
Feasibility of in situ trapping of selenium hydride in a DBD atomizer for ultrasensitive Se determination by atomic absorption spectrometry studied with a 75Se radioactive indicator
Jan
Kratzer
 *,
Stanislav
Musil
and
Jiří
Dědina
*,
Stanislav
Musil
and
Jiří
Dědina
Czech Academy of Sciences, Institute of Analytical Chemistry, Veveří 97, CZ-602 00 Brno, Czech Republic. E-mail: jkratzer@biomed.cas.cz
First published on 16th November 2018
Abstract
Preconcentration of selenium hydride in a planar dielectric barrier discharge (DBD) atomizer was optimized with detection by atomic absorption spectrometry. Addition of 3.5 mL min−1 O2 to the Ar plasma discharge resulted in the retention of selenium hydride in the optical arm of the DBD atomizer. Analyte release and subsequent atomization were achieved as soon as oxygen was switched off. Other parameters such as the discharge gas flow rate (75 mL min−1 Ar) and the discharge voltage (17 kV, corresponding power 14 W) were kept constant during the whole procedure. The use of a 75Se radiotracer combined with radiometry and autoradiography allowed proving efficient analyte trapping (>90%) in a small spot in the central part of the DBD optical arm whereas incomplete analyte release was found responsible for a preconcentration efficiency of 70%. The preconcentration method developed can be applied to real samples as demonstrated by accurate analysis of a GRUMO-K certified reference material achieving a detection limit of 12 pg mL−1 Se for a 300 s preconcentration period (sample volume 20 mL).
1 Introduction
Selenium and its compounds are essential for life, but its physiological function is ambivalent. Its toxic levels are less than an order of magnitude above those required for health.1 As a consequence, reliable and sensitive analytical methods for trace and ultratrace selenium determination in environmental, clinical or food samples are needed.Hydride generation (HG)2,3 is an expedient approach to efficient Se introduction into a spectrometric detector. High analyte introduction efficiency and matrix separation are achieved owing to the principle of HG. Chemical HG based on analyte reduction by NaBH4 to the corresponding hydride is the most common and effective approach to HG. Atomic absorption spectrometry (AAS) fulfils the criteria of a reliable and cheap detector to be coupled with HG. Externally heated quartz tube atomizers (QTAs) are the dominant atomizers employed in HG-AAS.2 However, dielectric barrier discharge (DBD) atomizers have been reported recently to be a good alternative to QTAs with AAS detection.4,5 Our recent study6 focusing on the comparison of analytical performance of QTA and DBD atomizers for Se determination by HG-AAS revealed that DBD atomizers can very well compete with QTA in terms of the detection limit (LOD) and sensitivity. Sensitivity and the LOD in QTA were 0.53 s ng−1 Se and 0.15 ng mL−1 Se, respectively, whereas only slightly worse sensitivity and LOD values of 0.32 s ng−1 Se and 0.24 ng mL−1 Se, respectively, have been achieved in DBD atomizers.6
However, a preconcentration step must be included to achieve a sufficiently low detection limit with HG-AAS for ultratrace determination of Se and other hydride forming elements. Preconcentration might take place in a special device (trap) placed downstream of the hydride generator and upstream of the atomizer or can be performed directly in the atomizer.
Hydride preconcentration has been tested employing graphite, quartz or metal surfaces as reviewed recently.7,8 Preconcentration in the atomizer is more advantageous keeping the distance between the preconcentration and detection areas as short as possible. In situ trapping in a graphite furnace is an example of in-atomizer preconcentration taking place directly in the optical axis of the spectrometer. It is the most common approach to hydride preconcentration with AAS detection.9 Analyte hydrides generated are retained in a graphite furnace heated to 200–600 °C, with the inner surface modified with Pd or Ir in the same manner as in liquid sampling, to be subsequently volatilized and detected at temperatures around 2500 °C. The facts that the preconcentration efficiency usually reaches 100% under optimum experimental conditions and the possibility of effortless automation of the procedure employing the autosampler arm for hydride introduction make this approach a number one choice. The best LODs reported for Se using this approach are around 1 pg mL−1.9 Preconcentration of Se hydride on the surface of a graphite rod as well as inside a cavity of this rod was also tested.10
Metal surfaces were reported as suitable materials for Se preconcentration with subsequent AAS detection as reviewed by Ataman7 and Dědina.8 The metals were a molybdenum strip modified by rhodium,11 tungsten modified by platinum,12 rhodium13,14 or gold15 and a non-modified gold surface.16
Preconcentration of hydride forming elements at a quartz surface has also been extensively studied. The first studies have employed water cooled quartz tubes inserted into the burner of a flame AAS (FAAS) instrument. These systems in various designs were used before to improve FAAS detection limits with liquid sampling of many elements as reviewed by Matusiewicz17 and Ataman.7 The nebulized analyte was trapped at the cooled quartz surface to be subsequently released as soon as the cooling water flow was stopped. Nevertheless, these systems are also compatible with HG as a sample introduction technique instead of liquid nebulization.
Lately, preconcentration of hydride forming elements in a QTA has also been tested. A compact quartz trap-and-atomizer device based on a modified QTA was designed in our laboratory. This device and the preconcentration procedure developed have been demonstrated to work universally for preconcentration of all important hydride forming elements. The preconcentration takes place in the inlet arm of the QTA to be controlled by both surface temperature and atmosphere composition. Analyte retention is achieved always under an oxygen-rich atmosphere, i.e. in oxygen excess over hydrogen produced as a side-product of HG, whereas analyte release is achieved in a hydrogen rich atmosphere when the oxygen flow is switched off and hydrogen either from a blank solution subjected to HG or from a gas cylinder is introduced. Apart from the atmosphere composition, the temperature of the quartz surface is also crucial typically with the trapping temperature being lower than that required for volatilization. Although the particular preconcentration conditions as well as the preconcentration efficiency are analyte dependent, the main features of the preconcentration procedure, i.e. trapping in oxygen excess over hydrogen while volatilization in hydrogen excess over oxygen, as well as apparatus design, are the same regardless of the analyte. A preconcentration efficiency close to 100% has been achieved for the hydrides of Sb, Bi,18 Pb19,20 and Sn21 proving the universality of both the device and the procedure. Moreover, preconcentration of Se and As hydrides was also found feasible using the same approach, but the preconcentration efficiency reached only 70% for Se and 50% for As.22 The incomplete preconcentration for As and Se hydrides in this arrangement can be explained by analyte losses during trapping as proven for Se22 or volatilization. Additionally, the drawback of this approach in the case of As and Se hydride preconcentration is the fact that the oxygen concentration changes significantly between trapping (10 mL min−1 O2) and volatilization steps (no addition of O2) in the heated optical arm of the QTA (900 °C) since oxygen shows a strong structured absorption below 200 nm at elevated temperature as demonstrated in our study.23 As a consequence, this interferes with detection of As at 193.7 nm or Se at 196.0 nm and the interference cannot be compensated for with deuterium background correction.23 This is why a delay period of 5 s was set in our previous work22 prior to analyte volatilization by H2 addition and subsequent detection by AAS, in which the oxygen flow was switched off while the preconcentration surface was already heated to volatilization temperature (in the absence of H2). This delay served to flush out oxygen from the system. Unfortunately, the preconcentrated analyte can be partly lost during this 5 s period. Other elements such as Sb, Bi, Pb or Sn are not affected by this interference from O2 since their detection is performed at wavelengths above 200 nm, namely 217.6 nm (Sb), 223.1 nm (Bi), 283.3 nm (Pb) and 286.3 nm (Sn),18–21 where oxygen absorption does not occur.
