
Characterization of inductively coupled plasma time-of-flight mass spectrometry in combination with collision/reaction cell technology – insights from highly time-resolved measurements†
Marcel
Burger‡
 *,
Lyndsey
Hendriks‡
,
Jérôme
Kaeslin‡
,
Alexander
Gundlach-Graham
,
Bodo
Hattendorf
and
Detlef
Günther
*,
Lyndsey
Hendriks‡
,
Jérôme
Kaeslin‡
,
Alexander
Gundlach-Graham
,
Bodo
Hattendorf
and
Detlef
Günther
Laboratory of Inorganic Chemistry, ETH Zürich, Vladimir-Prelog-Weg 1, CH-8093 Zürich, Switzerland. E-mail: burger@inorg.chem.ethz.ch
First published on 12th November 2018
Abstract
We present results from studies investigating the capabilities of inductively coupled plasma time-of-flight mass spectrometry (ICP-TOFMS) in combination with collision/reaction cell technology (CCT). Experiments were carried out using various sample introduction techniques including high- and low-dispersion laser ablation and microdroplet generation. Specifically, we investigated H2 as a reaction gas and He as a collision gas for their effects on reduction of background species and interferences, limits of detection (LODs), quantification capabilities, and structure of short transient signals. With H2 as reaction gas, argon-based ions were suppressed. Ar+ and Ar2+ signals could be attenuated to intensity levels in the hundreds and single-digit counts per second range, respectively. Selective suppression of Ar-based ions gives access to the most abundant isotopes of Ca and Se. Furthermore, the attenuation of the Ar+ signals allows the instrument to be operated without m/z-selective attenuation of the ion beam prior to TOF analysis, which enhances transmission of isotopes near m/z 40. It was also found that application of flow rates ≤4 mL min−1 of H2, He or mixtures of these gases results in collision-induced focusing, and enhances sensitivities by a factor of 1.5 to 2 and mass resolving power by up to 16%. Use of CCT enabled an improvement in the LOD for 40Ca of more than three orders of magnitude. The LOD for 80Se was improved by more than one order of magnitude. For many of the elements contained in NIST SRM 610, LODs were lower by a factor of two to four. However, for most elements, improvement in quantification accuracy was not observed. Experiments with microdroplet sample introduction demonstrated that the mass-dependent ion transit times between the ICP and the TOF extraction region are affected by the amount of He buffer gas in the collision cell. Changes in transit times, as well as signal broadening, were observed on a time scale of tens to hundreds of μs. The duration of individual aerosol plumes from low-dispersion laser ablation, however, remained practically unaffected from collisional effects and imaging at 100 Hz laser repetition rate with baseline-separated aerosol plumes is possible.
Introduction
Since its introduction by Houk and co-workers in 1980,1 inductively coupled plasma mass spectrometry (ICPMS) has become a well-established and widely used technique for element- and/or isotope-specific analyses in various research fields including geology,2–10 environmental- and materials sciences,11–14 forensics,15–17 archaeometry,18–21 and biology and medicine.22–25 ICPMS provides excellent sensitivities, low limits of detection (LODs), wide linear dynamic range, multi-isotope detection capabilities, and compatibility with a broad range of front-end separation and sample-introduction techniques, such as chromatographic methods,26 electro-thermal vaporization (ETV),27 microdroplet-generation (MDG)28 and laser ablation (LA).29Despite numerous advantages of ICPMS, spectral interferences are a major limitation for the accurate determination of many elements. Spectral overlaps in ICPMS occur primarily as singly and doubly charged atomic or molecular ions30,31 whose abundances depend on the sample composition, operating conditions of the ICP, and physical–chemical properties of the species involved.32,33 Mass separation of these spectral interferences is, generally, not possible on quadrupole-based ICPMS instruments because of their limited mass resolving power (MRP). On the other hand, sector-field ICPMS instruments34 can provide MRP up to 12′000,35 and so can be used to resolve many polyatomic interferences from atomic analyte ions. However, even a MRP of 12′000 can be insufficient when abundant isobars or molecular ions need to be separated from the isotopes of interest.
An alternative approach to resolve spectral interferences is to use ion molecule reactions36 or kinetic energy discrimination37 in a gas filled multipole ion guide upstream of the mass analyzer, as pioneered by Houk and co-workers.38 A gas filled ion guide can be used to attenuate interfering ions by selective charge- or atom-transfer reactions, collision-induced dissociation, or by kinetic energy discrimination prior to mass analysis. Ion-molecule reaction and collision cells have been implemented in quadrupole,39,40 sector-field41 and time-of-flight mass spectrometers.42,43 They are considered a cost-effective means for interference control, which may even exceed the performance offered by the high MRP of a sector-field instrument.
“Chemical resolution”44 originates from different reaction rates of the analyte isotope and the isobaric interference with the reactive gas. Reactions can either attenuate interferent-species abundance39,45,46 or mass-shift analyte isotopes to an unoccupied m/z channel.47,48 Mass-shift reactions are most effective if an m/z selection is carried out before the ions enter the reaction region. This reaction-type is not well-suited for multi-element analyses by ICP-TOFMS because the highly reactive gases required for such approaches usually result in a multitude of reaction products, which occur as new spectral interferences in the TOFMS spectrum. On the other hand, reaction with H2 has been shown to produce few additional interferences and can be successfully used in a dynamic reaction cell for multi-element applications.49 Reaction with H2 can be used to substantially attenuate Ar+ and Ar-based polyatomic ions, which, for example, allows for detection of Ca, Fe or Se at their most abundant isotopes with minimum spectral interference from the Ar-plasma background. The dominant reaction product is H3+, which does not interfere with the isotopes of interest in ICPMS. This characteristic can be especially useful for ICP-TOFMS applications in which attenuation of 40Ar+ is usually required to protect the detector. The attenuation is commonly done by either “ion-blanking”50,51 of the high abundant ions at the first space focus of the TOFMS drift tube or by using a “notch filter”52,53 upstream the TOFMS. Both methods however are not very selective with respect to m/z removal so that “blanking” or “notching” 40Ar+ also attenuates ions of nearby m/z. Furthermore, this physical signal attenuation does not allow for discrimination between the plasma background and analyte ions of the same nominal m/z. Using H2 as reaction gas causes selective removal of the Ar-based species, which alleviates the required beam attenuation and thus increases the useable range of the mass spectrum. Early experiments using a gas-filled hexapole ion guide with ICP-TOFMS42 showed that blanking of the Ar+ signal was not anymore required when H2 was used as reaction gas.
