DOI:
10.1039/C9JA00045C
(Technical Note)
J. Anal. At. Spectrom., 2019,
34, 1279-1286
Heart-cut 2DSEC-RP-LC-ICP-MS as a screening tool in metal-based anticancer research†
Received
1st February 2019
, Accepted 26th March 2019
First published on 26th March 2019
Abstract
In this work, we introduce a new screening tool for metal-based anticancer drugs, enabled by on-line heart-cut two-dimensional liquid chromatography combined with inductively coupled plasma mass spectrometry (2DLC-ICP-MS). This method employs ultrahigh performance size-exclusion chromatography (UHPLC SEC) in the first dimension and reversed phase liquid chromatography (RP-LC) in the second dimension. In this way, the quantification of free and intact metal-based anticancer drugs was addressed in biologically relevant matrices without otherwise necessary sample preparation steps such as protein removal and extraction of the drug. In a proof of principle study the half-life of oxaliplatin upon ex vivo incubation in serum and cellular extracts was assessed by repeated analysis every 25 min over several hours. This method showed good column recoveries for intact oxaliplatin (80% assessed by on-line isotope dilution) and limits of detection <3 nM. Oxaliplatin exhibited a half-life of 1.6 ± 0.1 h (N = 3) in serum. In the cellular extract prepared from the colorectal cancer cell line HCT116 oxaliplatin showed a prolonged half-life of 3.5 ± 0.3 h and 4.2 ± 0.4 h, assuming zero order or first order kinetics, respectively.
Introduction
The discovery of platinum drugs, which are also on the WHO list of “essential medicines”, dates back to 1969.1 Currently, about every second anticancer treatment scheme makes use of one of the three worldwide-approved platinum therapeutics, cisplatin, carboplatin or oxaliplatin. With regard to the mode of action, for a long time it has been assumed that their activity against cancer cells is solely based on DNA platination followed by stalling of replication and cell death. Over the last few years, however, it has become clear that metal-based anticancer drugs exert multifaceted cytotoxic activity,2,3 fueling the development of next generation metal-based anticancer agents. As a consequence, following chemical synthesis of a drug candidate and assessment of preliminary safety and efficacy profiles, serious efforts in analytical chemistry are required before its nomination as a clinical development candidate. Current optimization of lead compounds is accompanied by solution based experiments mimicking intracellular chemistry. Ex vivo studies using drug incubation in serum4 and cell lysates5,6 provided valuable information preceding in vivo and in vitro screening. Combined with mass spectrometric analysis (elemental and molecular mass spectrometry), these types of experiments emerged as a valuable tool supporting preclinical drug development and promoting the basic understanding of drug biomolecule interaction.1,3 For example, ex vivo incubation provided information about distribution of metal-based drugs in blood cells, plasma and ultra-filtrate,7 which is key to determining the pharmacokinetics of the drug, which in turn is key for ascertaining the optimum dosage. The assessment of biotransformation products is another crucial aspect of drug development, as distinctly different cytotoxicity/toxicity profiles arise.2,3 Again, in the past, incubation experiments together with advanced techniques provided useful information prior to clinical studies showing non-enzymatic bio-transformations such as hydrolysis product and/or adduct formation.8,9 The fact that metal-based anticancer drugs can be determined by their metal content following diverse separation or fractionation methods carried out either on-line or off-line proved to be advantageous in many preclinical studies. Powerful methods for the quantification of metallodrug–protein association and biotransformation were developed for different matrices such as human plasma, serum and cell lysates.4,5,10 Recently, a systematic study evaluated different methods for quantitative screening of protein adduct formation. These methods included separation methods/fractionation methods such as centrifugal ultrafiltration, size exclusion and turbulent flow chromatography in combination with ICP-MS.11 However, using these methods precluded accurately differentiating between the free intact drug, small-molecule transformation products and protein adducts. Tailored chromatographic separation for elemental speciation analysis of small molecular transformation products required sample preparation steps (e.g. centrifugal ultrafiltration or extraction) for removing the protein rich fraction of the samples.10 Only in this way was the selectivity offered by the one dimensional separation sufficient for the accurate quantification of the free intact drug versus transformation products. Evidently, the additional sample preparation step was time consuming (centrifugal ultrafiltration required up to 30 min) and prone to error.11 The procedures could not be generalized to a wide panel of metal-based drugs, as co-precipitation, species interconversion during preparation and recovery had to be validated for each compound individually. As a drawback, extraction using polar organic solvents hampered subsequent application of reversed phase chromatographic separation. Either an additional sample preparation step for removing organic solvents was implemented or hydrophilic interaction chromatography (HILIC)7 was employed resulting in poor limits of detection. In both cases, drug stability needed to be considered. In this work, we present a novel elemental speciation strategy for quantifying the free and intact drugs in biological matrices omitting off-line sample preparation. More specifically, we present an on-line combination of two orthogonal separations, i.e. size exclusion chromatography (SEC) and reversed phase liquid chromatography (RP-LC). In the past, two dimensional separation involving size exclusion addressed heart-cut SEC-ion chromatography SEC-(IC) and comprehensive 2DLC as established by fast column switching SEC-IC (monoliths).12–14 Both set-ups were dedicated to the determination of metallodrug–protein interaction and operated at low backpressures (a requirement for the implemented SEC). This pressure limitation of size exclusion columns was overcome with the introduction of UHPLC SEC, facilitating the combination with reversed phase separations as established in this work. The presented approach allowed separating the low molar mass fraction (LMF) eluted from the size exclusion in the second chromatographic dimension. In this way, kinetic studies of metal-based drug degradation in biologically relevant matrices are significantly improved. In a proof of principle study the half-life of oxaliplatin was investigated in plasma and cell extracts.