In situ trapping of hydrides in DBD atomizers has been reported very recently as a promising alternative to other in-atomizer preconcentration strategies. A simple procedure for in situ preconcentration of As24 and Sb25 hydrides in a planar DBD atomizer has been developed in our laboratory recently. Hydride analytes were quantitatively retained in the optical arm of a DBD atomizer when a few mL min−1 of oxygen was admixed with the Ar discharge gas while the analytes were completely and rapidly released as soon as the oxygen flow was switched off.24,25 This approach is thus analogous to the procedure discussed above for QTAs, having the same feature of trapping in the presence of oxygen and volatilization in its absence. No change in other experimental parameters such as DBD power (DBD temperature) or the Ar flow rate was necessary for volatilization making the preconcentration procedure extremely simple. A great advantage of preconcentration in DBD atomizers is the fact that it takes place almost at ambient temperature since the applied power is so low that the surface temperature of the DBD does not exceed 50 °C. Therefore, the oxygen structured absorption is negligible even for As and Se as analytes under these conditions, since oxygen absorption increases dramatically with temperature.23 Preconcentration of Bi in the same manner was also found feasible achieving 62% preconcentration efficiency under non-optimized conditions.26 A successful in situ preconcentration of arsenic in a tubular-shaped DBD atomizer with AFS detection has been described recently by Mao et al.27 also based on oxygen addition to the discharge in order to trap arsane achieving 100% preconcentration efficiency. Their procedure is slightly more complicated since the discharge power supply rate has to be changed between the trapping and volatilization steps as well as gas flows have to be redirected. To the best of our knowledge, the possibility of Se hydride preconcentration in a DBD atomizer has not been reported in the literature yet.
The aim of this work was to optimize the experimental conditions for Se hydride preconcentration in a DBD atomizer with AAS detection, quantify the preconcentration efficiency, assess its potential for analytical routine and study the mechanisms of analyte trapping and its subsequent release by using a radioactive indicator.
2 Experimental
2.1 Reagents
All reagents were of analytical reagent grade or higher purity. Deionized water (<0.1 μS cm−1, Ultrapure, Water, USA) was used to prepare solutions. Working Se standards were prepared fresh daily from 1000 μg mL−1 Se stock solution (Fluka) by dilution in 1 mol L−1 HCl (Merck). The blank solution consisted of 1 mol L−1 HCl. The reductant was a 0.5% (m/v) solution of NaBH4 (Sigma) in 0.4% (m/v) KOH (Merck) filtered after preparation and stored frozen. A groundwater reference material (trace elements/metals) Grumo-K (Eurofins, Denmark) with a defined Se content was employed to assess the accuracy and precision of the results. Hydrofluoric acid (38% (m/v), p.a., Spolchemie, Czech Republic) and nitric acid (65%, p.a., Lach-Ner, Czech Republic) were used to clean the DBD atomizers if necessary as well as to leach trapped Se prior to its radiometric determination. If explicitly stated, a surface modification (passivation) of the inner surface of the DBD atomizer was performed using a 5% solution of dimethyldichlorosilane (DMDCS) in toluene (Sylon CT solution produced by Supelco). Compressed gases of Ar (99.996%), H2 (99.95%) and O2 (99.5%) were produced by SIAD Czech, Ltd., Czech Republic. An aqueous solution of radioactive 75Se (half-life 119.8 day), declared as sodium selenite, was purchased from Lacomed Ltd., Czech Republic.2.2 Instrumentation
A GBC model SavantAA atomic absorption spectrometer (GBC, Australia) was employed without background correction. A Photron Se super lamp was operated at a 196.0 nm analytical line with a 1.0 nm spectral bandpass and a lamp current of 18 mA (boost current 23 mA).Radiometric measurements were performed using an automatic gamma counter (1480 Wizard 3, PerkinElmer, USA) equipped with a NaI(Tl) well-type crystal; a counting time of 60 s was used. The spatial distribution of 75Se in the DBD atomizer was assessed using image plate autoradiography. The atomizer was fixed on a sheet of paper and placed onto a photoplate sensitive to gamma radiation (Fuji, Japan). The radiograms were obtained with the help of a multiimager laser scanner system (FX ProPlus Molecular Imager, Bio-Rad, USA).
