
Cellular heterogeneity identified by single-cell alkaline phosphatase (ALP) via a SERRS-microfluidic droplet platform†
Dan
Sun
a,
Fanghao
Cao
ab,
Lili
Cong
a,
Weiqing
Xu
 a,
Qidan
Chen
b,
Wei
Shi
c and
Shuping
Xu
*a
a,
Qidan
Chen
b,
Wei
Shi
c and
Shuping
Xu
*a
aState Key Laboratory of Supramolecular Structure and Materials, Institute of Theoretical Chemistry, Jilin University, Changchun 130012, PR China. E-mail: xusp@jlu.edu.cn
bSchool of Chemical Engineering and New Energy Materials, Zhuhai College, Jilin University, Zhuhai 519041, PR China
cKey Lab for Molecular Enzymology & Engineering of Ministry of Education, Jilin University, Changchun 130012, PR China
First published on 19th December 2018
Abstract
Alkaline phosphatase (ALP) is a useful indicator for disease state diagnosis and clinical outcome. Investigation of ALP expression among cells is still challenging since ALP expression in a single cell is too low to be detectable. In our work, an ultrasensitive, high-throughput analytical method was applied for ALP determination in a single cell by using a surface-enhanced resonance Raman scattering (SERRS)-based microfluidic droplet technique. An ALP catalyzed substrate (5-bromo-4-chloro-3-indolyl phosphate, BCIP) was used to evaluate ALP activity in the cell within one droplet. When BCIP was incubated with cells, ALP can catalyze a hydrolysis reaction of colorless BCIP and oxidize the intermediate compound to form blue 5,5′-dibromo-4,4′-dichloro-1H,1H-[2,2′]biindolylidene-3,3′-dione (BCI), which is a resonant Raman-active species. The encapsulation of BCI in droplets is favorable for detecting extremely low levels of molecules due to an accumulation effect along with reaction time. The ALP concentration as low as 1.0 × 10−15 M can be successfully detected in a uniform droplet. In addition, cellular heterogeneity profiled by ALP expression on single-cell resolution was monitored with this SERRS-based microfluidic droplet technique. Ultrasensitive determination of ALP secreted from individual cells can help us to understand cell-to-cell heterogeneity.
Introduction
Single-cell analysis has been paid increasing attention because it provides an important way to understand and decipher many key biological processes and cell heterogeneity, which are the fundamental issues of early diagnosis, precision treatment and drug development for diseases. This desire becomes the driving force to promote the development of corresponding instruments, protocols, and methods for single-cell analysis. The fluorescence-activated flow cytometry1 based on end point measurements is one of the most widely used tools in high-throughput single-cell studies, as well as cell sorting due to its big data collection. Recently, microfluidic tools under precisely defined conditions as another high-throughput method for analyzing individual cells have been paid more and more attention.2–4 Monodisperse aqueous droplets derived from soft lithography are one of the hottest topics in microfluidics due to their wide applications in nanosphere lithography, drug microcapsules, microreactors, etc.5 This technique, achieved by compartmentalizing water via a continuous oil phase with a special channel, can also compartmentalize individual cells to investigate antibody production by single cells dating back to the 1950s.6 The picoliter droplet can be considered as a ‘nanolab’ for accommodating a single cell and all factors secreted from this cell in an isolated environment.7,8 Such a single-cell analytical platform exhibits remarkable merits beyond flow cytometry such as: (1) droplets can be collected from a microfluidic device and incubated off-chip,9 which allows hours or days of residence time to finish the whole cell physiological processes. (2) The platform can realize the drug dose by fusion10 or picoinjection11 at a well-defined moment. (3) Droplets can also be easily sorted from the rest of the droplet stream by the difference in size12 or activity.13 Also, real-time detection in microfluidic droplet systems is not restricted to fluorescence or chemiluminescence as compared to traditional flow cytometry methods.14 In addition, it can be widely combined with various probing techniques, such as absorption spectroscopy,15 electrochemisty,16 mass spectrometery,17 and Raman spectroscopy,18–20 which makes microfluidic droplet techniques more flexible and applicable for diverse detections.Surface-enhanced Raman scattering (SERS) spectroscopy is a highly sensitive bio-analytical method with superiority in pointing out the biomolecular vibration.21–23 However, SERS performs poorly in quantitative analysis of bio-samples, due to its difficulty in reproducing SERS enhancement. SERS signal variability is primarily derived from the lack of control over factors, such as the degree of aggregation, particle size and inhomogeneous distribution of analytes on the surface.24,25 The combination of SERS and micro-droplet technique provides a practical strategy for sensitive detection and reproducible measurement at a defined detection spot, since the aggregation time of metal nanoparticles and the mixing efficiency of the metal colloids and the probed solutions can be precisely controlled via the tunable flow rate and the designed channel structure.26 Thus, the SERS-based droplet microfluidic method has been considered as a promising platform for highly sensitive, quantitative detection of bio-samples.27,28
In the present study, for cell-to-cell heterogeneity and spatiotemporal determination of single living cells, a technique combining surface-enhanced resonance Raman scattering (SERRS) and droplet microfluidics was developed for the ultrasensitive analysis of alkaline phosphatase (ALP) activity. ALP is a zinc-containing homodimeric glycoprotein, which is able to remove the phosphate group on substrate molecules including nucleic acids, proteins and carbohydrates through hydrolyzing the phosphate monoester to generate phosphate ions and free hydroxyl groups. The process is considered as dephosphorylation. ALP exists in various tissues of the body and is a common indicator used for the diagnosis of biliary and skeletal diseases. The level of ALP within normal human serum was about 0.71–3.37 mM for adults.29 The abnormal level of ALP was closely related to various diseases including hepatic tuberculosis, obstructive jaundice, secondary liver cancer, osteomalacia, fracture healing, bone cell cancer and malignant bone metastasis.30–32 In this work, the live hepatocellular carcinoma (HepG2) cell line was chosen as a model. To detect ALP in a HepG2 cell in an isolated droplet, a substrate, 5-bromo-4-chloro-3-indolyl phosphate (BCIP), can be specifically hydrolysed by ALP and then oxidized by oxygen to form a blue-coloured indigo dye derivative (5,5′-dibromo-4,4′-dichloro-1H,1H-[2,2′]biindolylidene-3,3′-dione, BCI). It is found to exhibit high SERS-activity33 and a Raman resonance effect when excited under a 632.8 nm laser. The SERRS spectra of the catalytic product (BCI) were recorded by mixing the substrate with gold nanoparticles (AuNPs) and HepG2 cells in individual droplets, and the SERRS intensities obtained by measuring a large number of droplets were plotted to compare the ALP expression between normal liver cells and hepatocellular carcinoma. This method shows ultrahigh detection sensitivity from the SERRS effect, and the lowest detectable concentration of ALP achieved is 1.0 × 10−15 M in a uniform droplet. In order to identify distinct epigenetic states and characterize underlying patterns of variability, we distinguished the single cells from cell clusters according to the ALP contents. In addition, the effect of a drug (sorafenib, a drug for patients with advanced liver cancer34) on the dynamic expression of ALP was also investigated. This research platform that integrates micro-droplet technology with Raman spectroscopy can be used for non-destructive, real-time online, highly sensitive, and single-cell metabolite analysis.