A potential limitation for TOFMS detection is the mass-dependent transmission of the multipole ion guide together with ion loss through collisional scattering inside the cell. In sequential MS, the operating conditions of the ion guide can be adjusted in parallel with a mass scan to optimize transmission for different m/z. In TOFMS, variation of the transmission properties of the ion guide cannot practically be carried out within a single spectrum. Additionally, orthogonal extraction TOFMS exhibits a velocity-(mass-)dependent duty factor that discriminates against low m/z and this may become even more pronounced by collisional losses in the ion guide. An increasing number of collisions within the ion guide continuously reduces the kinetic energy of the ions. This collisional cooling can initially lead to higher transmission54 because lower radial kinetic energy can increase the fraction of ions that reach the exit of the pressurized cell (“collisional focusing”). However, at a certain gas pressure, the loss in the axial kinetic energy will cause a proportionally greater fraction of the ions to be removed from the beam and transmission drops. Thus, increasing the gas pressure inside the cell typically leads to an initial increase in signal intensities until a maximum is reached. Transmission maxima occur at higher flow rates for higher m/z ions because the heavier ions also carry a higher kinetic energy after extraction from the ICP and thus require more collisions before trapping becomes dominant. In addition to H2, He gas can be employed as a non-reactive collision gas. With He, ion cooling is more efficient and so lower gas pressure is required for collisional focusing. Favorable exothermic reactions inside the cell may then occur with similar efficiency at a lower partial pressure of a reactive gas.
A potential limitation in the performance achievable with reactive and non-reactive gases is the presence of reactive impurities like H2O (g) or ambient gas that can be present even in high-purity gases or enter the vacuum system through leaks. Even when using high purity H2 (99.9999%, 6N), molecular ions from water-, OH- and O-adduct ions can be formed at substantial levels.37,55 In multi-element analyses using an ICP-TOFMS, such molecular ions will give rise to additional spectral interferences. Indeed, m/z selection before the reaction region is pointless in view of maintaining the multi-isotope advantage of TOFMS in analytical applications.
In 2014, the icpTOF (Tofwerk AG, Thun, CH) was released, which is a new ICP-TOFMS instrument with a spectral-readout frequency of up to 33.3 kHz.56,57 The icpTOF instrument is based on an iCAPq (Thermo Fisher Scientific, Waltham, USA) and is the first commercial ICP-TOFMS equipped with quadrupole-based collision/reaction cell technology (CCT). Previous reports56,58 focused on the characteristics of the instrument without the CCT pressurized. The present work has investigated the impact of using H2 or He as reaction and/or collision gases with a specific focus on multi-element determinations. The impact of the pressurized cell was studied with respect to suppression of Ar-based background ions, multi-isotope sensitivities, and quantification in laser ablation applications. Furthermore, the time resolution of the TOFMS allowed for studying variations of ion transit times that occur on the time scale of <100 μs and are caused by collisions inside the pressurized CCT.
Experimental details
Experiments were carried out with the icpTOF instrument (TOFWERK AG, Thun, Switzerland) in combination with different sample introduction systems. Detailed description and characterization of these techniques can be found in previous publications.56,58 The icpTOF utilizes a multi-notch filter downstream the CCT to attenuate abundant ions at up to four m/z of the TOF-spectrum. Depending on the application, the respective m/z can be selected around a user-defined center mass by adjusting frequency and amplitude. The attenuation however also affects adjacent m/z. In normal operation using LA, only Ar+ ions need to be attenuated in order to avoid saturation of readout electronics and damage to the micro channel plate detector. In the following paragraphs, experimental parameters of the methodologies discussed in this work are summarized.Collision/reaction cell operation
The collision/reaction cell was pressurized with either He gas (99.999%, PanGas AG, Dagmersellen, Switzerland), H2 gas (99.9999%, PanGas AG, Dagmersellen, Switzerland) or mixtures of these gases. H2 and He were supplied to the instrument with mass flow controllers. Gas purifiers (Micro Torr, SAES Pure Gas, Inc., California) were used to eliminate impurities. In experiments requiring attenuation of the ion beam, the operating parameters of the multi-channel notch filter were adjusted to maintain amplitudes of background signals below ∼500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cps. Table 1 reports typical ICP-TOFMS operating conditions and collision/reaction cell settings for experimental setups discussed in this study.
000 cps. Table 1 reports typical ICP-TOFMS operating conditions and collision/reaction cell settings for experimental setups discussed in this study.
| High-dispersion LA | Low-dispersion LA | Microdroplet detection | |
|---|---|---|---|
| a n.d. not determined. b These values were determined when working with an unpressurized cell. | |||
| Injector diameter | 2.5 mm | 2.5 mm | 1.5 mm |
| RF power | 1400–1600 W | 1600 W | 1500 W |
| Plasma gas flow rate (Ar) | 17 L min−1 | 17 L min−1 | 17 L min−1 |
| Auxiliary gas flow rate (Ar) | 1.05 L min−1 | 0.79 L min−1 | 0.8 L min−1 |
| Carrier gas flow rate (He) | 0.95–1.05 L min−1 | — | — |
| Nebulizer gas flow rate (Ar) | 0.6–0.7 L min−1 | — | — |
| Carrier gas flow rate (Ar) | — | 0.68 L min−1 | — |
| Ablation gas flow rate (He) | — | 1.48 L min−1 | — |