2.3 Hydride generator
An in-house continuous flow hydride generator (see Fig. 1) was driven by a peristaltic pump (Ismatec, Switzerland) and mated to a 3 mL internal volume gas–liquid separator (GLS) having a forced outlet. The flow rates of the peristaltic pump have been set to 4.0 mL min−1 and 1.2 mL min−1 in the standard/blank channel and the reductant (NaBH4) channel, respectively. The switching between the standard and blank solution was realized using a manual 3-way valve. The carrier/discharge gas flow rate (Ar) was regulated using a mass flow controller (Omega Engineering, USA). If explicitly stated, the carrier/discharge gas consisted of a mixture of Ar and H2, both supplied from gas cylinders (SIAD Czech, Ltd). The flow rate of hydrogen evolved from the decomposition of NaBH4 was calculated to be approximately 15 mL min−1 and experimentally determined using a manual volumetric flow meter.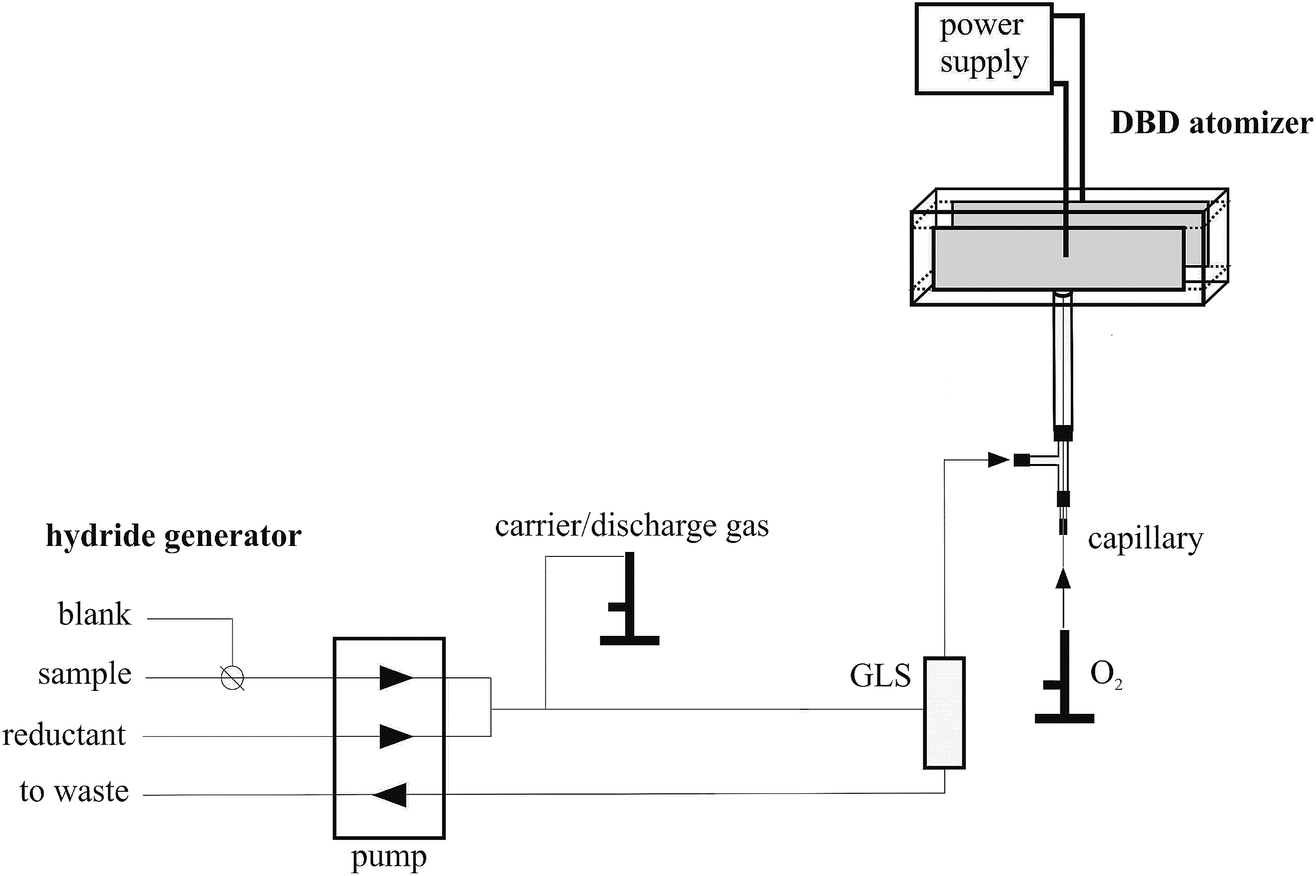 | ||
| Fig. 1 Scheme of the HG-DBD apparatus setup employed for Se hydride preconcentration. | ||
2.4 DBD atomizer
The design of the DBD atomizer was similar to that used in previous studies.6,24–26,28,29 Its optical arm, adjusted to the axis of the AAS spectrometer, was fused together from two quartz microscope slides (15 × 75 × 1 mm) and two quartz spacers (3 × 75 × 3 mm) purchased from UQG Optics Ltd., England. A quartz tube (20 mm long, 2 mm inner diameter (i.d.), 4 mm outer diameter (o.d.)) was sealed to the center of one of the quartz spacers and it served as an inlet arm. The whole DBD atomizer body was thus T-shaped, see Fig. 1 for the scheme of the atomizer. The plasma channel in the optical arm was 75 mm long with a 7 mm × 3 mm rectangular cross-section. Two copper electrodes (50 mm long; 5 mm wide; 0.15 mm thick) were glued to the central part of the outer surface of the microscope slides and their surfaces were covered with an insulating layer of epoxy resin to avoid surface discharge across the outer body of the chamber. In total, four pieces of DBD atomizers with identical design have been employed in this study. The DBD atomizer was coupled to a power source generating a sinusoidal waveform frequency (excitation frequency 28.5 kHz) and a high voltage transformer, both fabricated by Lifetech (Czech Republic) and described previously.6,24–26,28,29An extra T-connector made of polypropylene (1 mm i.d.) was placed ca. 3 cm upstream of the DBD atomizer to introduce oxygen into the DBD atomizer (see Fig. 1). A deactivated fused silica capillary (Supelco, Germany, 0.53 mm i.d.) was centered inside the inlet arm of the DBD atomizer with its tip aligned with the junction of the inlet arm with the optical arm. Oxygen was introduced through the capillary whereas all the gases from the GLS were introduced into the inlet arm (see Fig. 1). The oxygen flow rate was regulated using a mass flow controller (Omega Engineering, USA). If not stated otherwise 3.5 mL min−1 O2 was employed.
The same hydride generator and DBD atomizer configuration as described above were employed also for radiometric measurements (see Section 2.5.2. for details). The only difference was that PTFE plugs were integrated with two columns packed with granulated activated carbon (not displayed in Fig. 1) that were attached to both ends of the DBD atomizer's optical arm. These prevent the release of the radiotracer to the atmosphere. Moreover, they permitted quantification of the analyte fraction being transported outside the DBD atomizer during the preconcentration procedure, i.e. breaking through in the trapping step or being released in the volatilization step. AAS measurements could not be performed simultaneously with radiometric measurements since the superlamp's beam was blocked by PTFE plugs and columns with activated carbon.
2.5 Procedure
(i) Trapping step: selane was generated from a sample solution. A sample introduction time between 30 and 300 s was employed followed by introduction of the blank solution for 30 s. The DBD atomizer was supplied, if not stated otherwise, with a flow of 3.5 mL min−1 oxygen during the whole period of sample and blank introduction. 75 mL min−1 Ar was employed as the discharge gas and the DBD atomizer was supplied with 17 kV of sinusoidal voltage from the source.
(ii) Volatilization step: signal measurement was initiated at the beginning of the volatilization step immediately followed by switching off the oxygen flow while hydride generation from a blank solution was still running thus serving as a source of hydrogen required for analyte volatilization. 75 mL min−1 Ar was employed as the discharge gas and the DBD atomizer was supplied with 17 kV of sinusoidal voltage from the source in the same manner as in the trapping step. If explicitly stated, the sinusoidal voltage was increased to 30 kV. If explicitly stated, hydrogen from a cylinder was admixed with the Ar discharge gas during the volatilization step to increase the H2 fraction in the discharge gas. The total flow rate of Ar and H2 from gas cylinders was kept constant at 75 mL min−1. In order to achieve 100% H2 fraction in a single experiment, the Ar stream was completely replaced by hydrogen from the cylinder. Signals were recorded for 20 s by AAS.