Experimental
Reagents, chemicals and standards
A Pt standard (1002 ± 6 μg mL−1) was purchased from Inorganic Ventures, Christiansburg, Virginia, USA. A platinum spike enriched in 196Pt (abundance ∼97.25%, Science Technical Centre “Stable Isotopes” of State Scientific Centre of the Russian Federation–Institute of Physics and Power Engineering, Obninsk, Kaluga Region, Russia)15 was kindly provided by professor Stephan Hann at the Department of Chemistry, Division of Analytical Chemistry, University of Natural Resources and Life Sciences – BOKU, Vienna, Austria, and it was employed for species-unspecific quantification by on-line isotope dilution. Glucose and ammonium acetate were purchased from Sigma Aldrich (St. Louis, MO, USA). LC-MS grade water and MeOH were purchased from Honeywell (Muskegon, MI, USA). Oxaliplatin and fetal calf serum (FCS) were kindly provided by professor Christian R. Kowol at the Department of Inorganic Chemistry, University of Vienna, Vienna, Austria. The media and the reagents for cell culture were purchased from Sigma-Aldrich Austria and all the material such as dishes, plates and flasks were from StarLab, Germany unless indicated otherwise.
Cancer cell culture
Cells were grown as adherent monolayer cultures in 160 cm2 culture flasks (Iwaki, Japan) in McCoy's medium supplemented with 10% heat-inactivated fetal bovine serum (Biowest, USA) and 4 mM L-glutamine (Sigma-Aldrich, Austria) without antibiotics at 37 °C under a humidified atmosphere containing 5% CO2 and 95% air. Cells from the human colon cancer line, HCT116, acquired from ATCC (American Type Culture Collection, USA) were harvested from the culture flasks by trypsinization. The trypsinization was stopped by addition of fresh medium and the cell suspension was washed three-times by centrifugation (300 rcf, 3 min) and addition of PBS. Subsequently, the cell number in the suspension was determined and 30 × 106 cell aliquots were distributed in 5 mL Eppendorf tubes. Following this, the aliquots were centrifuged twice and the excess PBS was aspirated. Finally, the cell pellets were snap-frozen with liquid N2 and stored at −80 °C until analysis.
Ex vivo incubation, cell extraction and cytosolic incubation
Oxaliplatin was weighed and dissolved in glucose 5% (w/v) to reach a concentration of 1 mM. This stock solution was kept in ice and it was further diluted in glucose 5% (w/v) to reach a concentration of 10 μM and finally diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 in FCS (molar ratio of drug:albumin of ∼1:600, ∼8 times higher compared to clinical infusions16) to reach a concentration of 1 μM and incubated in the autosampler of a HPLC system at 37 °C and injected every 0.83 h for 2.5 h.
:10 in FCS (molar ratio of drug:albumin of ∼1:600, ∼8 times higher compared to clinical infusions16) to reach a concentration of 1 μM and incubated in the autosampler of a HPLC system at 37 °C and injected every 0.83 h for 2.5 h.
A lysis procedure using freeze/thaw cycles with NaCl as described elsewhere was implemented.10 Briefly, 100 μL of 15 mM NaCl was added in a 5 mL Eppendorf tube containing 30 × 106 cells of the human colon cancer line HCT116. The Eppendorf tube was frozen with liquid N2 and thawed at room temperature. This procedure was repeated two more times but in the last repetition 90 μL of 15 mM NaCl was added. Then it was centrifuged (Hermle Z446 K, 4350 rcf, 15 min, 4 °C) and the supernatant (cytosolic fraction) was transferred to a 1.5 mL Eppendorf tube whereas the pellet (nuclei fraction) was discarded. Since the 5 mL Eppendorf tube can be centrifuged only at a maximum speed of 4350 rcf in the Hermle Z446 K centrifuge, the 1.5 mL Eppendorf tube containing the cytosolic fraction was centrifuged again at a higher speed (Hermle Z366, 21380 rcf, 60 min, 4 °C) in order to separate the rest of the pellet (nuclei fraction) that could not be separated in the first centrifugation, and 290 μL of the supernatant (cytosolic fraction) was finally transferred to a HPLC vial (final biomolecule dilution 1:100). 10 μL of a fresh stock solution of oxaliplatin in glucose 5% (w/v) was added in the vial containing the cytosolic fraction to reach a final concentration of 2 μM. This oxaliplatin concentration was calculated assuming a cell volume of 10−12 L and an intracellular concentration corresponding to typical oxaliplatin incubation in a cell culture medium of 20 μM as explained elsewhere.10 The vial containing the oxaliplatin in the cytosolic extract was placed in the autosampler of a HPLC at 37 °C to inject it at different times. After the analysis, an aliquot of 50 μL was used for protein quantification with a BCA protein assay kit obtaining a concentration of 35.2 ± 0.7 mg protein per mL (corresponding to a molar ratio of ∼1:300 drug:biomolecule based on albumin molarity).
Instrumentation
ICP-MS.
An Agilent 8800 ICP-MS/MS (Agilent Technologies, Tokyo, Japan) was used with oxygen as the reaction gas for the monitoring of proteins via S32O16 (for efficient control of protein removal); data not shown. The Agilent MassHunter software package (Workstation Software, Version C.01.03, 2016) was employed for data treatment. The ICP-MS parameters are summarized in Table 1.
Table 1 ICP-MS operation parameters
| Nebulizer |
MicroMist |
| Spray chamber |
Scott double-pass |
| Nebulizer gas flow |
1.05 L min−1 |
| Aux. gas |
0.90 L min−1 |
| Plasma gas |
15 L min−1 |
| ICP RF power |
1550 W |
|
m/z measured |
195, 196, 48 |
| Reaction gas |
0.3 L min−1 |
Heart-cut UHPLC SEC-RP.