| 238U+/232Th+ | 1.25–1.47b | 1.31–1.43b | 0.98–1.10b |
| 248ThO+/232Th+ | 0.49–8.75b% | 0.68–2.44b% | 1.57–5.64b% |
| 137Ba++/137Ba+ | 0.13–0.70% | n.d. | n.d. |
| 156CeO+/140Ce+ | n.d. | n.d. | 1.19–4.71b% |
| CCT focus | 0.75 V | 1 V | 2.5 V |
| CCT entry | −100 to −50 V | −80 V | −100 V |
| CCT Amp | 260–300 V | 275 V | 250 V |
| CCT Amp Offset | 0–1.5 V | 1 V | 0 V |
| CCT bias | −1–2 V | 0.5 V | −2 V |
| CCT exit | −250 to −200 V | −250 V | −250 V |
| He gas flow rate | 0–10 mL min−1 | 0–3.5 mL min−1 | 0–8.5 mL min−1 |
| H2 gas flow rate | 0–10 mL min−1 | 0–10 mL min−1 | — |
| Multi-notch filter amplitude at m/z 40 | 0–0.6 V | 0–0.6 V | 0.7 V |
Laser ablation
LA was carried out on reference materials NIST SRM 610, NIST SRM 612 and USGS BCR-2G using a 193 nm ArF excimer laser ablation system (GeoLas C, Lambda Physik, Goettingen, Germany). High-dispersion LA was performed in a helium-filled, single-volume, cylindrical ablation chamber.59 The experiments were carried out with a 44 μm diameter circular spot and a laser repetition rate of 10 Hz (single-pulse signal duration of 1.5–2 s (FW0.01M)). Low-dispersion LA was carried out in a two-volume, large-format tube cell,60,61 using a 5 μm diameter circular laser spot and a laser repetition rate of 100 Hz (single-pulse signal duration of <10 ms (FW0.01M)). All experiments were executed in line-scanning mode with a scan speed of 5 μm s−1 (high-dispersion) or 50 μm s−1 (low-dispersion). A detailed description of the TOFMS data acquisition protocol, and data post-processing routines are reported elsewhere.58 Operating parameters were adjusted on a daily basis to obtain highest 238U+ sensitivity while maintaining similar 238U and 232Th sensitivities, and low oxide formation rates. High-dispersion LA-ICP-TOFMS data were acquired with a time resolution of 1 s, while low-dispersion LA-ICP-TOFMS data was acquired at a time resolution of 1 ms. In post-processing, TOF mass spectra were re-calibrated and baseline-subtracted prior to integration of the mass-spectral peaks. Time traces of integrated signal intensities were used for further processing.Discrete microdroplet introduction
A commercially available microdroplet generator (30 μm dispenser head MD-K-150-020 equipped with a control unit MD-E-3001 from Microdrop Technologies GmbH, Germany) was used. Droplets with diameters ranging from 25 to 30 μm (for a given set of parameters, droplet diameter variability is ∼1%) were produced at 50 Hz and transported into the ICP via a flow of helium and argon.28,53,62,63 A multi-element stock solution was prepared from single-element standard solutions (Merck AG, Darmstadt, Germany and Inorganic Ventures, Christiansburg, Virginia, USA). Dilutions were made with a solution of 2% HNO3 in ultra-high purity water (Millipore, Billerica, Massachusetts, USA), and were carried out gravimetrically using an electronic balance (Mettler AE240, Mettler-Toledo, Greifensee, Switzerland). Final concentrations were nominally 100 ng g−1 for each element.Results and discussion
Attenuation of background signal intensities using H2 as reaction gas
This study was carried out after optimization of operating parameters while ablating NIST SRM 610. Fig. 1 reports average ICP-TOFMS signal intensities recorded when no sample was introduced into the plasma (gas-blank signal intensities). The experiments were performed using the high-dispersion LA-ICP-TOFMS setup with different H2 gas flow rates through the reaction cell. In all three experiments, operating parameters of the ICP-TOFMS and collision/reaction cell settings (Table 1.) were kept constant, apart from the H2 gas flow rate and the multi-notch filter, which were adjusted to allow for highest intensities while still preventing detector saturation. Optimum notch filter settings were determined for each experiment separately following the routine described in the experimental section. Signal intensities at m/z 40 had to be attenuated when no H2 gas flow was applied. When H2 gas flow was increased to 1.5 mL min−1 and above, no signal attenuation was required. Even though complete elemental mass spectra were acquired, signal intensities are only presented for m/z range ≤100 to highlight the most pronounced background signals. However, background signal intensities across the whole elemental mass range have been studied for different H2 gas flow rates. A table reporting these results is provided in the ESI (Table A1†). | ||
| Fig. 1 Average signal intensities recorded for m/z range ≤100 when analyzing gas blank as a function of H2 gas flow rate through the reaction cell. Identical spectra are plotted on a linear y-axis (a) and a logarithmic y-axis (b). The multi-notch filter was used to attenuate signal intensity at m/z 40 when no H2 gas flow through the reaction cell was applied. No signal attenuation was required when H2 gas flow rates through the reaction cell were 2.5 mL min−1 and 5 mL min−1, respectively. | ||
Gas-blank analysis provides a measure for instrumental background, but does not necessarily represent background signal intensities observable at the time a given sample is analyzed. This is because the sample matrix can elevate the baseline. Nonetheless, gas-blank measurements are often used to estimate background signal intensities in LA experiments and this strategy was considered appropriate for the purpose of this work.64 In addition to high sensitivities, low background signal intensities are critical for low LODs.64–66 Here, when applying standard conditions (no H2 gas flow), most pronounced background signals are produced by Ar+ ions and argon-based molecular ions such as Ar2+, ArN+ and ArO+. Additionally, H2O+, N2+ and O2+ also have intense peaks. Due to moderate abundance sensitivity of the ICP-TOFMS, these background ions elevate the mass-spectral baseline on adjacent masses.