Measurements in on-line atomization mode (no preconcentration) were performed in the same apparatus. The analyte standard was introduced for 30 s followed by introduction of the blank solution for 30 s with no oxygen added to the capillary. The high voltage supply as well as carrier/discharge gas composition were kept the same as in the volatilization step of the preconcentration procedure, 17 kV and 75 mL min−1 Ar, respectively, if not stated otherwise. Signals were recorded for 90 s by AAS, and hydride generation from a blank solution was not switched off to provide a continuous supply of hydrogen from the decomposition of NaBH4 (15 mL min−1, see Section 2.3) to the DBD atomizer.
In both modes of operation, the peak area as well as the peak height response were evaluated. Averages from at least 5 replicate measurements were recorded; the peak area of the signals is plotted in the figures; error bars represent standard deviation (SD). To quantify the preconcentration efficiency, the peak areas of the signal in the volatilization step of the preconcentration mode were related to those obtained in the on-line atomization mode under strictly the same experimental conditions (standard concentration and sample introduction time, DBD high voltage, and discharge gas composition).
75Se tracer working solutions were employed instead of non-radioactive Se standard solutions used in AAS experiments. They were prepared from the commercial 75Se tracer solution by appropriate dilution with 1 mol L−1 HCl. Two kinds of 75Se tracer working solutions were employed in this study. A more diluted 75Se tracer working solution fortified by addition of the non-radioactive carrier (0.5 ng mL−1 Se) was used in the majority of the experiments yielding an estimated total Se(IV) concentration of 0.8 ng mL−1. The activity concentration of this 75Se tracer working solution was ca. 0.8 kBq mL−1 corresponding to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cpm mL−1 in radiometric measurements. This solution mimics very well typical analytical conditions for preconcentration regarding the mass of the analyte trapped (a few ng of Se).
000 cpm mL−1 in radiometric measurements. This solution mimics very well typical analytical conditions for preconcentration regarding the mass of the analyte trapped (a few ng of Se).
A less diluted 75Se tracer working solution without addition of the carrier (non-radioactive Se standard solution) was used for autoradiography measurements containing naturally abundant non-radioactive Se isotopes in addition to the radioactive 75Se, yielding an estimated total Se(IV) concentration of 1.5 ng mL−1. The activity concentration of this 75Se tracer working solution was ca. 4.0 kBq mL−1 corresponding to 85000 cpm mL−1 in radiometric measurements. Higher activity of the 75Se tracer working solution was necessary to obtain autoradiograms with good contrast while the Se concentration was still low enough to simulate preconcentration of a few ng of Se owing to the absence of non-radioactive carrier Se addition into this solution.
Columns with activated carbon were replaced by new ones between the trapping and volatilization steps in order to distinguish between (i) the Se fraction not trapped in the first step of the preconcentration procedure (breakthrough signal) and (ii) the Se fraction released outside the DBD atomizer in the volatilization step of the preconcentration procedure. Fractions of Se retained in the DBD atomizer either (iii) after the trapping step or (iv) the volatilization step were also quantified by radiometry. The fractions of Se (i) and (ii) were quantified by radiometry of the activated carbon absorber columns whereas activities (iii) and (iv) were determined by radiometry of the leachate (10% HNO3) from the DBD atomizer. From the experimental point of view analogous radiotracer experiments have been performed in our previous work30 where a more detailed description can be found.
3 Results and discussion
Experimental conditions for on-line atomization of selenium hydride in a planar DBD atomizer were optimized in our previous work.6 The best discharge gas was Ar at a flow rate of 75 mL min−1 while the DBD power was kept at 14 W which corresponds to 17 kV of sinusoidal voltage. No signal was measured in air as the discharge gas whereas transient signals were detected as soon as air was replaced by a stream of Ar in the presence of hydrogen evolved from a blank solution subjected to HG.6 These observations clearly indicated the retention of Se hydride in air discharge with subsequent release of Se species in an Ar–H2 atmosphere. Based on these facts and preliminary observations preconcentration of Se hydride in a planar DBD atomizer was optimized in this work.3.1 Optimization of preconcentration conditions
Effects of several experimental parameters on the Se preconcentration efficiency were investigated employing a univariate approach. These parameters include the flow rate of oxygen added into the Ar discharge gas in the trapping step, modification of the inner DBD surface using a passivation agent, and the high voltage delivered to the DBD electrodes in the volatilization step as well as the hydrogen fraction in the Ar discharge gas in the volatilization step. Finally, also the effect of preconcentration period on the preconcentration efficiency was studied.It has been demonstrated in a preliminary experiment that oxygen from air is responsible for no Se signal being observed in on-line atomization mode, employing 75 mL min−1 of air as a discharge gas. The same result, i.e. no Se signal in on-line atomization mode, is observed when 15 mL min−1 of O2 is admixed with 60 mL min−1 of Ar simulating the same O2 fraction in inert (Ar) gas as is contained in air, in which N2 acts as the inert gas. This is clear evidence that oxygen causes Se hydride retention in the DBD atomizer since the on-line atomization signal of Se can be detected when employing Ar or N2 as discharge gases, of course always mixed with hydrogen as a by-product of hydride generation. Since the Se signal re-appears as soon as oxygen is switched off, while the inert gas (Ar or N2) is introduced continuously into the DBD atomizer together with H2 evolved from a blank solution subjected to HG, Se hydride trapping in the DBD atomizer must occur. The sensitivity of on-line measurements is ca. two times higher in Ar compared to N2.6
As a consequence, the effect of oxygen flow rate admixed with the Ar discharge gas on the preconcentration efficiency was studied in detail with the result depicted in Fig. 2. The Ar flow rate (75 mL min−1) and the DBD high voltage (17 kV) were kept constant during the whole measurement employing a Se standard concentration of 1 ng mL−1. No preconcentration of Se hydride is obviously observed in the absence of oxygen, i.e. no volatilization signal was observed. These conditions are absolutely the same as those employed in on-line atomization mode. The peak shape and area of the signal corresponded to the values observed typically in the on-line atomization mode. This signal can be taken as a reference signal from which the preconcentration efficiency can be quantified. If 1.0 mL min−1 O2 is admixed with the Ar discharge gas a volatilization peak can be clearly detected corresponding to a preconcentration efficiency of ca. 35% (Fig. 2) while the signal recorded during trapping, thus corresponding to non-trapped Se, drops to one half of its original value observed when no oxygen has been added. The volatilization peak is much narrower (full width at half maximum ca. 1.8 s) than the signals recorded either during trapping or in on-line atomization mode (both having a baseline width of 30 s). Increasing the oxygen flow rate to 1.5 mL min−1 results in an increase of the volatilization signal achieving a preconcentration efficiency of 70% (Fig. 2). The signal of Se recorded during trapping cannot be detected for oxygen flow rates of 1.5 mL min−1 and higher indicating complete trapping of Se hydride. Slightly better preconcentration efficiencies of 75 and 78%, respectively, were achieved for 2.0 and 2.5 mL min−1 O2 as depicted in Fig. 2. A further increase in the O2 flow rate up to 7.5 mL min−1 cannot improve the preconcentration efficiency (see Fig. 2). An oxygen flow rate of 3.5 mL min−1 was employed as the optimum in further experiments.