Materials and methods
Fabrication of the microfluidic chip
Microfluidic flow chambers were fabricated by soft lithography.35 In detail, negative photoresist SU-8 2025 (Microchem) was spin-coated onto a clean silicon wafer and the SU-8 film was patterned through a transparency photomask via exposing it to a 385 nm LED array light source. Thus, a microchannel structure with the mask pattern was transferred to the SU-8 film. Then, Sylgard 184 poly(dimethylsiloxane) (PDMS) (Dow Corning, Midland, MI) mixed with a crosslinker (ratio as 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and degassed thoroughly was poured onto the photoresist patterns, and cured for at least 3 h at 80 °C. The PDMS replica was peeled off from the wafer. Before the PDMS was bonded to a glass slide, oxygen-plasma activation of both the PDMS and the glass surfaces was carried out.
:1) and degassed thoroughly was poured onto the photoresist patterns, and cured for at least 3 h at 80 °C. The PDMS replica was peeled off from the wafer. Before the PDMS was bonded to a glass slide, oxygen-plasma activation of both the PDMS and the glass surfaces was carried out.
They were bonded under high pressure and heating conditions. The microfluidic channels were then filled with an Aquapel solution (PPG Industries, Pittsburgh, PA) to improve their wettability toward the fluorinated oil. Polyethylene tubes with an inner diameter of 0.8 mm and an outer diameter of 1.6 mm were connected to the channels by syringes.
Cell culture and cell viability assay
HepG2 (human liver cancer cell carcinoma) and BNL.CL (a noncancer liver cell line) were both bought from Shanghai ATCC Cell Bank that has been issued the permission of the Human Research Ethics Committee of the country for manipulations of human cells. They were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Gibcos) supplemented with 10% fetal bovine serum (FBS, GIBCO) along with penicillin/streptomycin (1%, 1.0 g mL−1) and incubated in a humidified atmosphere with 5% CO2 for 37 °C.The WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium monosodium salt) assay was used to test the viability of cells incubated with different concentrations of AuNPs while BCIP was added at 3.0 mg mL−1 as an optimized concentration reported before.33 Firstly, the HepG2 cells were cultured in a 96-well plate for 24 h. Then, 5.0 μL of the 3.0 mg mL−1 BCIP solution and 5.0 μL of different concentrations of AuNPs were added into the culture medium for another 2 h. After that, 20.0 μL of the WST-1 solution was added to each well to incubate for 1 h. A microplate reader (Tecan Sunrise) was used to determine the cell viability by the fluorescence intensity of each well at a wavelength of 450 nm. HepG2 cells were incubated in standard culture medium as the control group.
Droplet formation and cell encapsulation
Cells were digested with trypsase (wt 0.25%) for 3 min to achieve a cell-suspension solution. Then trypsase was removed and the cells were re-suspended in fresh medium. They were centrifuged at 1500 rpm for 3 min and the growth medium was carefully removed. They were re-suspended in 840 μL of ice-cold complete DMEM, with 160 μL of OptiPrep medium. The cell density was adjusted to a desired value. For the purpose of single cell studies, we required that each droplet encapsulate one cell. The majority of the droplets contained no cell at all because of the encapsulation process following Poisson statistics according to David A. Weitz's research work.36 In our experiment, by controlling the cell density at 3.0 × 106 cells per mL, we obtained roughly 20% of droplets with a single cell, while less than 6% of droplets have two or more cells (Fig. S4 and related discussion are found in the ESI†). The chip and droplet formation were visualized using a microscope equipped with a CCD and a bright lamp, as shown in Fig. S1(a).†Fluorinert HFE-7500 fluorocarbon oil (3 M, St. Paul, MN) was used to suspend the drops. To stabilize the drops, a PFPE-PEG block-copolymer surfactant (Ranbiotech) was added to the suspending oil with a concentration of 2.0% (w/w). The surfactant can protect the drops against coalescence. For drop formation, the outer, carrier-oil flow rate was 1040 μL h−1 and the inner, cell suspension solution flow rate was set as 58 μL h−1. The droplets were collected in an oil-filled glass vial.
Calibration of ALP in droplets
The AuNPs used in the experiment were synthesized by the citrate reduction reaction.37 The achieved quasi-spherical shape AuNPs have a plasmonic band located at 530 nm. The BCIP solution (3.0 mg mL−1, 100 μL) was added into 800 μL of ALP solutions. The mixture was incubated at room temperature for 1 h, followed by addition of 100 μL of concentrated AuNPs (about 8.0 nM). 10 μL of the resulting solution was transferred onto a glass slide for SERS measurements.To establish a standard curve for determining ALP in droplets, the mixed solution including BCIP, AuNPs and different concentrations of ALP (1.0 × 10−10 M to 1.0 × 10−15 M in pH = 7.4 Tris-HCl buffer solution) was co-encapsulated into water-in-oil droplets. The emulsion was collected over 10 min in a glass vial respectively and then the packed droplets were re-injected into a microfluidic readout circuit and spaced out into single-droplet trains by infusion of oil, as shown in Fig. S1(d–f).† Then, the SERRS measurement at defined location points of the droplets was carried out using a Horiba Jobin Yvon Aramis spectrometer at 632.8 nm from a laser power of ∼7 mW and an accumulation time of 5 s per time.