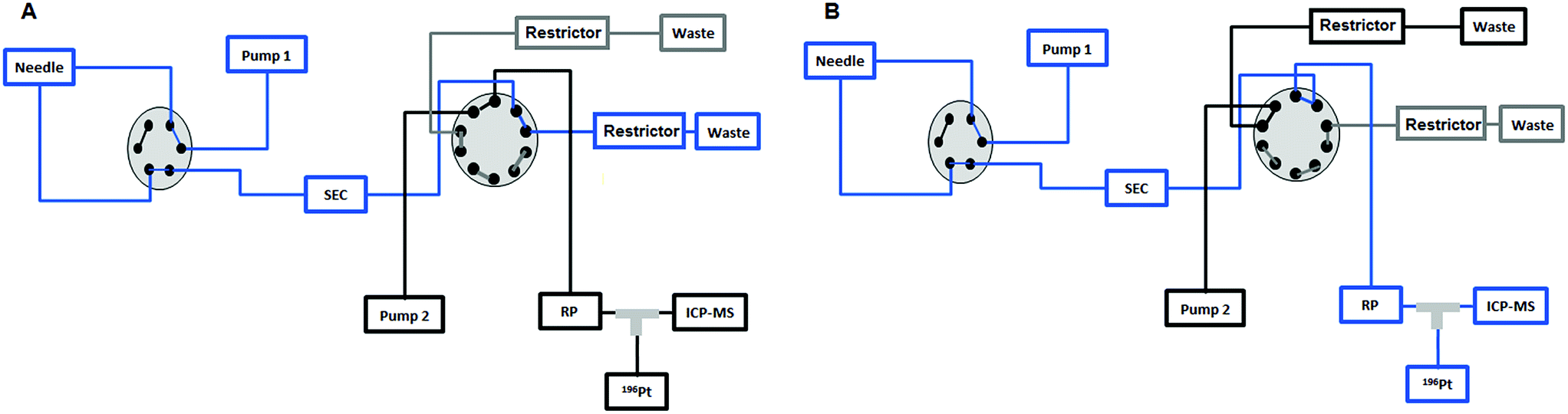
An Agilent 1260 Infinity Bio-Inert HPLC system (Agilent Technologies, Waldbronn, Germany) was employed for this setup. The sample was injected into a 6-port valve of the HPLC and sent to a 10-port valve (Agilent Technologies, Waldbronn, Germany) where the removal of the HMF was carried out by means of a UHPLC SEC column in the first 7.5 min and the LMF was sent to a RP-ICP-MS system by switching this 10-port valve after the 7.5 min. The chromatographic conditions and the method are shown in Table 2. The instrumental setup for the heart-cut UHPLC SEC followed by RP-ICP-MS is shown and explained in detail in Fig. 1.
Table 2 Chromatographic conditions for the heart-cut UHPLC SEC-RP method
| Chromatographic conditions |
|
See Fig. 1A and B for the position of the 10-port valve.
|
| SEC column |
Acquity UPLC Protein BEH SEC, 4.6 × 150 mm, 200 Å |
| 1.7 μm, 10–500 kDa, Waters |
| RP column |
Atlantis T3, 2.1 × 150 mm, 3 μm, Waters |
| Eluent pump 1 |
A 20 mM CH3COONH4, pH = 7 |
| Flow pump 1 |
250 μL min−1 |
| Eluent pump 2 |
A 20 mM CH3COONH4, pH = 7 |
| B 20 mM CH3COONH4, pH = 7, 80% MeOH (v/v) |
| Flow pump 2 |
250 μL min−1 |
| Injection volume |
2 μL |
| Temperature SEC |
37 °C |
| Temperature RP |
37 °C |
| Autosampler temperature |
37 °C |
| Chromatographic method |
| Time (min) |
Position 10-port valvea |
Pump 1 |
Pump 2 |
| A (%) |
A (%) |
B (%) |
| 0 |
A |
100 |
100 |
0 |
| 7.5 |
B |
100 |
100 |
0 |
| 11 |
A |
100 |
100 |
0 |
| 17.5 |
A |
100 |
80 |
20 |
| 21 |
A |
100 |
80 |
20 |
| 21.1 |
A |
100 |
100 |
0 |
| 25 |
A |
100 |
100 |
0 |
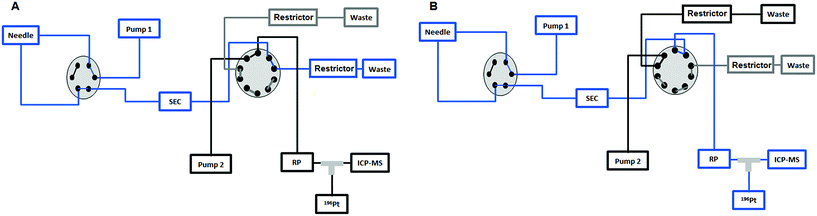 |
| | Fig. 1 (A) The sample is injected by means of the first pump from the 6-port valve of the Agilent 1260 Infinity Bio-Inert HPLC system to the UHPLC SEC column and to the external 10-port valve to send the high molar fraction (HMF) to the waste in the first 7.5 min. (B) After 7.5 min the 10-port valve is switched to send the low molar fraction (LMF) eluted from the UHPLC SEC column to the RP column. Then the valve is switched again at 11 min to position (A) and the second pump is used to separate the different species present in the LMF on the RP column. | |
Post-column isotope dilution.
The peristaltic pump of the ICP-MS instrument was employed with a mass flow of 98 mg min−1 to introduce a platinum spike enriched in 196Pt (abundance 97.25%) (0.82 μg Pt per L) after the RP column (Fig. 1) for species-unspecific quantification of oxaliplatin by on-line isotope dilution in order to calculate accurately the recovery by this heart-cut 2D-LC approach.
