamics, Lippincott Williams & Wilkins, Baltimore, MD, USA, 4th edn, 2011, pp. 183–215 Search PubMed.
- H. W. Frijlink, D. J. Touw and H. J. Woerdenbag, Biopharmaceutics, in Practical Pharmaceutics, ed. Y. Bouwman-Boer, V. Fenton-May and P. Le Brun, Springer International Publishing, Cham, Switzerland, 1st edn, 2015, pp. 323–346 Search PubMed.
- A. G. Oomen, C. J. M. Rompelberg, M. A. Bruil, C. J. G. Dobbe, D. P. K. H. Pereboom and A. J. A. M. Sips, Arch. Environ. Contam. Toxicol., 2003, 44, 281–287 CrossRef CAS PubMed.
- Y. Imura, Y. Asano, K. Sato and E. Yoshimura, Anal. Sci., 2009, 25, 1403–1407 CrossRef CAS PubMed.
- H. Kimura, T. Yamamoto, H. Sakai, Y. Sakai and T. Fujii, Lab Chip, 2008, 8, 741–746 RSC.
- A. A. Ahmad, Y. Wang, A. D. Gracz, C. E. Sims, S. T. Magness and N. L. Allbritton, J. Biol. Eng., 2014, 8, 9 CrossRef PubMed.
- K. Y. Shim, D. Lee, J. Han, N. T. Nguyen, S. Park and J. H. Sung, Biomed. Microdevices, 2017, 19, 37 CrossRef PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Lab Chip, 2012, 12, 2165–2174 RSC.
- D. Huh, H. J. Kim, J. P. Fraser, D. E. Shea, M. Khan, A. Bahinski, G. A. Hamilton and D. E. Ingber, Nat. Protoc., 2013, 8, 2135–2157 CrossRef CAS PubMed.
- H. J. Kim, H. Li, J. J. Collins and D. E. Ingber, Proc. Natl. Acad. Sci. U. S. A., 2015, 113, E7–15 CrossRef PubMed.
- G. Trujillo-de Santiago, M. J. Lobo-Zegers, S. L. Montes-Fonseca, Y. S. Zhang and M. M. Alvarez, Microphysiol. Syst., 2018, 2, 7 Search PubMed.
- Q. Ramadan, H. Jafarpoorchekab, C. Huang, P. Silacci, S. Carrara, G. Koklü, J. Ghaye, J. Ramsden, C. Ruffert, G. Vergeres and M. A. M. Gijs, Lab Chip, 2013, 13, 196–203 RSC.
- Y. Imura, K. Sato and E. Yoshimura, Anal. Chem., 2010, 82, 9983–9988 CrossRef CAS PubMed.
- M. B. Esch, G. J. Mahler, T. Stokol and M. L. Shuler, Lab Chip, 2014, 14, 3081–3092 RSC.
- I. Maschmeyer, A. K. Lorenz, K. Schimek, T. Hasenberg, A. P. Ramme, J. Hübner, M. Lindner, C. Drewell, S. Bauer, A. Thomas, N. S. Sambo, F. Sonntag, R. Lauster and U. Marx, Lab Chip, 2015, 15, 2688–2699 RSC.
- G. J. Mahler, M. B. Esch, R. P. Glahn and M. L. Shuler, Biotechnol. Bioeng., 2009, 104, 193–205 CrossRef CAS PubMed.
- Y. Imura, E. Yoshimura and K. Sato, Anal. Sci., 2012, 28, 197–199 CrossRef CAS PubMed.
- A. Choe, S. K. Ha, I. Choi, N. Choi and J. H. Sung, Biomed. Microdevices, 2017, 19, 4 CrossRef PubMed.
- D. W. Lee, S. K. Ha, I. Choi and J. H. Sung, Biomed. Microdevices, 2017, 19, 100 CrossRef PubMed.
- T. Bricks, J. Hamon, M. J. Fleury, R. Jellali, F. Merlier, Y. E. Herpe, J. M. Regimbeau, F. Bois and E. Leclerc, Biopharm. Drug Dispos., 2015, 293, 275–293 CrossRef PubMed.