Evaluation of ALP on cells by SERRS
To display the difference of ALP expression on different cell lines, we performed the analysis on two cell lines (HepG2 and BNL.CL2 cells) by staining ALP on the cell with AP Live Stain (500×) dye that highlights ALP in vivo with green fluorescence. Before that, the cells were cultured on coverslips in DMEM culture medium with 10% fetal bovine serum (FBS) at 37 °C in a 5% CO2 incubator for 24 h. Then, 2.0 μL of AP Live Stain (500×) dye was added into the plate over 10 min. Then the coverslips were washed three times with a PBS buffer (pH = 7.4) before taking the confocal fluorescence images.To evaluate the ALP expression on a single HepG2 cell, the suspended cells combined with BCIP (3.0 mg mL−1) and AuNPs (8.0 nM) were co-encapsulated into a water-in-oil droplet. By tuning the pumping rate and cell concentration, we isolated a single cell by one droplet. The achievement for cell isolation was nearly 20%. The collected droplets were incubated for 2 h (Fig. S5†) and then they were re-injected into the detection chip. The reason why we used a non-continuous chip is based on the consideration of flexibility. The bad units can be easily replaced and the inevitable experimental problems caused by clogging or leakage can be mitigated. The detection chip contains three parallel channels and each channel can store about 100 droplets, where the droplets are closely arranged without fusion due to the stabilization of the surfactant. The droplets containing single cells were easily found under a bright field microscope. When the laser was focused on the cell in the droplets, the SERRS signal of BCI in a single cell was collected and the activity of ALP in the individual cells was evaluated according to the signal intensity value of BCI (as shown in Fig. S1g†).
Results and discussion
Characterization of the enzymatic reaction substrate and product
We have proposed a high-throughput SERRS-based droplet microfluidic platform for sensitive analysis of the ALP activity on a single cell. Here, AuNPs, as a SERS signal enhancement agent, were fabricated. The achieved AuNPs have a plasmonic band located at 530 nm (Fig. 1c) and the size characterized by the transmission electron microscope (TEM) is about 40 nm (Fig. 1a and S3†). Such size of AuNPs can not only guarantee the SERS enhancement effect, but also keep high stability and mobility when they were incubated with cells in the cell culture medium. The cell viabilities of HepG2 cells incubated with 5.0 μL of the BCIP solution (3.0 mg mL−1) and 5.0 μL of different concentrations of AuNPs were evaluated after 2 h of incubation and the results shown in Fig. 1b demonstrate that our designed sensing method has weak cell cytotoxicity to HepG2 cells.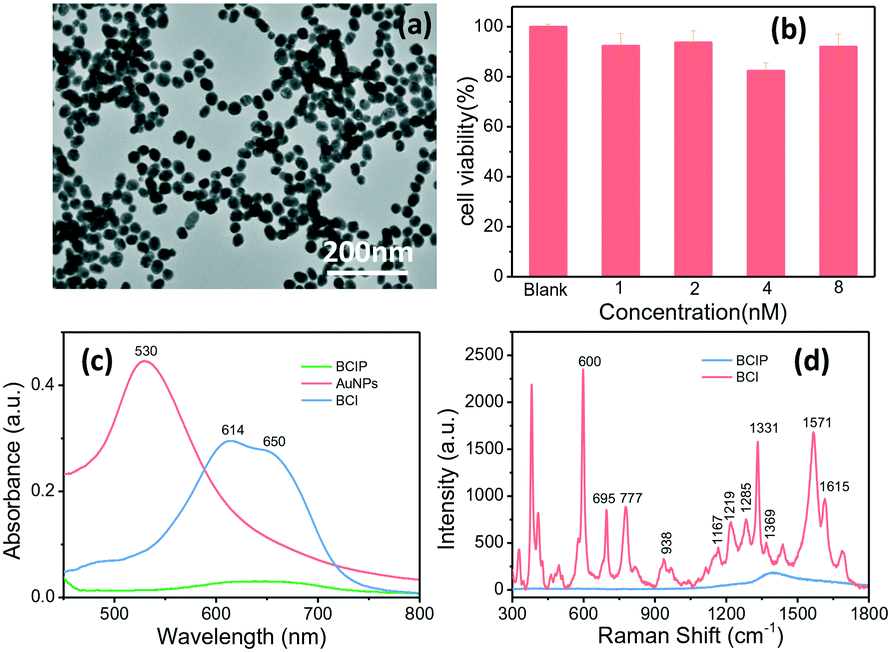 | ||
| Fig. 1 (a) TEM image of AuNPs. (b) Cell viability of HepG2 cells in a 96-well plate incubated with 5.0 μL of the 3.0 mg mL−1 BCIP solution and 5.0 μL of different concentrations of AuNPs for 2 h. HepG2 cells incubated with standard culture medium were used as the blank. (c) UV-vis spectra of the AuNPs and BCIP (3.0 mg mL−1) before and after reacting with ALP (1.0 × 10−10 M). (d) SERRS spectra of both BCIP and its enzymatic reaction product (BCI) under excitation at 632.8 nm. | ||
In order to evaluate the feasibility of BCIP for ALP determination, ultraviolet-visible (UV-vis) absorption measurements of BCIP (3.0 mg mL−1) before and after reacting with ALP (1.0 × 10−10 M) were carried out. As shown in Fig. 1c, BCIP exhibits no absorbance peaks in the visible range. However, the enzymatic reaction product (BCI) has a typical absorption peak at 614 nm, with a shoulder peak at about 650 nm, indicating that the colourless BCIP has been converted to the coloured BCI under ALP catalysis. It was known that resonating the electron state with a suitable laser wavelength would markedly induce its SERS signal, that is, the SERRS effect.38,39 Therefore, a 632.8 nm laser was used to excite BCI in our sensing system. The SERRS spectra of both BCIP and its enzymatic reaction product (BCI) were collected to explore the feasibility and potential interferences in ALP sensing by using BCIP as a substrate (Fig. 1d). The results indicate that the BCIP substrate gives no discernible SERS signal, except for a broad peak at 1300–1500 cm−1, which is assigned to the background scattering from the glass slide. Exposing BCIP to ALP induces a hydrolysis reaction of the phosphate moiety of BCIP and generates an enol intermediate compound, which is subsequently oxidized in air to generate the blue BCIP dimer (BCI) (Scheme 1). Fortunately, BCI produces an intense and identifiable SERRS spectrum, in which the strong vibrational bands of the enzymatic reaction product adsorbed onto AuNPs are observed at 600, 695, 777, 938, 1167, 1219, 1285, 1331, 1369, 1571, and 1615 cm−1 (Fig. 1d). The band assignments are provided in Table S1 in the ESI.† These observations demonstrate that BCI produced from the enzymatic reaction is SERRS-active, and the strongest Raman shift at 600 cm−1 that is assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CO–C bending vibration40 was chosen to quantify the ALP expression in the following part.