Results and discussion
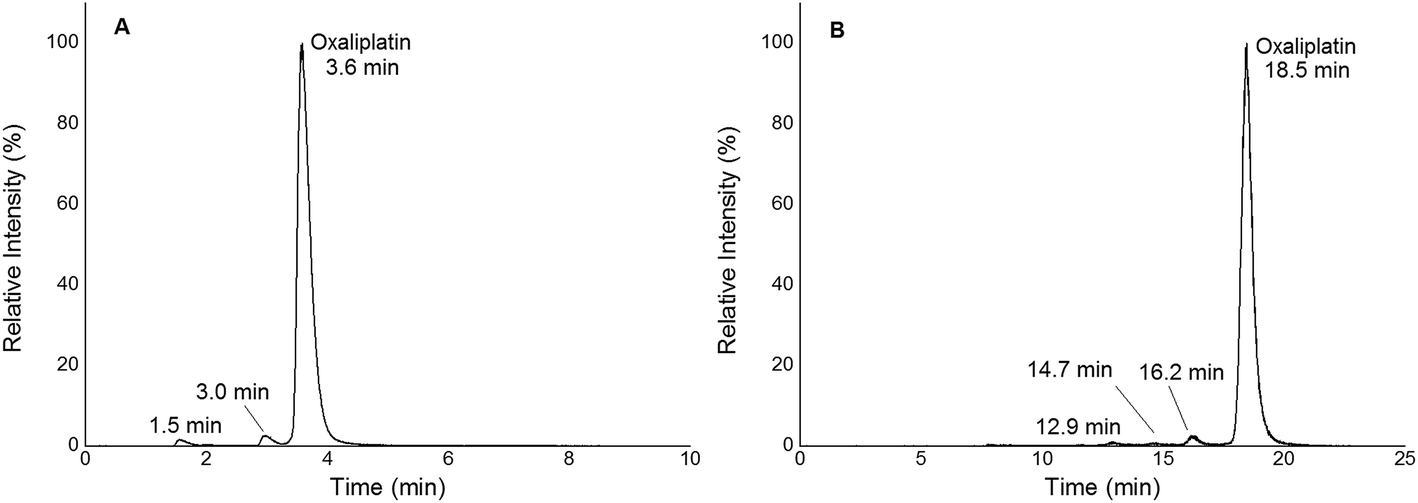
In this work, quantification of intact and free metal-based anticancer drugs in protein rich samples was addressed. More specifically, a heart-cut 2D-LC approach enabling combination with ICP-MS without off-line protein removal was validated for oxaliplatin. When analysing (pre)clinically relevant samples, 1D separation combined with ICP-MS detection required dedicated sample preparation. The 2DLC-ICP-MS method provided on-line separation of proteins by SEC. The SEC separation time could be reduced to a few minutes as a sub-2 μm particle UHPLC SEC stationary phase was implemented.17,18 The size range of the column was 500 kDa to 10 kDa, (exclusion limit 500 kDa) resulting in the complete elution of the high molar mass fraction (HMF) within 6 min when employing moderate flow rates of 250 μL min−1. Under these conditions oxaliplatin was eluted in the low molar mass fraction (LMF) within 9 min with a LOD of 2.7 nM (3σ criterion) and a peak width of 1.7 min (see ESI Fig. S1†). The separation range of size exclusion was selected in a way such that the LMF containing intact drugs and potential transformation products (e.g. hydrolysis products) were eluted within a short time. With the use of a post column 10-port valve, a heart cut 2D separation was established separating the LMF by RP-LC (see Fig. 1). The reversed phase separation was carried out using either a fully wettable C18 material or pentafluorophenyl phases allowing stacked injections (high volume) of metal-based drugs showing medium to high retention on the reversed phase.19,20 Due to the use of quaternary pumps, 3 μm-particle RP-LC stationary phases were selected, as imposed by the pressure limit of the pump. Reversed phase separations using polar embedded groups or other functionalized C18 material had been shown in the past to be suitable for the selective separation of oxaliplatin and its hydrolysis products.19,20 Using 1D RP-LC-ICP-MS or 2D UHPLC SEC-RP-ICP-MS for the analysis of intact oxaliplatin in 15 mM NaCl excellent separation of oxaliplatin from hydrolysis products could be observed (Fig. 2). After 2 h incubation time at 37 °C, >80% was still intact. The observed two-phase decay is in accordance with that in other publications.21
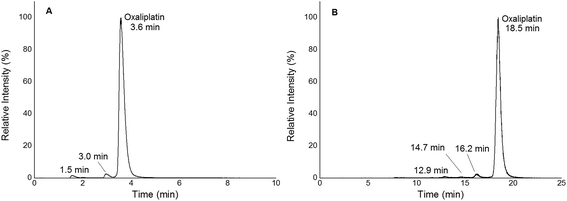 |
| | Fig. 2 (A) 1D RP-LC separation (column Accucore PFP, 2.1 × 150 mm, 2.6 μm) of 2 μM oxaliplatin in 15 mM NaCl incubated for 2 h at 37 °C; gradient adapted from elsewhere.19 The hydrolysis products were observed at 1.5 and 3.0 min. Oxaliplatin was observed at 3.6 min. (B) 2D SEC-RP-LC separation (Acquity UPLC Protein BEH SEC, 4.6 × 150 mm, 200 Å; Atlantis T3, 2.1 × 150 mm, 3 μm) of 1 μM oxaliplatin in 15 mM NaCl incubated for 2 h at 37 °C. The hydrolysis products were observed at 12.9, 14.7 and 16.2 min. Oxaliplatin was observed at 18.5 min. | |
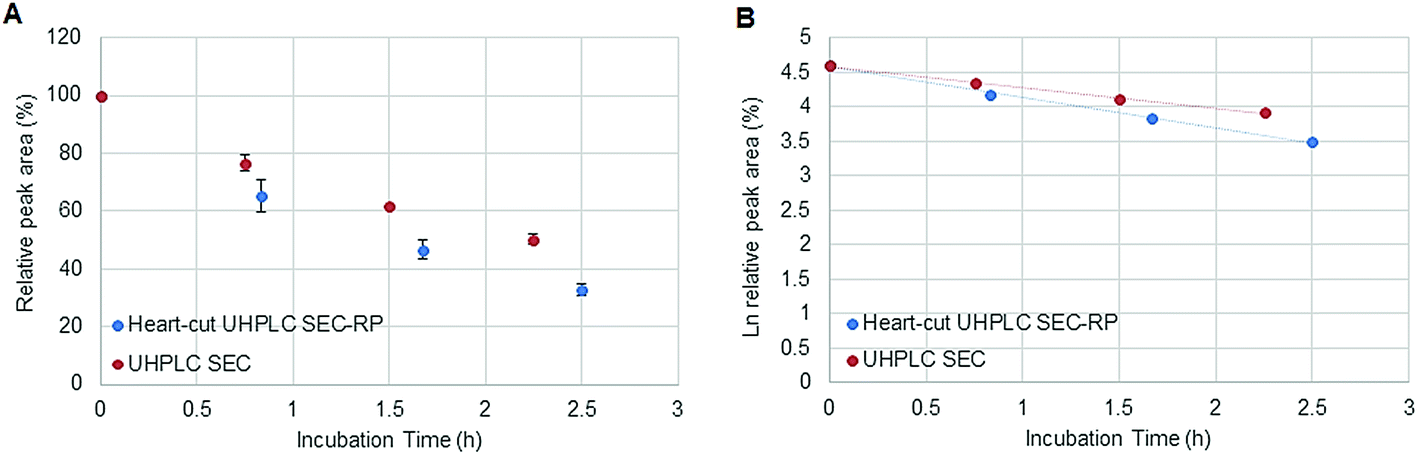