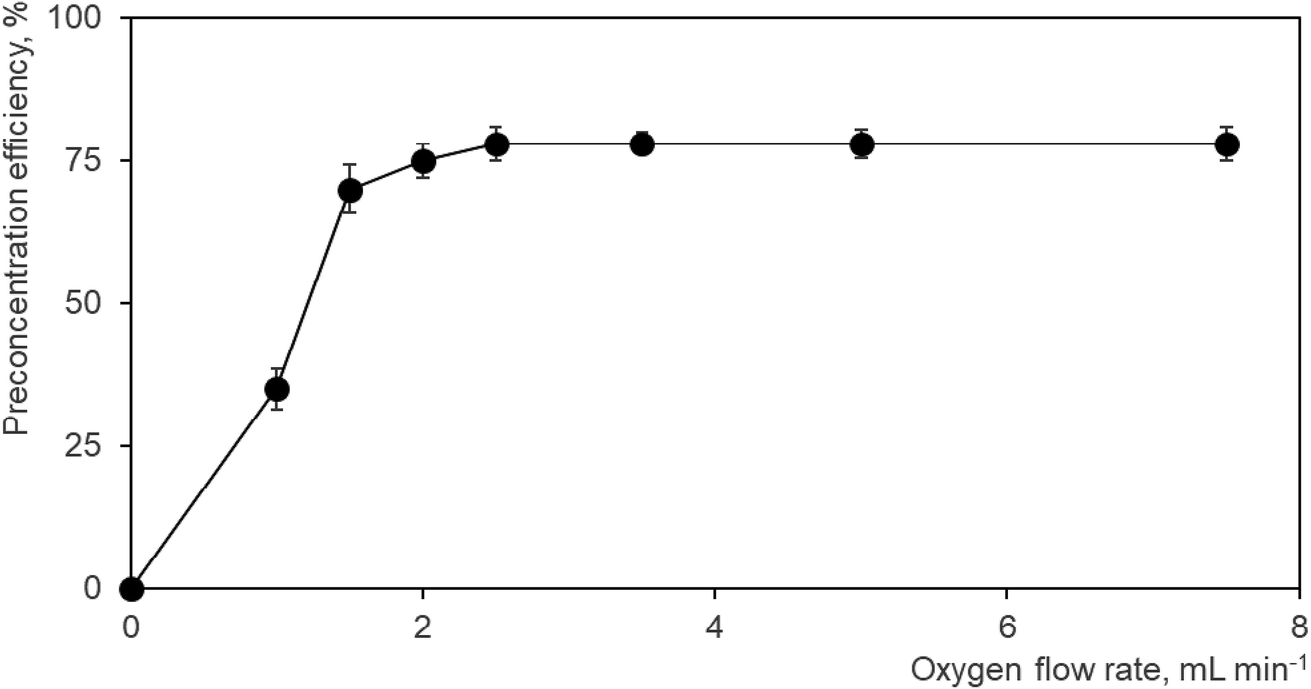 | ||
| Fig. 2 Effect of O2 addition during the trapping step to the Ar discharge gas on the Se preconcentration efficiency. 75 mL min−1 Ar, 17 kV (14 W), 1 ng mL−1 Se standard solution. | ||
Experimental parameters affecting the volatilization step have been optimized in an effort to improve the preconcentration efficiency. The DBD voltage in the volatilization step was increased from 17 kV (estimated power of 14 W) to 30 kV (estimated power of 26 W), but no effect of high voltage on the preconcentration efficiency was observed in this range. A preconcentration efficiency of 72 ± 2% was achieved at 17 kV while 75 ± 6% was observed at 30 kV. Unfortunately, further increase in high voltage was not possible due to the limitations of the sinusoidal power supply source. A DBD voltage of 17 kV was employed in further experiments.
Another parameter that may affect the efficiency of Se volatilization is the content of hydrogen in the discharge gas. Hydrogen has been clearly proven essential for on-line atomization of Bi28 and Se30 hydrides in DBD atomizers recently; this feature is believed to be valid universally for all hydride forming elements. Hydrogen presence is also necessary for analyte volatilization in the preconcentration procedure as demonstrated for As, Sb and Bi.24–26 Hydrogen produced during hydride generation from a blank solution, i.e. a hydrogen fraction of 17% in the Ar discharge gas, was found sufficient for complete volatilization of trapped Sb and As while a preconcentration efficiency of 60% was achieved for Bi.24–26 In this study, the effect of hydrogen fraction in the discharge gas on the preconcentration efficiency of Se was found negligible since a preconcentration efficiency of 78 ± 3% was found for 17% H2 fraction in Ar (as produced by NaBH4 decomposition), while 81 ± 3% preconcentration efficiency was observed employing 25% H2 fraction in Ar and a preconcentration efficiency of 75 ± 6% was achieved in pure hydrogen, i.e. corresponding to 100% H2 fraction. For the sake of simplicity hydrogen produced by NaBH4 decomposition (17% H2 fraction) was used in further experiments.
The effect of inner DBD surface modification by DMDCS on the preconcentration efficiency was investigated. This passivation agent was believed to decrease the affinity of the trapped analyte to the quartz surface. As a consequence, easier and more efficient Se volatilization was expected. A Se preconcentration efficiency of 71 ± 2% was found in a DMDCS-treated DBD atomizer while it reached 71 ± 3% in the same atomizer piece but without surface modification. This result indicates no positive effect of DMDCS modification on the preconcentration efficiency. Thus, no surface modification was further employed.
The effect of preconcentration period on the preconcentration efficiency has been studied. Preconcentration periods of 30 s, 120 s and 300 s were investigated employing Se standard solutions with 1.0 ng mL−1 Se, 0.25 ng mL−1 Se and 0.1 ng mL−1 Se, thus keeping the mass of Se hydride generated constant. Preconcentration efficiencies of 72 ± 2%, 64 ± 5% and 60 ± 6% have been achieved for 30 s, 120 s and 300 s preconcentration indicating no significant changes in preconcentration efficiency at various preconcentration periods. Longer preconcentration periods enable LOD improvement but the 300 s preconcentration period was the longest studied to maintain a reasonable sample throughput.