C–CO–C bending vibration40 was chosen to quantify the ALP expression in the following part.
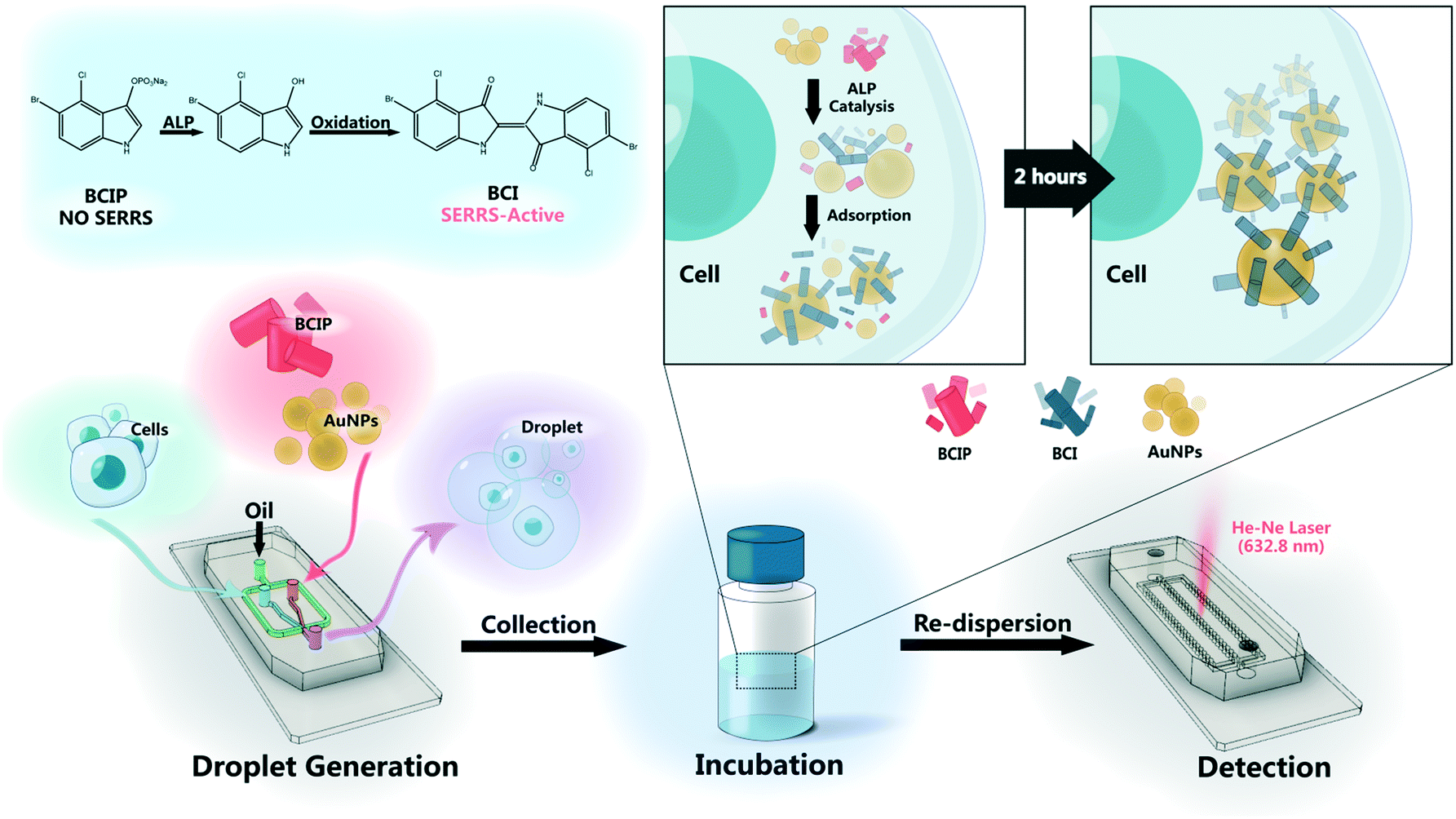 | ||
| Scheme 1 Overview of the work flow of the droplet-based microfluidic SERRS method for single-cell encapsulation and detection of secreted ALP. Single cells (3.0 × 106 cells per mL) are encapsulated in droplets together with 3.0 mg mL−1 of the BCIP solution and 8.0 nM of AuNPs. After collection and incubation, the expression of ALP by single cells is quantified by SERRS. | ||
Calibration of ALP in droplets
The background signal of PDMS may interfere with the microfluidic chip-based Raman detection system when the analytes have weak Raman activity. To check whether the ALP on-chip Raman analysis system is compatible with the PDMS microfluidic chip, the spectra of PDMS, a blank drop in the chip channel, and BCIP in the droplets before and after reacting with ALP were collected and compared. The backgrounds from PDMS and fluorinated carrier oil HFE-7500 (including surfactant PFPE-PEG) can be clearly observed in their Raman spectra. As shown in Fig. 2A, there are two strong Raman characteristic peaks observed for PDMS at 490 and 709 cm−1. However, the Raman spectra of blank droplets in the chip show no new peaks compared to PDMS. It might be that the spectrum of the fluorinated carrier oil has been overwhelmed by the strong scattering of PDMS. Although the PDMS background exists, the SERS spectrum of the product BCI contains almost all the characteristic peaks which are strong enough and identifiable in the range of 1300–1500 cm−1 when measured on the droplet-based microfluidic chip, especially the strongest peak at 600 cm−1. | ||
| Fig. 2 (A) Raman spectra of individual PDMS (1) and blank drops measured in the PDMS chip channel (2), and SERS spectra of BCIP in the droplets before (3) and after reacting (4) with ALP. (B) SERRS spectra of the enzymatic reaction products at different ALP concentrations measured in droplets. (a)–(f) refer to ALP concentrations of 1.0 × 10−15, 1.0 × 10−14, 1.0 × 10−13, 1.0 × 10−12, 1.0 × 10−11 and 1.0 × 10−10 M, respectively, while 3.0 mg mL−1 of the BCIP solution was kept for all trials. A reaction time of 2 h was used. A laser wavelength of 632.8 nm and an acquisition time of 5 s were used for SERRS measurements. (C) A plot of the ratio of I600cm−1/I709cm−1 of BCI along with the concentration of ALP. | ||
Then, the quantification of ALP in droplets was carried out by recording the SERRS spectra of ALP at different concentrations ranging from 1.0 × 10−10 to 1.0 × 10−15 M after incubation with BCIP in pH 7.4 Tris-HCl buffer for 2 h (Fig. 2B). The SERRS intensity of BCI increases as the enzyme concentration, suggesting that more BCI have been produced in the presence of high concentrations of ALP. Using the characteristic peak of PDMS at 709 cm−1 as an internal standard, the values of I600cm−1/I709cm−1 with the ALP concentrations are plotted and fitted by a non-linear model (Fig. 2C). The limit of detection is calculated to be 1.0 × 10−15 M (signal/noise = 3), indicating that the technique allows for an ultrasensitive detection of ALP in the microfluidic droplets. The reasons behind such high sensitivity in our method can be explained by three aspects: (1) the gold nanoparticles amplify the Raman scattering intensity of molecules adsorbed on their surface. (2) We incorporated the probed molecules in resonance with the 632.8 nm laser used in our droplet-based microfluidic system, maximizing the resonant interaction at the metal/molecule interface, which results in markedly higher SERS intensities. (3) Acting as small volume reactors, the droplets can not only reduce the noise and increase the detection signal-to-noise ratio but also provide a narrow space, where the enzymatic reaction product can be gradually accumulated to quickly reach a detectable level.