By increasing the amount of H2 in the CCT, Ar+ signals could be attenuated to intensity levels in the range of several hundreds of counts per second. Ar2+ signals could be suppressed by more than four orders of magnitude and reach single digit counts per second levels when a H2 gas flow rate of 5 mL min−1 is used. Reactions involved in the removal of these species include H atom transfer.67 The transformation of Ar+ to ArH+ causes the signals at m/z 37 and m/z 41 to dominate the spectrum acquired, for example when using a H2 gas flow rate of 2.5 mL min−1. At higher H2 gas flow rates, ArH+ is increasingly lost through proton transfer.67
Analyte signals as a function of H2 and He gas flow rate
Fig. 2a and c report sensitivities for selected isotopes as a function of H2 and He gas flow rates through the CCT cell, respectively. Reported isotopes (27Al, 55Mn, 89Y, 141Pr, 238U) were selected to cover a large proportion of the elemental m/z range. Data were acquired with the high-dispersion LA-ICP-TOFMS setup. In both cases, sensitivities follow the same general trend: an initial increase in sensitivity is observed with increasing gas flow rates before sensitivities start to decrease. Here we found that for both gases tested, 55Mn sensitivities were highest at a flow rate of 1 mL min−1. With respect to the sensitivities observed under standard conditions (no gas flow), they were increased by 28% (H2) and 84% (He). When pressurizing the reaction cell with H2, 27Al and 238U sensitivities were highest when the gas flow rates were 0.5 mL min−1 and 1.5 mL min−1, respectively. In contrast to sensitivities obtained with an unpressurized cell, they were enhanced by 11% (27Al) and 2% (238U). When using He, 27Al and 238U sensitivities maximized at gas flow rates of 0.5 mL min−1 and 3.5 mL min−1. The sensitivities were increased by 3% (27Al) and 73% (238U). These increases in sensitivities are attributed to collisional focusing.54m/z-dependent sensitivity decrease at higher gas flow rates indicates the relevance of scattering processes. Gas flow rates at which scattering processes become dominant are dependent on the type of cell gas and differ for low-mass and high-mass elements because low-mass ions are slowed down faster in scattering processes. | ||
| Fig. 2 Sensitivities and selected intensity ratios as a function of H2 gas flow rate through the reaction cell (a). Limits of detection calculated for different calcium isotopes as a function of H2 gas flow rate through the reaction cell (b). For every H2 gas flow setting, voltages on the multi-notch filter were adjusted to maintain highest possible sensitivities while still preventing detector saturation. For H2 gas flow rates ≥1.5 mL H2 min−1, no signal attenuation was applied. Sensitivities and selected intensity ratios as a function of He gas flow rate through the collision cell (c). Mass resolving power and abundance sensitivity as a function of He gas flow rate through the collision cell (d). Multi-notch filter settings were held constant during these experiments. Signal intensity at m/z 40 had to be attenuated at all times. All experiments were carried out on NIST SRM 610 using a 44 μm diameter circular spot and a laser repetition rate of 10 Hz. Experiments were performed in line-scanning mode applying a scan speed of 5 μm s−1. | ||
238U+/232Th+ intensity ratios increased steadily with increasing gas flow rates. Increases from 1.25 to 1.36 and from 1.31 to 1.47 were observed in experiments with H2 and He, respectively. Without a pressurized cell the 238U+/232Th+ intensity ratio is considered an indicator for particle evaporation and dissociation, similar to 232Th16O+/232Th+ intensity ratios. Working with a pressurized cell and keeping laser and plasma settings constant causes the 238U+/232Th+ intensity ratio to increase in parallel with flow rate. This observation would indicate that Th+ is removed at a higher rate from the ion beam population, by either reactions with impurities in the gas or through scattering. The latter is not too likely however due to the fact that U and Th collision cross sections and axial energies are deemed very similar. Reactions with residual water on the other hand are a likely candidate causing the abundance of atomic Th+ in the CCT to decrease faster than U+. This is however only in part supported by the evolution of the 232Th16O+/232Th+ intensity ratios. Upon pressurizing the cell, the ThO+ abundance first rises sharply, followed by an intermediate minimum and a continuous increase after a flow rate near 4 mL min−1 (H2) or 1.5 mL min−1 (He). The reason for the intermediate minimum in 232Th16O+/232Th+ intensity ratios is not clear at this stage. Similar trends were observed for the abundances of ThOH+ species. In both experiments, the 238U16O+/238U+ and the 238U16O1H+/238U+ intensity ratios showed comparable behavior. The trend in the 238U+/232Th+ intensity ratio suggests that Th+ is continuously removed from the spectra as the flow rate increases. This would indicate that initially only a small fraction of Th+ is sufficiently thermalized to efficiently react to ThO+. Under these conditions the increase in pressure first leads to higher scattering losses of the molecular ions, causing an intermediate decay of their transmission. At sufficiently high pressures, the majority of Th+ may become sufficiently thermalized to react to ThO+ and their production overcompensates scattering losses.
Abundance ratio of doubly charged and singly charged species was determined by monitoring signal intensities from 137Ba++ and 137Ba+ at m/z 68.5 and m/z 137, respectively. For both gases, 137Ba++/137Ba+ intensity ratios were following the same general trend: an initial increase is observed with increasing gas flow rates before the relative abundance of the doubly charged ion starts to decrease. In experiments with H2, doubly charged intensity ratios were increasing from 0.46% (no H2 gas flow) to 0.70% (3.5 mL min−1 H2). In experiments with He the doubly charged formation rate was increasing from 0.33% (no He gas flow) to 0.67% (1.5 mL min−1). The intermediate maximum of the 137Ba++/137Ba+ intensity ratios observed with both gases indicates that the transmission of Ba++ initially increases as the cell is pressurized. This could be explained by the higher kinetic energy the doubly charged ions have gained in the electrostatic ion optics before entering the CCT. After sufficient thermalization however, reactions rates of Ba++ need to be expected to exceed those of the singly charged Ba+, explaining the continuous decay of their ion signal intensities at elevated gas flow.
Limits of detection as a function of H2 gas flow rate and isotope selection
Fig. 2b reports limits of detection achievable for various Ca isotopes as a function of H2 gas flow rate through the reaction cell. Lowest limits of detection were obtained for 40Ca with a H2 gas flow rate of 3 mL min−1. LOD is 0.33 mg kg−1, which is more than one order of magnitude better than limits of detection achievable with other Ca isotopes in CCT mode. In comparison to LODs achievable under standard conditions (no H2 gas flow), improvements by more than three orders of magnitude are observed. These improvements in LOD are a consequence of selective attenuation of Ar+ signals with H2, allowing to avoid operation of the notch filter attenuation near m/z 40. Increasing LODs with higher H2 gas flow rates are a result of decreasing sensitivities (Fig. 2a). Additionally, LODs for various Se isotopes were investigated as a function of H2 gas flow rate and a similar trend was observed. Lowest LOD was obtained for 80Se+ with a H2 gas flow rate of 3.5 mL min−1. LOD is 0.95 mg kg−1. This corresponds to a four-fold improvement when compared to the lowest LOD obtainable under standard conditions (77Se, LOD = 4.1 mg kg−1). Detailed results are reported in the ESI (Fig. A1b†). Moreover, LODs as a function of H2 gas flow rate have been studied for selected isotopes across the entire elemental m/z range. Results are summarized in Fig. A5† and show that the benefit of H2 gas flow is not restricted to isotopes that interfere with background species. For example, a four-fold improvement in LOD was observed for 238U and 89Y at H2 gas flow rates of 5 mL min−1 and 3.5 mL min−1, respectively. LODs for 27Al were lowest without the application of an H2 gas flow. This most probably results from collision-induced signal attenuation even at low H2 gas flow rates. The LOD for 27Al was worse by a factor of two when an H2 gas flow rate of 3.5 mL min−1 was applied. The influence of H2 gas flow rates on analytical sensitivities and background intensities as well as their combined effect on LODs are provided in Fig. A2 in the ESI.†Mass resolving power and abundance sensitivity as a function of He gas flow rate
Fig. 2d reports mass resolving power (MRP) for selected isotopes as a function of He gas flow rate through the collision cell. MRP is defined as m/Δm where m represents the mass of the isotope considered and Δm is defined as the full peak width at 50% of its height (FWHM). In this study, we found that MRP can be slightly improved when He is added as collision gas. For example, mass resolving power of 141Pr and 238U are enhanced by 16% and 13% at 5 and 6 mL min−1 He, respectively. This improvement is due to the collision-mediated reduction of the kinetic energy spread of the ions. The same effect is also observed in experiments with H2. The MRP of 238U is enhanced by 4% with a H2 gas flow rate of 2.5 mL min−1.An experiment in which ICP-TOFMS operating parameters and collision/reaction cell settings were optimized prioritizing peak shape over sensitivity showed that mass resolving power for 238U can exceed 4000. In comparison to results reported here, 238U sensitivities were decreased by 7%.