Fig. 1 depicts the novel heart-cut 2D-LC separation. At 7.5 min the installed switching-valve directed the SEC flow for 3.5 min towards the second dimension, where the intact metal-based drug was retained. Backpressures of 350 bar were observed when both columns were connected in series. Restrictors established a constant backpressure upon valve switching. For this purpose a HPLC column (Atlantis T3, 2.1 × 150 mm, 3 μm, Waters) was installed in the flow path of pump 2 whereas for pump 1 a narrow bore capillary (inner diameter 0.127 mm) was introduced (Fig. 1). Table 3 summarizes the key analytical figures of merit for intact oxaliplatin quantification obtained using the novel 2DLC-ICP-MS set-up. As can be readily observed, oxaliplatin recovery and measurement repeatability was fit for purpose despite the high injection volume. The recovery was assessed by on-line isotope dilution. The limits of detection (2.5 nM, 3σ criterion) were comparable to those for RP-LC-ICP-MS analysis only (1.3 nM, 3σ criterion) and were significantly improved as compared to typical HILIC-ICP-MS limits of detection (13 nM, 3σ criterion; see ESI Table S1 and Fig. S2†). Oxaliplatin was quantified by on-line isotope dilution compensating for signal suppression with RP-LC gradients. By using the 2D-LC set-up excellent separation of the low molar mass biotransformation products could be obtained (Fig. 3). For a proof of principle study, ex vivo incubations of oxaliplatin in fetal calf serum were investigated by 2DLC-ICP-MS. In this model, the drug concentration of 1 μM resembled drug concentrations found in the plasma of chemotherapy patients.16 The decay of intact oxaliplatin was monitored over 2.5 hours. For the investigated period the observed decay resembled first order kinetics (linear regression for first order R2 = 0.9970). A half-life of 1.6 ± 0.1 h was calculated for three independent incubation experiments. Comparative experiments by using UHPLC SEC only (Fig. 4) revealed a half-life of 2.3 ± 0.1 h (N = 3) (Fig. 5). This higher value can be attributed to the poor chromatographic selectivity of UHPLC SEC regarding the separation of biotransformation products in the LMF. The obtained results were in good agreement with those in previously reported studies based on ultrafiltration and metal quantification. Using comparable drug concentrations, a half-life of 2.21 ± 0.72 h was reported for oxaliplatin in human plasma.22 In clinical samples of patients undergoing chemotherapy even faster clearance was observed due to the high degree of binding to erythrocytes.16
Table 3 2D SEC-RP-ICP-MS analytical figures of merit assessed for intact 1 μM oxaliplatin in glucose 5% (w/v) and in FCS for the first injection (incubation time 0 h). The recovery was assessed by on-line isotope dilution
| Matrix |
Recovery (%) n = 3 |
RSD retention time (%) n = 3 |
LOD (nM) 3σ criterion |
LOD (μg L−1 Pt) 3σ criterion |
| Glucose |
80 ± 1 |
0.49 |
2.53 |
0.58 |
| FCS |
80 ± 2 |
0.50 |
2.19 |
0.50 |
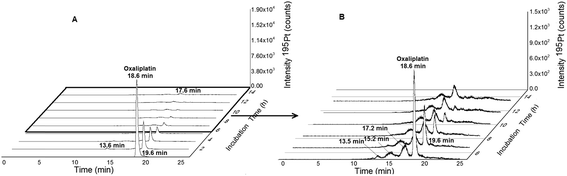 |
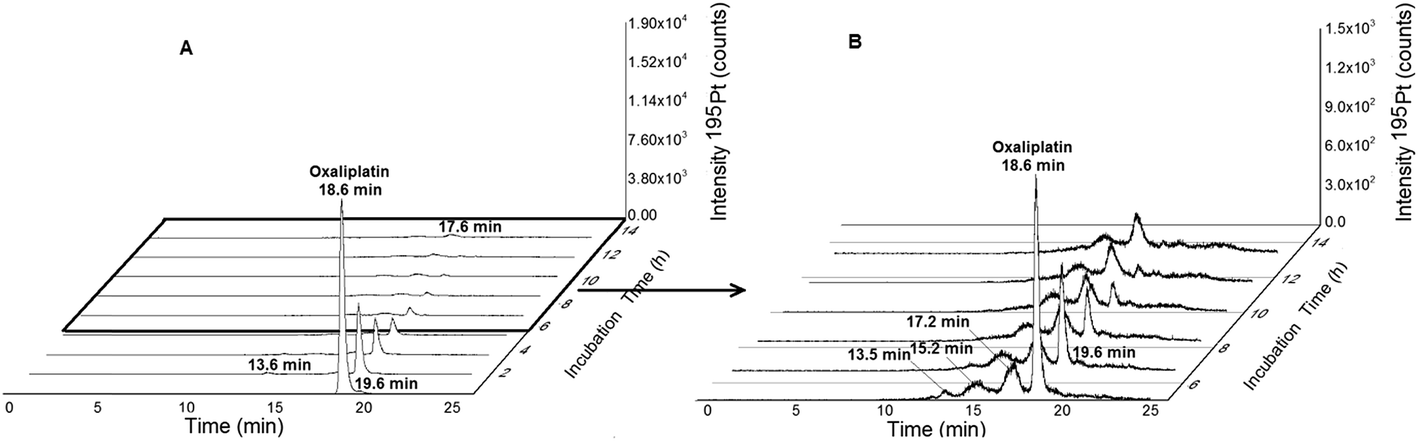
| | Fig. 3 (A) 2DLC-ICP-MS separation (columns Acquity UPLC Protein BEH SEC, 4.6 × 150 mm, 200 Å and Atlantis T3, 2.1 × 150 mm, 3 μm, Waters at a flow rate of 250 μL min−1) of oxaliplatin (10 μM previously dissolved in glucose 5% (w/v) and finally diluted 1:10 in FCS to a final concentration 1 μM) incubated in serum measured at 37 °C for 14 h. (B) Incubation time 5–14 hours. | |
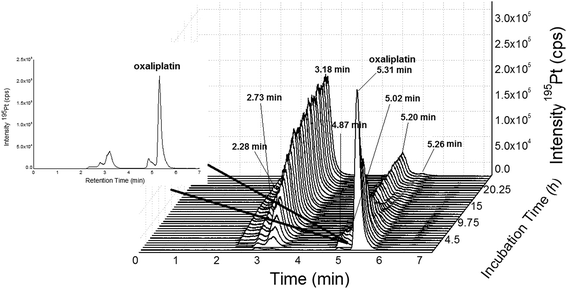 |