Monitoring the ALP expression on different cell lines
The applicability of the SERRS sensing system for the detection of ALP in single living cells was evaluated. BCIP, AuNPs and the suspended cells were mixed prior to the formation of discrete, water-in-oil droplets. In order to obtain the highest single-cell encapsulation efficiency, the starting cell concentration was optimized and strictly fixed at 3.0 × 106 cells per mL (as seen in Fig. S4†). As shown in Fig. 3a, plenty of uniform and monodisperse water-in-oil droplets were obtained. Many of them encapsulate single cells (as indicated by the yellow arrow). These droplets were collected on an oil phase surface in a vial. BCIP has a high diffusion rate and can rapidly diffuse to the cell within the droplet. The ALP expressed by the cell catalyzes BCIP to produce a hydrolysed intermediate, which can be directly oxidized by dissolved oxygen to form BCI. The continuously generated BCI can easily diffuse and attach to the surface of AuNPs. We focused the laser spot on the cells in the droplets and collected the SERRS signal from BCI. Thus, we can trace the SERRS of BCI to identify ALP, and the accumulation effect within a single isolated droplet over time allows for an ultrasensitive detection of ALP.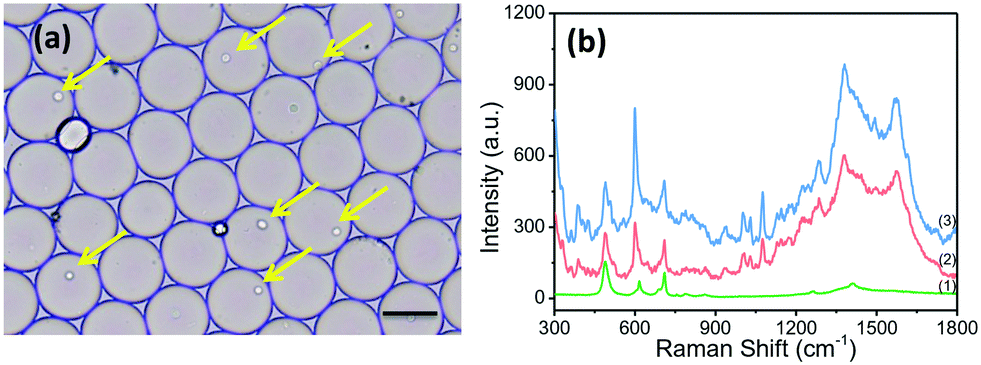 | ||
| Fig. 3 (a) Single cell encapsulated in an individual droplet. The cell-bearing drops are highlighted by the yellow arrows. Scale bar represents 100 μm. (b) Mean SERS spectra of AuNPs incubated with HepG2 cells in drop (1) and BCIP and AuNPs incubated with different cells: BNL.CL2 cells (2) and HepG2 cells (3). All spectra are averaged from at least forty spectra (from at least 10 cells and 4 locations from an individual cell). λex = 632.8 nm, t = 5 s and accumulation = 2 times. | ||
By using our platform, cancer cells and normal cells can be distinguished even at the single cell level. The spectra of HepG2 cells without the ALP-catalyzed substrate (BCIP) in the drop were also measured, and show no new peaks except for the SERS peaks of PDMS itself (curve (1) in Fig. 3b). HepG2 and BNL.CL2 cells were selected as the phosphatase positive controls and phosphatase negative controls, respectively, because of their different expression levels of ALP.32 We found that the SERRS intensity at 600 cm−1 of BCI measured from HepG2 cells after 2 h of treatment with 3.0 mg mL−1 of BCIP and 8.0 nM of AuNPs is at least one-fold stronger than that from BNL.CL2 cells under the same conditions (as shown in Fig. 3b), which indicates a relatively lower expression level of ALP within the normal cell. To further prove the differences in the expression of ALP between HepG2 cells and BNL cells, we conducted confocal fluorescence microscopic assays with the assistance of a fluorescent alkaline phosphatase (AP) live stain dye that can specifically identify ALP and yield green fluorescent precipitates.41 The fluorescence confocal imaging of cells mainly revealed the differences in ALP expression between HepG2 and BNL.CL2 cells, which can actualize the visualization of ALP expression within the normal and cancer cells. The confocal fluorescence images of HepG2 cells that have been treated with 2.0 μL of AP live stain for 10 min clearly show green fluorescence (Fig. S6†). Meanwhile, weak fluorescence is observed from BNL.CL2 cells under the same conditions. The fluorescence imaging results are in agreement with our SERRS data and previous publication32 which supports that HepG2 has a higher ALP expression than BNL.CL2.