Abundance sensitivity68 was determined by monitoring 209Bi+ intensities at m/z 209 and m/z 210. It was found that it can be increased through application of He gas flow rates ≤3 mL min−1. Abundance sensitivity maximizes before sensitivity and mass resolving power for 209Bi because increasing He gas causes broadening at the base of mass spectral peaks on the heavy mass side. This effect is not well represented in the determination of MRP.
Quantification capabilities for Ca as a function of H2 gas flow rate and isotope selection
Fig. 3a and b report quantitative results obtained for Ca determination in NIST SRM 612 and USGS BCR-2G, respectively. The experiments were carried out with the high-dispersion LA-ICP-TOFMS setup and results are displayed as a function of H2 gas flow rate through the reaction cell and isotope selection. In both cases, NIST SRM 610 and 29Si+ were selected as external reference material and internal standard, respectively. Due to highly similar matrix composition of NIST SRM 610 and NIST SRM 612, external calibration with NIST SRM 610 is considered matrix-matched. For quantification of USGS BCR-2G, external calibration with NIST SRM 610 is considered non-matrix-matched. | ||
| Fig. 3 Quantified Calcium concentration in NIST SRM 612 for different H2 gas flow rates through the reaction cell and isotope selection (a). Quantified calcium concentration in USGS BCR-2G for different H2 gas flow rates through the reaction cell and isotope selection (b). The error bars represent the standard deviation calculated from three individual measurements in both cases. The solid red lines indicate the reference value for calcium concentration in NIST SRM 612 and USGS BCR-2G. The dashed red lines limit the uncertainty range associated with these reference values. The dashed blue lines mark 5% deviation from the reference concentration. Experiments were carried out using a 44 μm diameter circular laser spot and a laser repetition rate of 10 Hz. They were performed in line-scanning mode with a scan speed of 5 μm s−1. In both cases, NIST SRM 610 and 29Si were selected as external reference material and internal standard, respectively. In both experiments, 40Ca+, 42Ca+ and 43Ca+ intensities were not detectable above background levels without application of a H2 gas flow. | ||
Under standard conditions (no H2 gas flow), Ca concentration in NIST SRM 612 and USGS BCR-2G could only be determined from 44Ca+ intensities. 40Ca+, 42Ca+ and 43Ca+ signals were not detected above background levels.
Independent of isotope selection, accuracy and precision of Ca determination in NIST SRM 612 follow a similar trend when plotted against H2 gas flow rate. For H2 gas flow rates ≤2.5 mL min−1, accuracy and precision of quantification are improved. At higher H2 gas flow rates opposite behavior is observed. This is attributed to pronounced collision-induced signal loss at higher H2 gas pressure inside the reaction cell. We found that a H2 gas flow rate of 2.5 mL min−1 allowed most accurate quantification. Ca concentration determined from 40Ca intensities deviated by 1.3% from the reference value reported in the GeoReM database.69 For experiments carried out on USGS BCR-2G we found that application of small H2 gas flow rates also leads to improved accuracy and precision of quantified results. While concentrations determined from 42Ca, 43Ca and 44Ca signal intensities showed best agreement with reference values in experiments that used a H2 gas flow rate of 1.5 mL min−1, most accurate quantification based on 40Ca signal intensities was observed when a H2 gas flow rate of 5 mL min−1 was applied. The deviation from the reference value was 3.5% in that case. An interference of Ca+ intensities with MgO+, MgOH+, AlO+ and AlOH+ is likely and could explain the overestimation of Ca concentration in USGS BCR-2G. These interferences are most pronouncedly affecting concentrations determined based on intensities of low abundant isotopes 42Ca+, 43Ca+ and 44Ca+ and are considered to become more dominant with increasing H2 gas flow rate. At the moment it is not clear why Ca determinations based on 40Ca+ intensities are more accurate at higher H2 gas flow rates. The difference in the Ca+/Ar+ ratio prevailing in experiments carried out with NIST SRM glasses and USGS BCR-2G as well as reduced tailing of K+ signals at higher H2 gas flow rates are possible causes. In both samples, access to 40Ca intensities provides an improvement in LOD by more than three orders of magnitude (Fig. 2b). Quantification of Se in NIST SRM 612 was also investigated as a function of H2 gas flow rate and isotope selection. Results are reported in the ESI (Fig. A1a†).
Furthermore, quantification capabilities have been tested for a broad selection of elements in NIST SRM 612 and USGS BCR-2G when a H2 gas flow rate of 2.5 mL min−1 was applied. This H2 gas flow rate has been selected because it represents a compromise between background attenuation and collision induced signal loss. For most elements, we found no pronounced difference between quantification capabilities observable under standard conditions (no H2 gas flow) and with a H2 gas flow rate of 2.5 mL min−1 (Fig. A3 and A4†). Concentrations determined in experiments with an unpressurized and a pressurized cell were both in good agreement with reference values. For 43% (no H2 gas flow) and 36% (2.5 mL min−1 H2) of the elements tested, the determined concentrations were within the uncertainties of the NIST SRM 612 preferred values. In both experiments, 70% of the elements under investigation showed relative deviations from NIST SRM 612 preferred values that were smaller than 5%. For 62% (no H2 gas flow) and 69% (2.5 mL min−1 H2) of the elements considered, the determined concentrations were within the uncertainties of the USGS BCR-2G preferred values. In both experiments, 62% of the elements showed relative deviations from USGS BCR-2G preferred values that were smaller than 5%. However, for certain elements we observed less accurate quantification as a function of H2 gas flow rate (Fig. A6†). Specifically, P, K and Sc quantifications were less accurate when the reaction cell mode with H2 was used. This behavior was observed in experiments carried out on both standard reference materials. Spectra recorded in context of these experiments show that 31P, 39K and 45Sc signal intensities are pronouncedly affected by interferences. Based on spectral fitting, the interferences at m/z 31, m/z 39 and m/z 45 were as assigned to hydride- and hydroxide species, namely 30Si1H, 38Ar1H and 28Si16O1H. For the three isotopes discussed here, we found that impact of interfering species is more pronounced the higher the H2 gas flow rate.