- J. M. Prot, L. Maciel, T. Bricks, F. Merlier, J. Cotton, P. Paullier, F. Y. Bois and E. Leclerc, Biotechnol. Bioeng., 2014, 111, 2027–2040 CrossRef CAS PubMed.
- European Directorate for the Quality of Medicines and HealthCare, The European Pharmacopoeia, 8.6., 2016, pp. 5117–5123 Search PubMed.
- C. H. M. Versantvoort, A. G. Oomen, E. Van de Kamp, C. J. M. Rompelberg and A. J. A. M. Sips, Food Chem. Toxicol., 2005, 43, 31–40 CrossRef CAS PubMed.
- M. Minekus, M. Alminger, P. Alvito, S. Ballance, T. Bohn, C. Bourlieu, F. Carrière, R. Boutrou, F. M. Corredig, D. Dupont, C. Dufour, L. Egger, M. Golding, S. Karakaya, B. Kirkhus, S. Le Feunteun, U. Lesmes, A. Macierzanka, A. Mackie, S. Marze, D. J. McClements, O. Ménard, I. Recio, C. N. Santos, R. P. Singh, G. E. Vegarud, M. S. J. Wickham, W. Weitschies and A. Brodkorb, Food Funct., 2014, 5, 1113–1124 RSC.
- D. Dupont, M. Alric, S. Blanquet-Diot, G. Bornhorst, C. Cueva, A. Deglaire, S. Denis, M. Ferrua, R. Havenaar, J. Lelieveld, A. R. Mackie, M. Marzorati, O. Menard, M. Minekus, B. Miralles, I. Recio and P. Van den Abbeele, Crit. Rev. Food Sci. Nutr., 2018, 1–17 CrossRef CAS PubMed.
- A. P. Walczak, R. Fokkink, R. Peters, P. Tromp, Z. E. Herrera Rivera, I. M. C. M. Rietjens, P. J. M. Hendriksen and H. Bouwmeester, Nanotoxicology, 2013, 7, 1198–1210 CrossRef CAS PubMed.
- A. P. Walczak, E. Kramer, P. J. M. Hendriksen, P. Tromp, J. P. F. G. Helsper, M. van der Zande, I. M. C. M. Rietjens and H. Bouwmeester, Nanotoxicology, 2014, 5390, 453–463 Search PubMed.
- A. P. Walczak, E. Kramer, P. J. M. Hendriksen, R. Helsdingen, M. van der Zande, I. M. C. M. Rietjens and H. Bouwmeester, Nanotoxicology, 2015, 9, 886–894 CrossRef PubMed.
- A. D. Stroock, S. K. W. Dertinger, A. Ajdari, I. Mezić, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647–651 CrossRef CAS PubMed.
- M. A. Ianovska, P. P. M. F. A. Mulder and E. Verpoorte, RSC Adv., 2017, 7, 9090–9099 RSC.
- C. B. Furlund, E. K. Ulleberg, T. G. Devold, R. Flengsrud, M. Jacobsen, C. Sekse, H. Holm and G. E. Vegarud, J. Dairy Sci., 2013, 96, 75–88 CrossRef CAS PubMed.
- B. Wang, Y. P. Timilsena, E. Blanch and B. Adhikari, Crit. Rev. Food Sci. Nutr., 2017, 1–17 Search PubMed.
- B. Wang, Y. P. Timilsena, E. Blanch and B. Adhikari, Int. Dairy J., 2017, 64, 22–30 CrossRef CAS.
- L. Stryer, Biochemistry, W.H. Freeman and Co., New York, NY, USA, 3rd edn, 1988, pp. 187–190 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- P. de Haan, J. P. S. H. Mulder and E. Verpoorte, Manuscript in preparation.
- CIBA-GEIGY, Wissenschaftliche Tabellen Geigy, Teilband Körperflüssigkeiten, Ciba-Geiby AG, Basel, Switzerland, 8th edn, 1977, pp. 113–137 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01080c |
| This journal is © The Royal Society of Chemistry 2019 |