Difference in ALP expression within single or cluster cells
ALP activity is routinely used as a diagnostic indicator of several human diseases, including bone diseases and liver diseases (hepatitis and liver cancer).31 However, current methods yield ensemble profiles that are insensitive to cell-to-cell variation. To accurately describe and eventually elucidate the underlying causes of these heterogeneities requires the analysis of single cells in large enough numbers to correctly represent the ALP population. We took HepG2 cells as an example and the ALP expression in the cells was analysed by using the microfluidic droplet-based SERRS platform. Droplets were collected in an oil-filled vial to begin incubation for the enzymatic reaction. After 2 h, the packed droplets were re-injected into the microfluidic readout chip. The droplets pass through a laser focused in the channel, and the SERRS signal from each droplet containing a single cell was collected for analysis. The ALP activities in single and cluster cells are shown in Fig. 4(a) and (b). The average level of ALP activity in 200 samples for single HepG2 cells in droplets is almost identical to that for bulk cells in a plate (1.458 vs. 1.400). However, in comparison with cell clusters, the expression of ALP on the individually incubated cells varied in a larger range, the standard deviation (SD) of which (0.217) is twice that of the cluster cells (0.102). The lower cell-to-cell difference for the group cells implies that when these cells grow close together they can provide nutrition and communicate with each other, and thus they tend to have a synchronous evolution. But, for the droplet-isolated cells, cell-to-cell communication is prohibited, which can make the cells undergo different and asynchronous stages, causing large heterogeneity.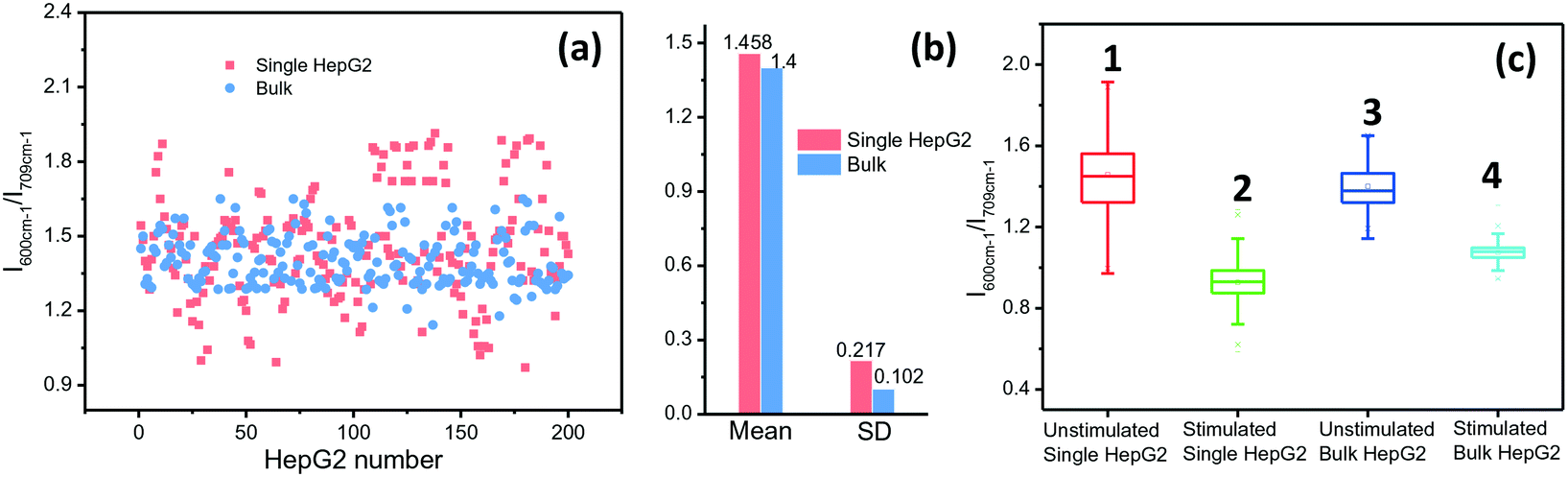 | ||
| Fig. 4 (a) Scattered plots of the SERS intensities of BCI at 600 cm−1 from the single-cell encapsulated microdroplets and bulk cells on the coverslips. (b) Means and standard deviations of 200 samples for single HepG2 cells in contrast to bulk cells. (c) A box-and-whisker plot is shown, where the whiskers represent the minimum and maximum of all of the data. The bottom and top of the box are the first and third quartiles, and the band inside the box is the median. 200 samples of each type were analysed. | ||
To further prove that individual cells differ from cluster cells in ALP expression, we used sodium orthovanadate (Na3VO4) as an inhibitor of ALP to stimulate cells for 30 min, which can adjust the ALP expression in the cells. The SERRS intensities of BCI produced from cells treated with Na3VO4 and untreated cells were then measured and compared. The statistic results of 200 cells show that the average level of ALP activity on the droplet-isolated cells is still close to that on the bulk cells (column 2 vs. column 4, Fig. 4c). However, the droplet-isolated cells after the Na3VO4 stimulation (noted as the error in column 2) still exhibit a larger fluctuation in ALP expression than the bulk cells (noted as the error in column 4), which proves again that the individually grown cells with no cell-to-cell communication display a larger cellular heterogeneity.