3.2 Radiotracer experiments
Experiments employing a 75Se radioactive indicator have been performed under the optimum preconcentration conditions found in Section 3.1 in order to quantify the trapping and volatilization efficiencies independently of AAS measurements as well as to investigate spatial distribution of the analyte in the apparatus during preconcentration and better understand the processes behind.The Se fraction detected by radiometry on activated carbon absorber columns in the trapping step of the preconcentration procedure, i.e. Se breakthrough signal, reached only 1.3 ± 0.9% (n = 6) of Se generated indicating satisfactorily low analyte losses during trapping. On the contrary the trapped Se fraction, quantified by radiometric determination of 75Se in the leachate of the optical arm, reached 92% proving effective retention of Se hydride in the optical arm of the DBD atomizer. Moreover, autoradiography has shown the spatial distribution of the analyte trapped in the optical arm of the DBD atomizer. As depicted in Fig. 3, it is trapped in the central part of the DBD's optical arm, in a small clearly bordered spot (ca. 1.5 cm wide) located symmetrically on both sides of the downstream part of the inlet arm. Both the radiometry and autoradiography revealed efficient analyte trapping.
 | ||
| Fig. 3 Spatial distribution of 75Se in a planar DBD atomizer after trapping (left panel, exposure time 7 hours) and volatilization (right panel, exposure time 50 hours). 75 mL min−1 Ar, 17 kV (14 W), 3.5 mL min−1 O2 added in the trapping step. Carrier-free 75Se tracer solution employed (8.2 kBq, 1.5 ng mL−1 Se, 2.6 ng Se absolute per experiment). | ||
The autoradiogram of the DBD atomizer after the volatilization step is also depicted in Fig. 3 showing clear re-distribution of the analyte compared to the situation after the trapping step. The analyte spot is much more diffuse and broader – expanding towards the end of the optical arm. The asymmetry of the spot is an artifact given by the presence of the columns with activated carbon. The manually prepared columns inherently suffer from an unbalanced resistance to the gas flow resulting in an asymmetry in the flow pattern. The fraction of Se released outside of the DBD atomizer and absorbed in the activated carbon columns was 21 ± 3% (n = 5) of Se introduced into the system. This fraction is obviously part of the volatilized analyte. The fraction of Se leached from the optical arm of the atomizer, 72 ± 13% (n = 5), is the quantification of the analyte shown in the autoradiogram after the volatilization step (Fig. 3). The sum of Se found in absorber columns and in the HNO3 leachate provides a reasonably good mass balance of the introduced analyte. However, it should be highlighted that the fraction of Se found in the leachate includes analyte (i) volatilized from the trapping spot in the central part of the optical arm but subsequently deposited on the inner surface of the atomizer (Fig. 3) and (ii) non-volatilized from its original position in the center of the optical arm (compare both autoradiograms in Fig. 3). The non-volatilized analyte is responsible for a preconcentration efficiency below 100%. In conclusion, the radiotracer experiments confirmed an efficient trapping of the analyte (>90% Se) whereas the volatilization step was found incomplete with a significant fraction of Se remaining non-volatilized.
3.3 Analytical performance
Four different DBD atomizers with the same planar design have been tested employing the optimum preconcentration conditions found in Section 3.1 to assess the reproducibility of preconcentration among various pieces and day to day repeatability for a given piece. Each of the DBD atomizers was investigated on at least two days within a week. A great variety in preconcentration efficiencies among the pieces was observed; the minimum preconcentration efficiency was as low as 47% while the maximum efficiency reached 77%. The average preconcentration efficiency for all pieces on all days was quantified as 63 ± 11%. The variability in preconcentration efficiencies found among the pieces was much greater than its day to day repeatability in a given piece. The variability in preconcentration efficiencies among the pieces can be most likely attributed to differences in the quality of the inner quartz surface that might affect preconcentration, especially the volatilization step of the procedure which is not complete, thus being less robust than the trapping step, as discussed in Section 3.2. The sensitivity in on-line atomization mode did not significantly differ among individual atomizers. On all days it was quantified as 0.40 ± 0.05 s ng−1 Se.The atomizer piece with the best performance showed a preconcentration efficiency (30 s preconcentration period) of 71 ± 3% on one day and 73 ± 5% on three consecutive days, respectively. This piece was selected for evaluation of the analytical performance of the preconcentration procedure. Its sensitivity in the on-line atomization mode reached 0.35 ± 0.01 s ng−1 Se. This value is in accord with the sensitivities of 0.38 ± 0.02 s ng−1 Se and 0.32 ± 0.01 s ng−1 Se reported in our previous studies6,30 dealing with Se hydride atomization in planar DBD atomizers. A sensitivity of 0.25 ± 0.02 s ng−1 Se in the preconcentration mode with a 30 s preconcentration period was found, whereas for a preconcentration period of 300 s it reached 0.22 ± 0.02 s ng−1 Se. Calibration curves were measured in the range from 0.25 to 2.0 ng mL−1 Se for a 30 s preconcentration period and 0.025 to 0.20 ng mL−1 for a 300 s preconcentration period, respectively. Both of them were linear in this range. The LOD found for a 300 s preconcentration period was 12 pg mL−1 Se (240 pg Se absolute), 20 times better than 240 pg mL−1 Se reported in the same system and the atomizer without preconcentration.6 The accuracy and precision of the measurements in the preconcentration mode were verified by Se determination in a GRUMO K certified reference material. Its certified value of 0.77 ± 0.40 ng mL−1 Se is in agreement with the determined value of 0.74 ± 0.01 ng mL−1 Se. The typical analyte signal obtained in the preconcentration mode is depicted in Fig. 4.