| | Fig. 4 SEC-ICP-MS of oxaliplatin (Acquity UPLC Protein BEH SEC, 4.6 × 150 mm, 200 Å, 1.7 μm, 10–500 kDa) at a flow rate of 400 μL min−1; 50 mM CH3COONH4, pH = 6.8 at a temperature of 37 °C; 10 μM oxaliplatin diluted 1:10 in FCS (final concentration 1 μM) and incubated for 24 h at 37 °C. Oxaliplatin shows binding to albumin (3.18, 2.73 and 2.28 min). The free drug could be observed at 5.31 min. The other peaks (5.20, 5.02, 4.87 and 6.60 min) could be due to binding to other biomolecules in the LMF or degradation products of oxaliplatin. The peak at 5.20 min could not be completely separated from the peak of oxaliplatin at 5.31 min. | |
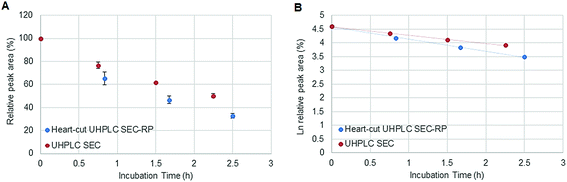 |
| | Fig. 5 (A) Stability of oxaliplatin in FCS assessed using the 2D-setup compared to UHPLC SEC in N = 3 biological replicates and (B) the logarithmic plot of oxaliplatin relative area versus incubation time. Linear regression analysis of the data plot showed that for the investigated period the decay showed first order kinetics. A half-life of 2.3 ± 0.1 and 1.6 ± 0.1 h was found for UHPLC SEC and heart-cut UHPLC SEC-RP, respectively. | |
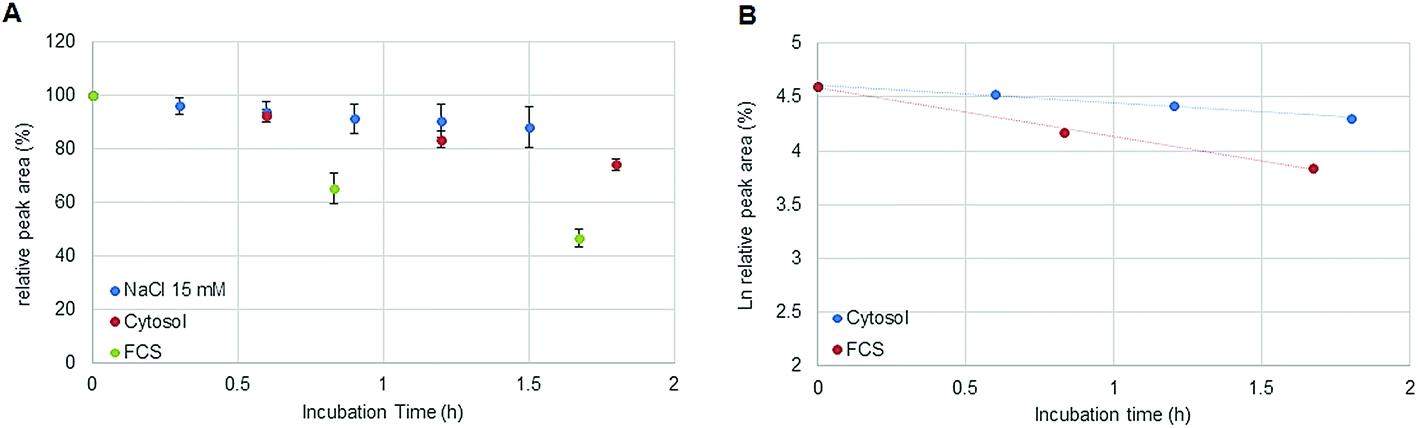
In the field of metal-based anticancer drugs, only a few studies addressing elemental speciation analysis in cancer cell models exist.5,6,10 As the metal concentrations in preclinically relevant settings are in the low sub-nM range, these experiments were hampered by the moderate limits of detection offered by elemental speciation approaches (especially in the case of HILIC-ICP-MS23). Accordingly, simplified model experiments such as drugs exposed to cell lysates contributed to the understanding of intracellular drug chemistry. In this study, the novel 2DLC-ICP-MS method was employed to address the kinetics of oxaliplatin degradation in the cytosolic preparation of a colon cancer model. 2 μM oxaliplatin was incubated with a cell lysate resulting in an estimated molar drug/biomolecule ratio of 1:300. Assuming a volume of 1 pL for a single cell, the cytosol of 30 × 106 cells was diluted by a factor <100 in 15 mM NaCl. The estimated ratio was calculated considering the protein concentration (expressed as molar albumin concentration) of the cell preparation. The decay of oxaliplatin was monitored in 15 mM NaCl in parallel to the cellular preparation over 1.8 hours. As a drawback, in some cases, precipitation was observed in the highly concentrated cell extract after several hours at 37 °C limiting the investigation time to 2 hours. Oxaliplatin is known to be activated by forming hydrolysis products in 15 mM NaCl.21 The experiments in this work show that the oxaliplatin transformation in cells is governed by interaction partners such as proteins and other biomolecules and not only by the reduced intracellular NaCl concentration. Due to the limited investigation period, no unambiguous kinetic model could be found. Assuming zero (Fig. 6A) and first (Fig. 6B) order kinetics (R2 = 0.9977 and R2 = 0.9922 for zero and first order kinetics, respectively) a half-life of 3.5 ± 0.3 h and 4.2 ± 0.4 h was estimated, respectively. For the oxaliplatin in NaCl stabilities >80% in 2 h of incubation at 37 °C and two-phase decay were observed as shown in other publications.21 The difference in half-life between serum and cell incubations can be explained by protein interactions in the protein rich fetal calf serum. In fact, UHPLC SEC-ICP-MS measurements of both sample matrices exposed to >24 hours revealed 90% of Pt in the HMF versus 85% Pt in the LMF, for serum incubation and cell incubation, respectively.