Analyte signals as a function of total gas flow rate and volumetric fraction of H2 in H2/He gas mixtures
Fig. 4 reports normalized sensitivities as a function of the volumetric fraction of H2 in H2/He gas mixtures. Data is presented for selected isotopes and two experiments in which the total gas flow rate through the collision/reaction cell was held constant at 1 mL min−1 and 3 mL min−1, respectively. Normalized sensitivities were calculated by dividing sensitivities observed when applying a given set of instrumental conditions by sensitivities observed under standard conditions (no gas flow in the CCT).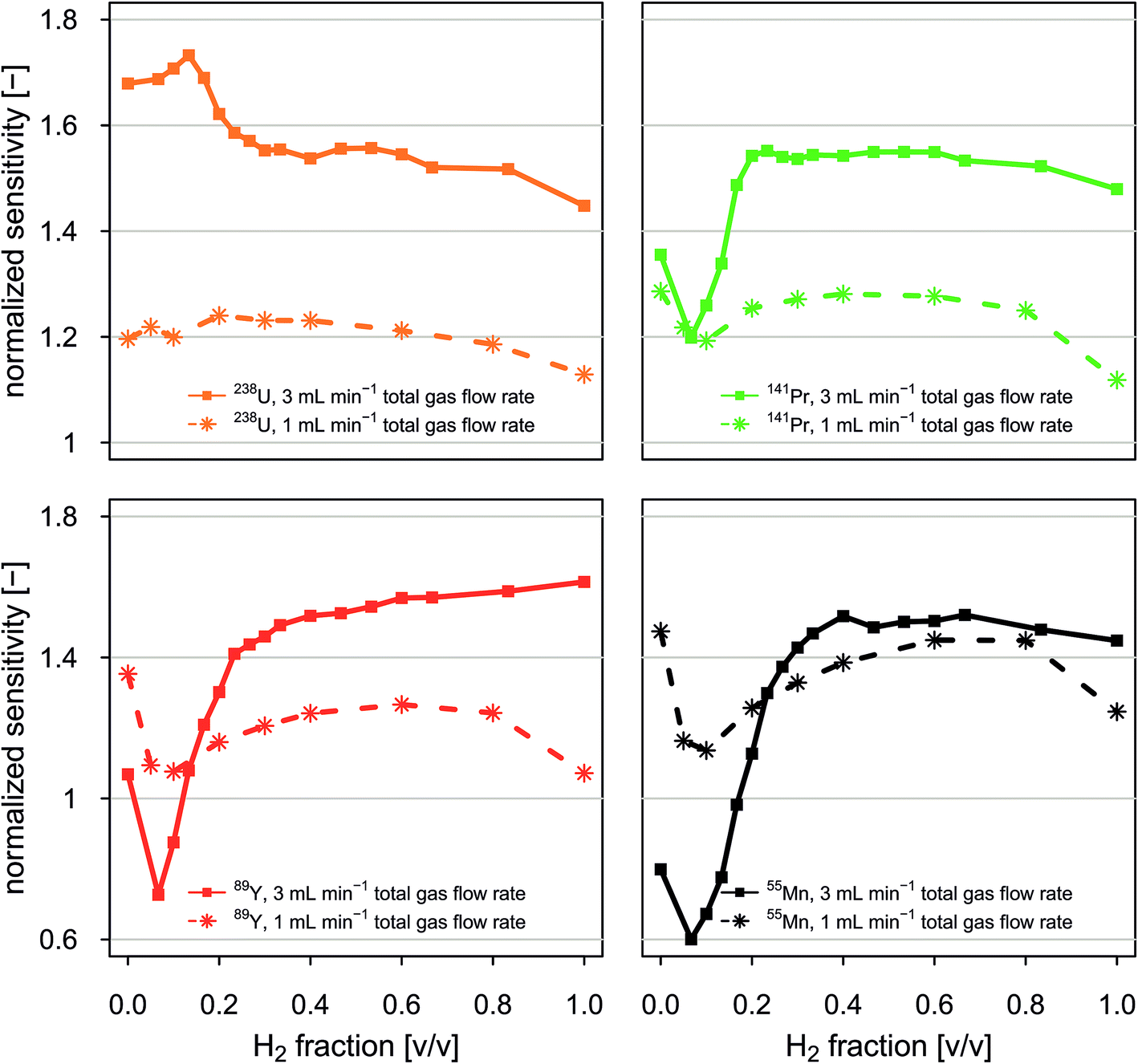 | ||
| Fig. 4 Normalized sensitivities for selected isotopes as a function of volumetric fraction of H2 in H2/He gas mixtures. Data is reported for two experiments in which the total gas flow rate through the collision/reaction cell was held constant at 1 mL min−1 and 3 mL min−1, respectively. Sensitivities are normalized to the sensitivities observable when experiments are carried out without application of collision/reaction cell technology. All experiments were carried out on NIST SRM 610 using a 44 μm diameter circular spot and a laser repetition rate of 10 Hz. Experiments were performed in line-scanning mode applying a scan speed of 5 μm s−1. Multi-notch filter settings were kept constant. | ||
Our data shows that normalized sensitivities depend on both the total gas flow rate and the volumetric mixing ratio between H2 and He. For the total gas flow rates investigated, data recorded for low- to medium-mass elements such as 55Mn, 89Y and 141Pr shows qualitatively similar dependence on volumetric mixing ratio between H2 and He. Sensitivities of high-mass elements such as 238U show a different behavior. We found that for the total gas flow rates tested, susceptibility of sensitivities to changes in composition of the gas mixture is higher for low-mass elements than for high-mass elements. For example, in an experiment with a constant gas flow rate of 3 mL min−1, normalized 238U sensitivities varied by 28%, while normalized 55Mn sensitivities varied by 92%.
Combination of total gas flow rate and volumetric fraction of H2 in a H2/He gas mixture that allows highest sensitivities is different for different elements. Here we found that normalized 238U sensitivities are highest when a total gas flow rate of 3 mL min−1 is applied and the H2 fraction is 13%. Normalized 141Pr sensitivities were maximum when a total gas flow rate of 3 mL min−1 was supplied and the H2 fraction of the gas mixture was in the range from 20% to 60%. Normalized 55Mn sensitivities were highest when a total gas flow rate of 3 mL min−1 was used. The H2 fraction was optimum in the range from 40% to 65%.