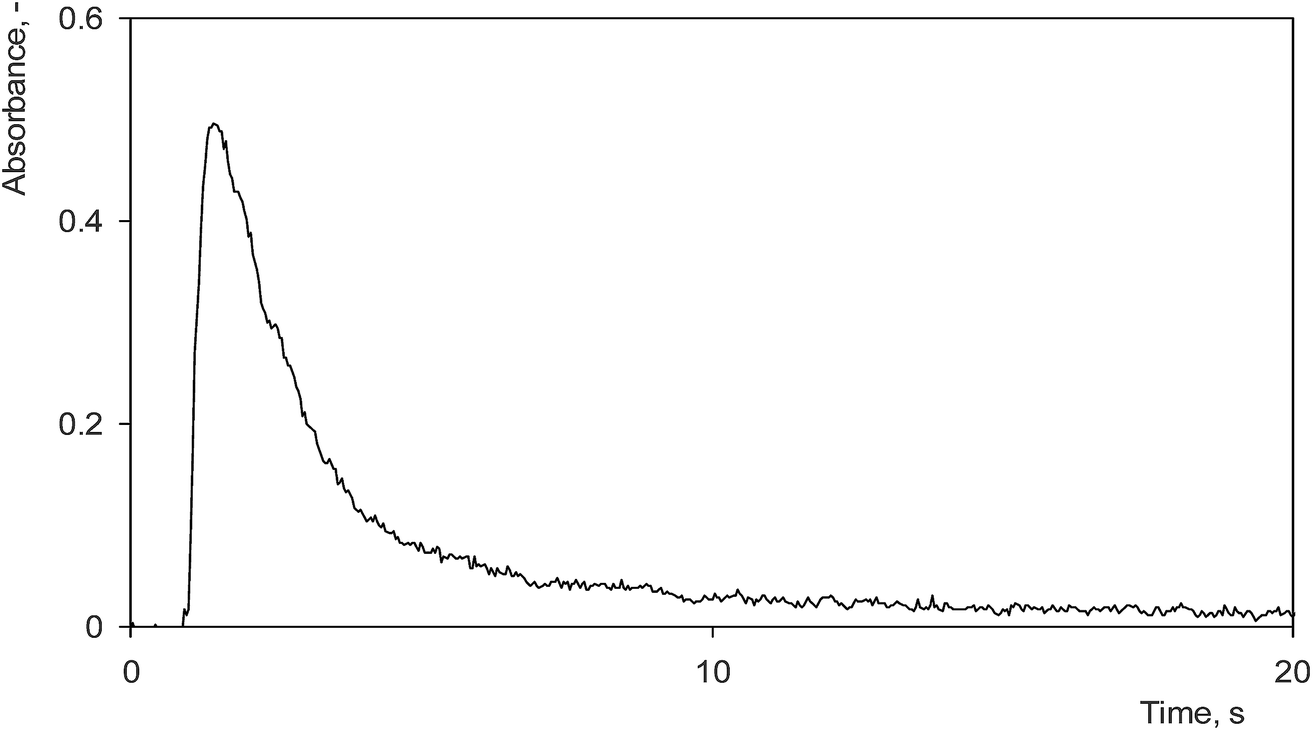 | ||
| Fig. 4 Typical signal of Se after preconcentration in a planar DBD atomizer. 75 mL min−1 Ar, 17 kV (14 W), 3.5 mL min−1 O2 added in the trapping step, 1 ng mL−1 Se standard solution, 30 s preconcentration period (2 ng Se absolute). | ||
The performance of the method developed based on in situ trapping of Se hydride in a planar DBD atomizer with subsequent AAS detection was compared to those of other preconcentration approaches employed for ultratrace Se determination by HG with AAS/AFS detection reported in the literature. The results are summarized in Table 1 comparing either the concentration or absolute limits of detection and the preconcentration efficiency depending on data available. As can be seen from Table 1 the preconcentration efficiency reported ranges from 30 to 100% whereas LODs span from 0.8 to 270 ng L−1 Se (from 8 to 30000 pg Se absolute). With a preconcentration efficiency of 70% and LOD of 12 pg mL−1 Se the performance of the proposed preconcentration procedure ranks among the average.
| Preconcentration surface | Surface modification | LOD, pg mL−1 | LOD pg | Preconcentration efficiency, % | Ref. |
|---|---|---|---|---|---|
| Metals | |||||
| W coil | Rh | 50 | 125 | — | 13 |
| W coil | Rh | 35 | 53 | — | 14 |
| W coil | Au | 39 | 1053 | — | 15 |
| W tube atomizer | Pt | 270 | 270 | 100 | 12 |
| Mo strip | Rh | — | — | 30 | 11 |
| Au wire/AFS detection | — | 5 | 250 | 100 | 16 |
|
|||||
| Graphite | |||||
| Graphite furnace | Pd, Ir | 0.8–1.4 | 8–70 | 100 | 9 and 31–33 |
| Graphite rod (cavity) | Ir | — | — | 70 | 10 |
|
|||||
| Quartz | |||||
| Water cooled tube (in FAAS) | — | 3000 | 30000 |
— | 34 |
| Quartz atomizer | — | — | — | 70 | 22 |
| DBD quartz atomizer | — | 12 | 240 | 70 | This work |
4. Conclusion
Preconcentration of Se hydride in a novel quartz dielectric barrier discharge (DBD) plasma atomizer was optimized and studied by means of a 75Se radioactive indicator. The simplicity of the developed procedure and instrumentation together with user friendliness belongs to the benefits of this approach. The preconcentration procedure proposed is applicable to routine use as demonstrated by the analysis of a certified reference material. This holds true at least for samples with ultratrace Se content in a simple matrix. An interference study, essential for the assessment of applicability to more complicated sample matrices, has not been carried out in this work. An emphasis was put on understanding the processes during analyte trapping and volatilization. The procedure is limited by the volatilization efficiency (reaching only 70%) that unfortunately cannot be further improved since no higher voltage of the DBD power supply source can be applied. As a consequence, our future effort will be focused on the development of Se preconcentration procedures having the potential to offer 100% Se preconcentration efficiency.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported by the Czech Science Foundation under contract 18-01116S, of the Czech Academy of Sciences, and the Institute of Analytical Chemistry (Institutional Research Plan no. RVO: 68081715). This work used instruments provided by the C4Sys infrastructure. The authors are obliged to MSc. Ondřej Duben for performing some preliminary experiments focusing on Se trapping.References
- Handbook of Elemental Speciation II, ed. R. Cornelis, John Wiley&Sons Ltd, Chichester, 2013 Search PubMed.
- J. Dědina and D. L. Tsalev, in Hydride Generation Atomic Absorption Spectrometry, AnonymousWiley and Sons, Chichester, 1995 Search PubMed.
- J. Dědina, in Generation of Volatile Compounds for Analytical Atomic Spectroscopy, ed. R. A. Meyers, John Wiley, Chichester, 2011 Search PubMed.
- S. Brandt, A. Schütz, F. D. Klute, J. Kratzer and J. Franzke, Dielectric barrier discharges applied for optical spectrometry, Spectrochim. Acta, Part B, 2016, 123, 6–32 CrossRef CAS.
- S. Liu, Y. L. Yu and J. H. Wang, Advances in discharge-based microplasmas for the analysis of trace species by atomic spectrometry, J. Anal. At. Spectrom., 2017, 32, 2118–2126 RSC.
- O. Duben, J. Boušek, J. Dědina and J. Kratzer, Dielectric barrier discharge plasma atomizer for hydride generation atomic absorption spectrometry – performance evaluation for selenium, Spectrochim. Acta, Part B, 2015, 111, 57–63 CrossRef CAS.
- O. Y. Ataman, Vapor generation and atom traps: atomic absorption spectrometry at the ng/L level, Spectrochim. Acta, Part B, 2008, 63, 825–834 CrossRef.
- J. Dědina, Atomization of volatile compounds for atomic absorption and atomic fluorescence spectrometry: on the way towards the ideal atomizer, Spectrochim. Acta, Part B, 2007, 62, 846–872 CrossRef.
- H. Matusiewicz and R. E. Sturgeon, Atomic spectrometric detection of hydride forming elements following in situ trapping within a graphite furnace, Spectrochim. Acta, Part B, 1996, 51, 377–397 CrossRef.