Dynamic expression of ALP in response to sorafenib
Single HepG2 cell behaviours in drops were further analysed when treated with sorafenib to adjust their ALP expression. Sorafenib is such a new multi-target antitumor drug that it is the first choice for patients with advanced liver cancer.42Fig. 5a–d summarize the SERRS spectra of BCI in response to single HepG2 cells that had been treated with sorafenib for different durations. The decreasing SERRS intensities at 600 cm−1 illustrate that the ALP levels reduce with the extension of the treatment time (Fig. 5a and b). Moreover, HepG2 cells treated with different concentrations of sorafenib were measured and the SERRS spectra are shown in Fig. 5c and d. It can be observed that the SERRS intensity of BCI changes continually with the increase of the sorafenib concentration and shows a negative correlation. To further evaluate the expression of ALP in the HepG2 cells treated with sorafenib, confocal fluorescence microscopic assays were conducted and the results in Fig. S7† show that the green fluorescence inside HepG2 cells labelled with AP live stain gradually weakens with the increase in the concentration of sorafenib, as well as the extension of the treatment time, which is consistent with the conclusions of our SERRS results (Fig. 5a–d).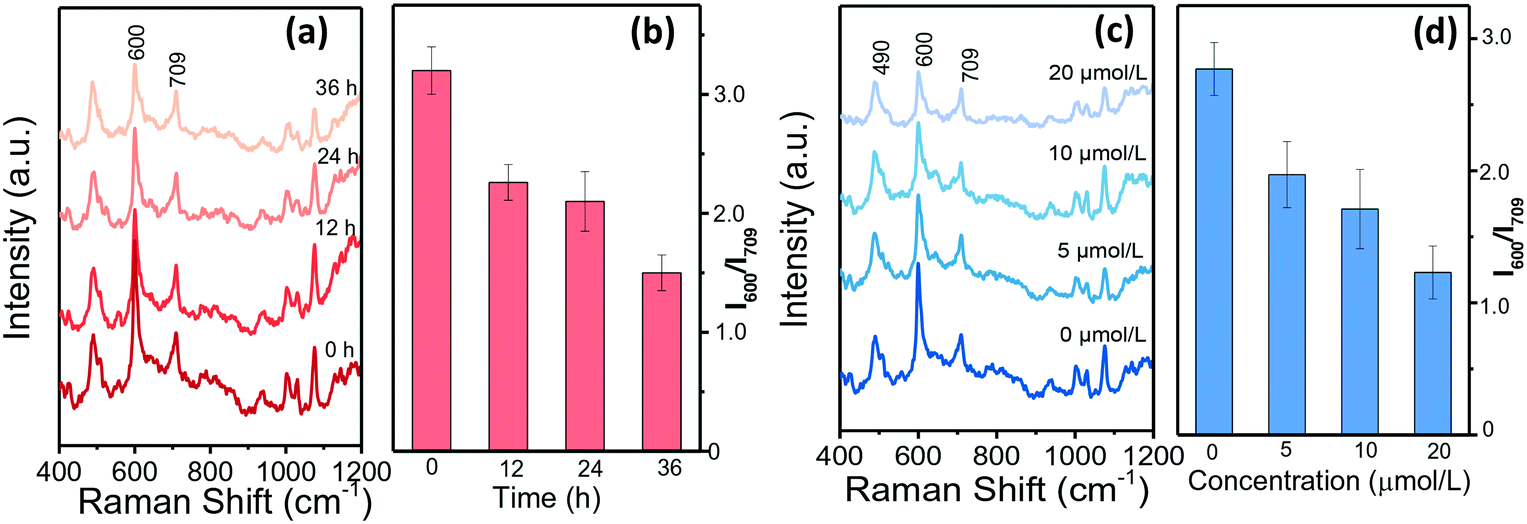 | ||
| Fig. 5 Mean SERRS spectra of BCIP and AuNPs in response to HepG2 cells that (a and b) have been treated with sorafenib (10 μmol L−1) for 0, 12, 24, and 36 h (from bottom to top), respectively and (c and d) different sorafenib concentrations (from bottom to top: 0, 5, 10, and 20 μmol L−1) for 24 h. | ||
Conclusions
In summary, we presented a method for studying low-abundance ALP secreted by individual cells using a microfluidic droplet-based SERRS technique. We first engineered a co-flow drop-maker module in which the suspended cells were mixed with a solution containing BCIP as an enzymatic substrate and AuNPs as a signal enhancement agent in milliseconds to encapsulate individual cells in drops. Then, the differences of cell lines (HepG2 and BNL.CL2) were compared in their ALP expression and it was found that normal liver cells show about 40% lower ALP expression relative to hepatocellular carcinoma (HepG2). In addition, we observed that the ALP activity of the droplet-isolated HepG2 cells fluctuates in a larger range than that of the cluster cells, though the overall ALP expression of these two cases is similar. The drug effects of inhibitors (sodium orthovanadate and sorafenib) on the ALP activity of HepG2 cells can be dynamically traced by this proposed droplet-based SERRS method and show a typical concentration-dependence. The drops are ideal micro-reactors and can be precisely sized to contain one individual cell. The introduction of SERRS into the microdroplets greatly improves the sensitivity of the detection due to surface enhancement resonance effects. From our work, we can sort the cells with abnormal expression of ALP, to realize the early diagnosis of a disease interrelated with ALP. Moreover, we would apply the SERS-droplet platform to investigate the reason behind the abnormal expression of ALP of cells in future, which will be helpful for studying ALP-related diseases. Consequently, we believed that our method of monitoring ALP expression within single cells paves the way for deciphering the heterogeneity and complexity of cell populations in cancer and has potential value for clinical applications. In addition, this microfluidic droplet-based SERRS technique would have potential application value for analysis of low abundance protein expression based on enzymatic catalytic reactions or immunoassays. Therefore, we will use this technique to further investigate of low abundance proteins from the single cell level in the future.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 21573087 and 21573092). We are grateful to Dr. Chongyang Liang, Jilin Univ., for his useful advice about the experiment design.References
- H. M. Shapiro, in Practical Flow Cytometry, John Wiley & Sons, Inc., 2005, pp. 73–100 Search PubMed.
- B. El Debs, R. Utharala, I. V. Balyasnikova, A. D. Griffiths and C. A. Merten, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11570–11575 CrossRef CAS PubMed.
- P. Mary, A. Chen, I. Chen, A. R. Abate and D. A. Weitz, Lab Chip, 2011, 11, 2066–2070 RSC.
- R. Novak, Y. Zeng, J. Shuga, G. Venugopalan, D. A. Fletcher, M. T. Smith and R. A. Mathies, Angew. Chem., Int. Ed., 2011, 50, 390–395 CrossRef CAS PubMed.
- D. Duffy, J. McDonald, O. Schueller and G. Whitesides, Anal. Chem., 1998, 70, 4974–4984 CrossRef CAS.
- G. J. V. Nossal and J. Lederberg, Nature, 1958, 181, 1419–1420 CrossRef CAS.
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS.
- O. J. Miller, A. E. Harrak, T. Mangeat, J. C. Baret, L. Frenz, B. E. Debs, E. Mayot, M. L. Samuels, E. K. Rooney, P. Dieu, M. Galvan, D. R. Link and A. D. Griffiths, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 378–383 CrossRef CAS PubMed.
- D. J. Eastburn, A. Sciambi and A. R. Abate, Anal. Chem., 2013, 85, 8016–8021 CrossRef CAS PubMed.
- M. Chabert, K. D. Dorfman and J. L. Viovy, Electrophoresis, 2005, 26, 3706–3715 CrossRef CAS PubMed.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef CAS PubMed.
- H. N. Joensson, M. Uhlen and H. A. Svahn, Lab Chip, 2011, 11, 1305–1310 RSC.
- J. C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850–1858 RSC.
- H. Song and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 14613–14619 CrossRef CAS.
- M. E. Dolega, S. Jakiela, M. Razew, A. Rakszewska, O. Cybulski and P. Garstecki, Lab Chip, 2012, 12, 4022–4025 RSC.
- Z. Y. Han, W. T. Li, Y. Y. Huang and B. Zheng, Anal. Chem., 2009, 81, 5840–5845 CrossRef CAS PubMed.
- H. Wang, B. B. Chen, M. He and B. Hu, Anal. Chem., 2017, 89, 4931–4938 CrossRef CAS PubMed.
- M. P. Cecchini, J. Hong, C. Lim, J. Choo, T. Albrecht, A. J. deMello and J. B. Edel, Anal. Chem., 2011, 83, 3076–3081 CrossRef CAS.
- E. Chung, R. Gao, J. KO, N. Choi, D. W. Lim, E. K. Lee, S. I. Chang and J. Choo, Lab Chip, 2013, 13, 260–266 RSC.
- M. P. Cecchini, A. Wiener, V. A. Turek, H. Chon, S. Lee, A. P. Ivanov, D. W. McComb, J. Choo, T. Albrecht, S. A. Maier and J. B. Edel, Nano Lett., 2013, 13, 4602–4609 CrossRef CAS.
- S. Laing, L. E. Jamieson, K. Faulds and D. Graham, Nat. Rev. Chem., 2018, 1, 0060 CrossRef.
- C. Zong, M. X. Xu, L. J. Xu, T. Wei, M. Ma, X. S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- G. Kuku, M. Altunbek and M. Culha, Anal. Chem., 2017, 89, 11160–11166 CrossRef CAS PubMed.
- K. R. Strehle, D. Cialla, P. Rosch, T. Henkel, M. Kohler and J. Popp, Anal. Chem., 2007, 79, 1542–1547 CrossRef CAS.
- R. Gao, N. Choi, S. I. Chang, S. H. Kang, J. M. Song, S. I. Cho, D. W. Lim and J. Choo, Anal. Chim. Acta, 2010, 681, 87–91 CrossRef CAS PubMed.
- I. Jahn, O. Žukovskaja, X.-S. Zheng, K. Weber, T. Bocklitz, D. Cialla-May and J. Popp, Analyst, 2017, 142, 1022–1047 RSC.
- C. D. Syme, C. Martino, R. R. Yusyana, N. M. S. Sirimuthu and J. M. Cooper, Anal. Chem., 2012, 84, 1491–1495 CrossRef CAS.
- K. Kantarovich, I. Tsarfati, L. A. Gheber, K. Haupt and I. Bar, Biosens. Bioelectron., 2010, 26, 809–814 CrossRef CAS.
- G. L. Li, H. L. Fu, X. J. Chen, P. W. Gong, G. Chen, L. Xia, H. Wang, J. M. You and Y. N. Wu, Anal. Chem., 2016, 88, 2720–2726 CrossRef CAS PubMed.
- W. H. Fishman, N. R. Inglis, S. Green, C. L. Anstiss, N. K. Gosh, A. E. Reif, R. Rustigia, M. J. Krant and L. L. Stolbach, Nature, 1968, 219, 697–699 CrossRef CAS PubMed.
- P. H. Lange, J. L. Millan, T. Stigbrand, R. L. Vessella, E. Ruoslahti and W. H. Fishman, Cancer Res., 1982, 42, 3244–3247 CAS.
- S. J. Wu, Y. X. Lin, H. Ye, X. Z. Xiong, F. Y. Li and N. S. Cheng, Int. J. Surg., 2016, 36, 143–151 CrossRef.
- C. M. Ruan, W. Wang and B. H. Gu, Anal. Chem., 2006, 78, 3379–3384 CrossRef CAS PubMed.
- G. K. Abou-Alfa, L. Schwartz and S. Ricci, J Clin Oncol, 2006, 2, 4293–4330 CrossRef PubMed.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS.
- S. Köster, F. E. Angilè, H. Duan, J. J. Agresti, A. Wintner, C. Schmitz, A. C. Rowat, C. A. Merten, D. Pisignano, A. D. Griffiths and D. A. Weitz, Lab Chip, 2008, 8, 1110–1115 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- J. R. Lombardi and R. L. Birke, J. Chem. Phys., 2012, 136, 144704 CrossRef PubMed.
- J. R. Lombardi and R. L. Birke, Acc. Chem. Res., 2009, 42, 734–742 CrossRef CAS PubMed.
- T. Karapanayiotis, S. E. J. Villar, R. D. Bowen and H. G. M. Edwards, Analyst, 2004, 129, 613–618 RSC.
- U. Singh, R. H. Quintanilla, S. Grecian, K. R. Gee, M. S. Rao and U. Lakshmipathy, Stem Cell Rev. Rep., 2012, 8, 1021–1029 CrossRef PubMed.
- A. L. Suo, M. X. Zhang, Y. Yao, L. M. Zhang, C. Huang, K. J. Nan and W. G. Zhang, Med. Oncol., 2012, 29, 1827–1836 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data including chemicals and reagents, band assignments, generation and collection of droplets, distributions of BCI and Au NPs in cells, particle size, the probability of a droplet encapsulating a single cell, distributions of BCIP and AuNPs on cells in droplets, confocal fluorescence microscopic images of different cell lines and dynamic expression of ALP in response to sorafenib. See DOI: 10.1039/c8lc01006d |
| This journal is © The Royal Society of Chemistry 2019 |