
Adipose-on-a-chip: a dynamic microphysiological in vitro model of the human adipose for immune-metabolic analysis in type II diabetes†

Abstract
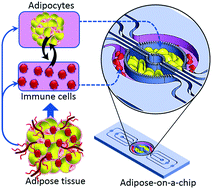
Infiltration of immune cells into adipose tissue is associated with chronic low-grade inflammation in obese individuals. To better understand the crosstalk between immune cells and adipocytes, in vivo-like in vitro models are required. Conventionally transwell culture plates are used for studying the adipocyte–immune cell interaction; however, the static culture nature of this approach falls short of closely recapitulating the physiological environment. Here we present a compartmentalized microfluidic co-culture system which provides a constant-rate of nutrient supply as well as waste removal, resembling the microvascular networks of the in vivo environment. Human adipocytes and U937 cells were co-cultured in close proximity in an enclosed system. The porous barrier between the adjacent compartments comprises an array of microchannels, which enables paracrine interaction between cells in adjacent compartments and improved perfusion-based long term cell feeding. Human pre-adipocytes were fully differentiated into adipocytes on the chip and remained viable for several weeks. Upon co-culturing with immune cells, adipocytes showed a tendency to develop insulin resistance. The immune-metabolic correlation has been studied by monitoring adiponectin and IL-6 expression, as well as glucose uptake upon treatment with insulin. Our microfluidic system can be potentially used to develop physiologically relevant adipose tissue models to study obesity-associated diseases such as insulin resistance and type 2 diabetes and therefore, facilitate drug development to treat these diseases.
- This article is part of the themed collection: Organ-, body- and disease-on-a-chip systems
 Please wait while we load your content...
Please wait while we load your content...