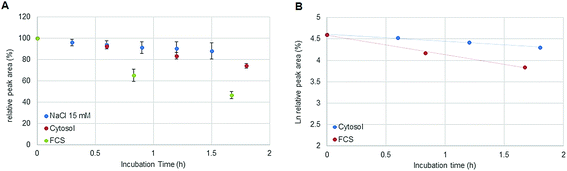 |
| | Fig. 6 (A) Stability of oxaliplatin in 15 mM NaCl assessed by RP-ICP-MS with the column Accucore PFP, 2.1 × 150 mm, 2.6 μm, Thermo Scientific and stability of oxaliplatin in the cytosolic extract in N = 3 biological replicates assessed using the 2D-setup employing the column Accucore PFP, 2.1 × 150 mm, 2.6 μm, Thermo Scientific compared to the previous investigated stability in FCS. The decay observed in the cytosol was different from the decay observed in 15 mM NaCl and in FCS showing a linear regression for zero order kinetics with a half-life of 3.5 ± 0.3 h although a (B) logarithmic plot of oxaliplatin relative area versus incubation time was also observed to be linear for the investigated period for first order kinetics, as it was observed for the incubation in FCS, obtaining a half-life of 4.2 ± 0.4 h. | |
Conclusions
Clinically established metal-based anticancer drugs either in their intact form or as activated transformation products bind to erythrocytes, plasma proteins, amino acids and nucleic acids. The extent of these key interactions ultimately determines the cytotoxic active fraction of the drug reaching the target DNA. In the case of candidate metal-based anticancer drugs, DNA is not always the final target. Despite this, biotransformation and interaction with cellular building blocks need to be carefully evaluated. Accordingly, the drug half-life in different (pre)clinically relevant matrices is an important parameter, which can be approximated by ex vivo incubation experiments. As a prerequisite, the intact and free drug has to be accurately quantified in biological complex matrices following different time points of drug exposure. In this work, we introduce a method enabling the quantification of the metal-based drug in serum samples and cell extracts without the need for a priori sample preparation. Samples are exposed to drugs at a fixed temperature (37 °C autosampler) and injected at defined time intervals on a heart-cut 2DLC-ICP-MS system. In a proof of principle study, the kinetics of oxaliplatin degradation in serum and cancer cell lysates was investigated. However, this approach is applicable to any metallodrug showing a significant retention on reversed phase chromatography.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge Professor Stephan Hann at the Department of Chemistry, Division of Analytical Chemistry, University of Natural Resources and Life Sciences – BOKU, Vienna, Austria for providing the enriched 196Pt for post-column species-unspecific isotope dilution and Professor Christian R. Kowol at the Department of Inorganic Chemistry, University of Vienna, Vienna, Austria for providing oxaliplatin and FCS.
References
- B. Michalke, Platinum speciation used for elucidating activation or inhibition of Pt-containing anti-cancer drugs, J. Trace Elem. Med. Biol., 2010, 24, 69–77 CrossRef CAS PubMed.
- D. Gibson, Metal Anticancer Compounds processing The mechanism of action of platinum anticancer agents—what do we really know about it?, Dalton Trans., 2009, 48, 10681–10689 RSC.