- B. Dočekal, Trapping of hydride forming elements within miniature electrothermal devices. Part 2. Investigation of collection of arsenic and selenium hydrides on a surface and in a cavity of a graphite rod, Spectrochim. Acta, Part B, 2004, 59, 497–503 CrossRef.
- B. Dočekal, S. Gucer and A. Selecká, Trapping of hydride forming elements within miniature electrothermal devices: part I. investigation of collection of arsenic and selenium hydrides on a molybdenum foil strip, Spectrochim. Acta, Part B, 2004, 59, 487–495 CrossRef.
- B. Dočekal and P. Marek, Investigation of in situ trapping of selenium and arsenic hydrides within a tungsten tube atomizer, J. Anal. At. Spectrom., 2001, 16, 831–837 RSC.
- F. Barbosa, S. Souza and F. Krug, In situ trapping of selenium hydride in rhodium-coated tungsten coil atomic absorption spectrometry, J. Anal. At. Spectrom., 2002, 17, 382–388 RSC.
- S. S. de Souza, J. Santos, F. J. Krug and J. Barbosa, Exploiting in situ hydride trapping in tungsten coil atomizer for Se and As determination in biological and water samples, Talanta, 2007, 73, 451–457 CrossRef PubMed.
- I. Kula, Y. Arslan, S. Bakirdere and O. Y. Ataman, A novel analytical system involving hydride generation and gold-coated W-coil trapping atomic absorption spectrometry for selenium determination at ng l-1 level, Spectrochim. Acta, Part B, 2008, 63, 856–860 CrossRef.
- X. Guo and X. Guo, Determination of ultra-trace amounts of selenium by continuous flow hydride generation AFS and AAS with collection on gold wire, J. Anal. At. Spectrom., 2001, 16, 1414–1418 RSC.
- H. Matusiewicz, Atom trapping and in situ preconcentration techniques for flame atomic absorption spectrometry, Spectrochim. Acta, Part B, 1997, 52, 1711–1736 CrossRef.
- J. Kratzer and J. Dědina, Stibine and bismuthine trapping in quartz tube atomizers for atomic absorption spectrometry – method optimization and analytical applications, Spectrochim. Acta, Part B, 2008, 63, 843–849 CrossRef.
- J. Kratzer, Ultratrace determination of lead by hydride generation in-atomizer trapping atomic absorption spectrometry: optimization of plumbane generation and analyte preconcentration in a quartz trap-and-atomizer device, Spectrochim. Acta, Part B, 2012, 71–72, 40–47 CrossRef CAS.
- J. Kratzer, S. Musil, M. Vobecký and J. Dědina, Hydride generation – in-atomizer collection of Pb in quartz tube atomizers for atomic absorption spectrometry – a 212Pb radiotracer study, J. Anal. At. Spectrom., 2013, 28, 344–353 RSC.
- L. Průša, J. Dědina and J. Kratzer, Ultratrace determination of tin by hydride generation in-atomizer trapping atomic absorption spectrometry, Anal. Chim. Acta, 2013, 804, 50–58 CrossRef PubMed.
- J. Kratzer and J. Dědina, Arsine and selenium hydride trapping in a novel quartz device for atomic-absorption spectrometry, Anal. Bioanal. Chem., 2007, 388, 793–800 CrossRef CAS PubMed.
- J. Kratzer, B. Dočekal, U. Heitmann and J. Dědina, Spectral interferences of oxygen and water molecules in hydride generation atomic absorption spectrometry with quartz atomizers: comparison of preconcentration and on-line atomization modes for As and Se determination, J. Anal. At. Spectrom., 2011, 26, 2230–2237 RSC.
- P. Novák, J. Dědina and J. Kratzer, Preconcentration and Atomization of Arsane in a Dielectric Barrier Discharge with Detection by Atomic Absorption Spectrometry, Anal. Chem., 2016, 88, 6064–6070 CrossRef PubMed.
- P. Zurynková, J. Dědina and J. Kratzer, Trace determination of antimony by hydride generation atomic absorption spectrometry with analyte preconcentration/atomization in a dielectric barrier discharge atomizer, Anal. Chim. Acta, 2018, 1010, 11–19 CrossRef PubMed.
- J. Kratzer, J. Boušek, R. E. Sturgeon, Z. Mester and J. Dědina, Determination of Bismuth by Dielectric Barrier Discharge Atomic Absorption Spectrometry Coupled with Hydride Generation: Method Optimization and Evaluation of Analytical Performance, Anal. Chem., 2014, 86, 9620–9625 CrossRef CAS PubMed.
- X. Mao, Y. Qi, J. Huang, J. Liu, G. Chen, X. Na, M. Wang and Y. Qian, Ambient-Temperature Trap/Release of Arsenic by Dielectric Barrier Discharge and Its Application to Ultratrace Arsenic Determination in Surface Water Followed by Atomic Fluorescence Spectrometry, Anal. Chem., 2016, 88, 4147–4152 CrossRef CAS PubMed.
- J. Kratzer, O. Zelina, M. Svoboda, R. E. Sturgeon, Z. Mester and J. Dědina, Atomization of Bismuthane in a Dielectric Barrier Discharge: A Mechanistic Study, Anal. Chem., 2016, 88, 1804–1811 CrossRef CAS PubMed.
- M. Straka, S. Burhenn, K. Marschner, S. Brandt, U. Marggraf, J. Dědina, J. Franzke and J. Kratzer, Novel designs of dielectric barrier discharge hydride atomizers for atomic spectrometry, Spectrochim. Acta, Part B, 2018, 146, 69–76 CrossRef CAS.
- J. Kratzer, S. Musil, K. Marschner, M. Svoboda, T. Matoušek, Z. Mester, R. E. Sturgeon and J. Dědina, Behavior of selenium hydride in heated quartz tube and dielectric barrier discharge atomizers, Anal. Chim. Acta, 2018, 1028, 11–21 CrossRef CAS PubMed.
- S. N. Willie, R. E. Sturgeon and S. S. Berman, Hydride generation atomic absorption determination of selenium in marine sediments, tissues and seawater with in situ concentration in a graphite furnace, Anal. Chem., 1986, 58, 1140–1143 CrossRef CAS.
- R. E. Sturgeon, S. N. Willie and S. S. Berman, Hydride generation-graphite furnace atomic absorption spectrometry: new prospects, Fresenius' J. Anal. Chem., 1986, 323, 788–792 CrossRef CAS.
- R. E. Sturgeon, S. N. Willie, G. I. Sproule, P. T. Robinson and S. S. Berman, Sequestration of volatile element hydrides by platinum group elements for graphite furnace atomic absorption, Spectrochim. Acta, Part B, 1989, 44, 667–682 CrossRef.
- H. Matusiewicz and M. Krawczyk, On-line hyphenation of hydride generation with in situ trapping flame atomic absorption spectrometry for arsenic and selenium determination, Anal. Sci., 2006, 22, 249–253 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |