
An integrated device for the rapid and sensitive detection of the influenza hemagglutinin†

Abstract
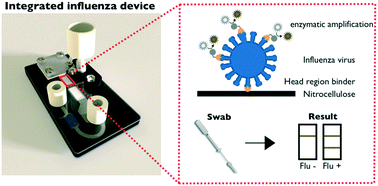
Influenza is a viral respiratory tract infection responsible for up to 5 million cases of severe infection and nearly 600 000 deaths worldwide each year. While treatments for influenza exist, diagnostics for the virus at the point of care are limited in their sensitivity and ability to differentiate between subtypes. We have developed an integrated two-dimensional paper network (2DPN) for the detection of the influenza virus by the surface glycoprotein, hemagglutinin. The hemagglutinin assay was developed using proteins computationally designed to bind with high affinity to the highly-conserved sialic acid binding site. The integrated 2DPN uses a novel geometry that allows automated introduction of an enzymatic amplification reagent directly to the detection zone. This assay was integrated into a prototype device and demonstrated successful detection of clinically relevant virus concentrations spiked into 70 μL of virus-free pediatric nasal swab samples. Using this novel geometry, we found improved assay performance on the device (compared to a manually-operated dipstick method), with a sensitivity of 4.45 × 102 TCID50 per mL on device.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