Admixing small amounts of He gas to a H2 gas flow is advantageous because it leads to a signal enhancement through collisional cooling while H2 suppresses various major background species. Moreover, attenuation of Ar-based species with H2 is more effective when the ions are thermalized. Fig. A7† shows that signal to background ratios can be enhanced through admixture of small amounts of He gas to a constant H2 gas flow rate of 1.5 mL min−1. This effect is most pronouncedly visible for isotopes that are interfered with Ar based species. For example, the signal to background ratios for 40Ca+ and 80Se+ are doubled and improved by a factor of five when He gas is admixed at flow rates of 0.5 and 2 mL min−1, respectively. At the same time, signal to background ratios for 44Ca+ and 82Se+ are improved by a factor of about 1.5.
Depending on the analytical question, optimum conditions have to be defined separately. For example, if maximum sensitivity for selected elements is critical, gas mixtures and total gas flow rates should be adjusted such that highest sensitivities for these isotopes can be achieved. Applying H2 gas flow rates ≥1.5 mL min−1 is interesting for experiments in which low LODs for elements such as K, Ca or Se are required. At H2 gas flow rates ≥1.5 mL min−1 no notch-filter attenuation of signal intensities at m/z 40 is required, which improves transmission of ions with m/z close to 40 such as 39K and 41K. However, this minimum H2 gas flow rate has been inferred from experiments carried out with H2 only. If H2 is used in combination with He, the minimum H2 gas flow rate needed to achieve sufficient attenuation of the Ar+ signals is expected to be lower, but has not been determined here.
Influence of collision/reaction gas flow rate on temporal structure of signals produced by discrete sample introduction
In this study, microdroplets are used as an alternative sample introduction system for ICPMS and result in discrete ICPMS signals of about 300 μs in duration. Thanks to the narrow size distribution of the droplets, the time required to undergo complete desolvation, atomization, excitation and ionization is highly reproducible from one droplet to another. This makes this discrete sample introduction system an ideal tool for investigating fundamental ICP processes.70 Here, we use microdroplets to study the effects of CCT conditions on signal structure and duration.Isotopic signals from single microdroplets containing a 100 ng g−1 multi-element solution were detected with a time resolution of 30.3 μs. Averaged signal intensities were calculated from signal intensities of 2000 individual microdroplets (Fig. 5). Slight shifts in the arrival time of the isotopes can be observed. However, the order of the observed shifts is not dictated by refractory or volatile properties of the elements,71 but by their masses. The time scale of these shifts is in the μs range as can be seen from Table 2. The observation that temporal shifts occur, illustrates that ions with different m/z travel at different speeds after they have been extracted from the plasma.
 | ||
| Fig. 5 Averaged transient signal intensities calculated from signal intensities of 2000 individually detected microdroplets of a 100 ng g−1 multi-element solution (a). Data was recorded with 30.3 μs time resolution and an unpressurized cell. Dashed lines mark the time at which different isotopic intensities maximize. Isotopic signal intensities are normalized to their maximum value for better visibility of temporal shifts (b). | ||
| Isotope | 27Al+ | 48Ti+ | 64Zn+ | 89Y+ | 109Ag+ | 115In+ | 139La+ | 140Ce+ | 184W+ | 232Th+ | 238U+ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Shift [μs] | 0 | 30.3 | 60.6 | 60.6 | 90.9 | 90.9 | 90.9 | 90.9 | 121.2 | 121.2 | 151.5 |
Application of a He gas flow through the collision cell affects structure of signals detected from individual microdroplets. With increasing He gas flow rates, the signals are stretched. For example, the 238U+ transient signal width is extended from 120 μs to 600 μs when the He gas flow rate is increased from 2.5 to 8.5 mL min−1. This observation can be explained with the increase in time it takes the ions to reach the TOF extraction region. Even though the ions' kinetic energy distribution is reduced when they exit the pressurized cell, their temporal dispersion is increased once they reach the TOF extractor. This is because the speed they are travelling with is reduced when compared to experiments carried out with an unpressurized cell. Furthermore, the sequence of shifts in arrival time to the TOF analyzer changes as a function of He gas flow rate. Indeed, with increasing flow of He through the CCT, a pronounced cooling effect takes place, which affects the light mass elements to a greater extent than the heavier ones. By gradually increasing the He flow, one can observe that the light mass ions are more slowed down, thereby the shifts are gradually reduced until all shifts are eliminated (He gas flow rate of 4 mL min−1). By increasing the He flow above 4 mL min−1, the light ions are slowed so much that the m/z-dependent shift sequence is reversed. In Fig. 6, three He gas flow rates were selected to showcase these three cases; the temporal shifts can first be observed with a He gas flow rate <4 mL min−1, then a He gas flow rate of 4 mL min−1 can be used to eliminate temporal shifts and finally a He gas flow rate of ≥6.5 mL min−1 causes the sequence of shifts to be reversed. In order to make use of the high time resolution of the icpTOF, the data presented in this section was acquired with the trigger option, where a fixed number of 31 spectra can be continuously read out. Hence, due to peak-tailing at higher He gas flow rates, data reported in Fig. 6 had to be acquired with a time resolution of 60.6 μs. Consequently, at half the time resolution as in experiments reported in Fig. 5, some of the shifts disappeared, as they are not resolved anymore, i.e.140Ce+ and 238U+ appear now simultaneously.
 | ||
| Fig. 6 Signal duration and shifts in arrival times of ions as a function of He gas flow rate through the collision cell. Data is presented for selected isotopes and He gas flow rates of 2.5 mL min−1 (a), 4 mL min−1 (b) and 6.5 mL min−1 (c). Signal intensities are normalized to their maximum value. Data was recorded with 60.6 μs time resolution. Dashed lines indicate the time at which different isotopic intensities maximize. Please note the difference in the timescale of the three subplots. | ||
Effects of collision/reaction gas flow rate on signal structure have also been investigated in context of low-dispersion LA experiments. Applicability of collision/reaction cell technology to low-dispersion LA-ICP-TOFMS imaging has been evaluated. No pronounced signal broadening was observed when low-dispersion LA was carried out with various flow rates of H2 and He (Fig. A8†). Transient signal width of less than 10 ms (FW0.01M) was achievable from 5 μm diameter circular laser spots. With 1 ms time resolution, no shifts between intensity maxima of different isotopes were detectable. This is expected because ablated aerosol transients are comparably long and changes in signal structure that occur on a timescale of tens to hundreds of μs are inconsequential in experiments that are performed with 1 ms time resolution.