- M. Groessl and C. G. Hartinger, Anticancer metallodrug research analytically painting the “omics” picture--current developments and future trends, Anal. Bioanal. Chem., 2013, 405, 1791–1808 CrossRef CAS PubMed.
- A. R. Timerbaev, Recent progress of ICP-MS in the development of metal-based drugs and diagnostic agents, J. Anal. At. Spectrom., 2014, 29, 1058–1072 RSC.
- T. Falta, P. Heffeter, A. Mohamed, W. Berger, S. Hann and G. Koellensperger, Quantitative determination of intact free cisplatin in cell models by LC-ICP-MS, J. Anal. At. Spectrom., 2011, 26, 109–115 RSC.
- Y. Kasherman, S. Sturup and D. Gibson, Is glutathione the major cellular target of cisplatin? A study of the interactions of cisplatin with cancer cell extracts, J. Med. Chem., 2009, 52, 4319–4328 CrossRef CAS PubMed.
- T. Falta, G. Koellensperger, A. Standler, W. Buchberger, R. M. Mader and S. Hann, Quantification of cisplatin, carboplatin and oxaliplatin in spiked human plasma samples by ICP-SFMS and hydrophilic interaction liquid chromatography (HILIC) combined with ICP-MS detection, J. Anal. At. Spectrom., 2009, 24, 1336–1342 RSC.
- E. Michelucci, G. Pieraccini, G. Moneti, C. Gabbiani, A. Pratesi and L. Messon, Mass spectrometry and metallomics: A general protocol to assess stability of metallodrug–protein adducts in bottom-up MS experiments, Talanta, 2017, 167, 30–38 CrossRef CAS PubMed.
- D. Esteban-Fernández, M. M. Gómez-Gómez, M. W. Linscheid, E. Moreno-Gordaliza, M. L. Mena and I. Moraleja, A shotgun approach for the identification of platinum–protein complexes, Anal. Bioanal. Chem., 2015, 407, 2393–2403 CrossRef PubMed.
- G. Hermann, P. Heffeter, T. Falta, W. Berger, S. Hann and G. Koellensperger, In vitro studies on cisplatin focusing on kinetic aspects of intracellular chemistry by LC-ICP-MS, Metallomics, 2013, 5, 636–647 RSC.
- L. Galvez, S. Theiner, M. Grabarics, C. R. Kowol, B. K. Keppler and S. Hann, Critical assessment of different methods for quantitative measurement of metallodrug–protein associations, Anal. Bioanal. Chem., 2018, 410, 7211–7220 CrossRef CAS PubMed.
- M. Sulyok, S. Hann, C. G. Hartinger, B. K. Keppler, G. Stingeder and G. Koellensperger, Two dimensional separation schemes for investigation of the interaction of an anticancer ruthenium(III) compound with plasma proteins, J. Anal. At. Spectrom., 2005, 20, 856–863 RSC.
- S. Hann, T. Falta, K. Boeck, M. Sulyok and G. Koellensperger, On-line fast column switching SEC × IC separation combined with ICP-MS detection for mapping metallodrug–biomolecule interaction, J. Anal. At. Spectrom., 2010, 25, 861–866 RSC.
- A. Martinčič, M. Cemazar, G. Sersa, V. Kovač, R. Milačič and J. Ščančar, A novel method for speciation of Pt in human serum incubated with cisplatin, oxaliplatin and carboplatin by conjoint liquid chromatography on monolithic disks with UV and ICP-MS detection, Talanta, 2013, 116, 141–148 CrossRef PubMed.
- Z. Stefánka, S. Hann, G. Koellensperger and G. Stingeder, Investigation of the reaction of cisplatin with methionine in aqueous media using HPLC-ICP-DRCMS, J. Anal. At. Spectrom., 2004, 19, 894–898 RSC.
- M. A. Graham, G. F. Lockwood, D. Greenslade, S. Brienza, M. Bayssas and E. Gamelin, Clinical Pharmacokinetics of Oxaliplatin: A Critical Review, Clin. Cancer Res., 2000, 6, 1205–1218 CAS.
-
Ultra Performance Liquid Chromatography, UPLC: New Boundaries for the Chromatography Laboratory, Waters, 2004 Search PubMed.
- M. E. Swartz, UPLC: An Introduction and Review, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 1253–1263 CrossRef CAS.
- S. Hann, Z. Stefánka, K. Lenz and G. Stingeder, Novel separation method for highly sensitive speciation of cancerostatic platinum compounds by HPLC-ICP-MS, Anal. Bioanal. Chem., 2005, 381, 405–412 CrossRef CAS PubMed.
- S. Theiner, M. Grabarics, L. Galvez, H. P. Varbanov, N. S. Sommerfeld, M. Galanski, B. K. Keppler and G. Koellensperger, The impact of whole human blood on the kinetic inertness of platinum(IV) prodrugs-an HPLC-ICP-MS study, Dalton Trans., 2018, 47, 5252–5258 RSC.
- E. Jerremalm, M. Hedeland, I. Wallin, U. Bondesson and H. Ehrsson, Oxaliplatin degradation in the presence of chloride: Identification and cytotoxicity of the monochloro monooxalato complex, Pharm. Res., 2004, 21, 891–894 CrossRef CAS.
- W. Kern, J. Braess, B. Böttger, C. C. Kaufmann, W. Hiddemann and E. Schleyer, Oxaliplatin pharmacokinetics during a four-hour infusion, Clin. Cancer Res., 1999, 5, 761–765 CAS.
-
D. Pröfrock, Coupling Techniques and Orthogonal Combination of Mass Spectrometric Techniques. Biomedical and pharmaceutical applications, in Metallomics: analytical techniques and speciation methods, ed. B. Michaelke, Wiley-VCH, 2016, pp. 17–67 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ja00045c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Mate
Rusz
b,
Michael A.
Jakupec
a,
Mate
Rusz
b,
Michael A.
Jakupec