Conclusion
In this work, we evaluate capabilities of ICP-TOFMS in combination with collision/reaction cell technology. The data presented were acquired using various sample introduction schemes including high- and low-dispersion LA and microdroplet generation. Specifically, we have studied the effects of H2 as reaction gas and He as collision gas on LODs, quantification capability and signal structure.We found that application of small flow rates of H2, He or mixtures of these gases improve sensitivity for intermediate and high m/z. The magnitude of this collisional focusing effect and the gas flow rate at which it maximizes depends on the m/z of the analyte. In this study, we observed sensitivity enhancements by a factor of 1.5 to 2 across a large proportion of the elemental m/z range.
H2 gas can be used to selectively suppress background species such as Ar+ and Ar2+ to intensity levels of several hundreds and single digit counts per second, respectively. Addition of small amounts of He increases the collisional cooling and can enhance the efficiency of H2-mediated attenuation of Ar+ and Ar2+ signals.
Access to most abundant isotopes of Ca and Se improves LODs for these elements by three orders of magnitude and a factor of four, respectively. Overall, when using H2 in the CCT, an up to four-fold improvement in LODs was also observed for various other isotopes. For most elements considered, accuracy of quantification was shown to be unaffected by the use of collision/reaction gases. However, due to formation of hydride-, oxide and hydroxide molecular ions, quantification for P, K and Sc was worse when the experiments were carried out in reaction gas mode with H2.
Spectral acquisition frequency available in TOFMS allows investigation of effects induced by application of collision- and/or reaction gases with a time resolution of 30.3 μs. From experiments with microdroplets, we determined m/z-dependent shifts in the arrival time of ions at the TOF extraction region. Through collisional cooling, these shifts could be eliminated at a He gas flow rate of 4 mL min−1 or reversed at higher He gas flow rates. Signal broadening and shifts occurred on the tens to hundreds of μs time scale. These changes in signal structure are inconsequential in low-dispersion LA experiments because the ablated aerosol transient is comparatively long.
A H2 gas flow rate of ≥1.5 mL min−1 allows operation of the instrument without application of ion attenuation with the notch filter. In this respect an instrument design without multi-notch filter but a permanent reaction gas mode with H2 seems promising.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge support from Dr Olga Borovinskaya, Dr Martin Tanner and Dr Mike Cubison from TOFWERK AG. Roland Mäder and the ETH mechanical workshop are gratefully acknowledged for custom manufacturing the adaptors for the microdroplet- and laser ablation sample introduction systems. Research leading to these results has received funding from ETH Zürich and the SNF project no. 200021 162870/1. Dr Alexander Gundlach-Graham likes to acknowledge financial support through the Marie-Curie International Incoming Fellowship from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 624280.References
- R. S. Houk, V. A. Fassel, G. D. Flesch, H. J. Svec, A. L. Gray and C. E. Taylor, Anal. Chem., 1980, 52, 2283–2289 CrossRef CAS
.
- G. A. Jenner, H. P. Longerich, S. E. Jackson and B. J. Fryer, Chem. Geol., 1990, 83, 133–148 CrossRef CAS
- S. M. Eggins, J. D. Woodhead, L. P. J. Kinsley, G. E. Mortimer, P. Sylvester, M. T. McCulloch, J. M. Hergt and M. R. Handler, Chem. Geol., 1997, 134, 311–326 CrossRef CAS
- Y. Amelin, D.-C. Lee, A. N. Halliday and R. T. Pidgeon, Nature, 1999, 399, 252–255 CrossRef CAS
- A. Gerdes and A. Zeh, Earth Planet. Sci. Lett., 2006, 249, 47–61 CrossRef CAS
- J. Košler, H. Fonneland, P. Sylvester, M. Tubrett and R.-B. Pedersen, Chem. Geol., 2002, 182, 605–618 CrossRef
- A. Audetat, D. Günther and C. A. Heinrich, Science, 1998, 279, 2091–2094 CrossRef CAS PubMed
- C. A. Heinrich, T. Pettke, W. E. Halter, M. Aigner-Torres, A. Audétat, D. Günther, B. Hattendorf, D. Bleiner, M. Guillong and I. Horn, Geochim. Cosmochim. Acta, 2003, 67, 3473–3497 CrossRef CAS
- D. Rubatto, Chem. Geol., 2002, 184, 123–138 CrossRef CAS
- D. Tabersky, K. Nishiguchi, K. Utani, M. Ohata, R. Dietiker, M. B. Fricker, I. M. de Maddalena, J. Koch and D. Günther, J. Anal. At. Spectrom., 2013, 28, 831–842 RSC
- D. Pröfrock and A. Prange, Appl. Spectrosc., 2012, 66, 843–868 CrossRef PubMed
-
R. Cornelis, J. A. Caruso, H. Crews and K. G. Heumann, Handbook of elemental speciation II: species in the environment, food, medicine and occupational health, John Wiley & Sons, 2005 Search PubMed
- A. Praetorius, A. Gundlach-Graham, E. Goldberg, W. Fabienke, J. Navratilova, A. Gondikas, R. Kaegi, D. Günther, T. Hofmann and F. von der Kammer, Environ. Sci.: Nano, 2017, 4, 307–314 RSC
- J. W. Olesik and P. J. Gray, J. Anal. At. Spectrom., 2012, 27, 1143–1155 RSC
- A. Ulrich, C. Moor, H. Vonmont, H.-R. Jordi and M. Lory, Anal. Bioanal. Chem., 2004, 378, 1059–1068 CrossRef CAS PubMed
- B. L. Batista, J. L. Rodrigues, V. C. de Oliveira Souza and F. Barbosa, Forensic Sci. Int., 2009, 192, 88–93 CrossRef PubMed
- Y. Suzuki, R. Sugita, S. Suzuki and Y. Marumo, Anal. Sci., 2000, 16, 1195–1198 CrossRef CAS
- M. Resano, E. García-Ruiz and F. Vanhaecke, Mass Spectrom. Rev., 2010, 29, 55–78 CAS
-
R. J. Speakman and H. Neff, Laser ablation ICP-MS in archaeological research, UNM Press, Albuquerque, 2005 Search PubMed
-
P. Degryse, in Isotopic Analysis, ed. F. Vanhaecke and P. Degryse, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 373–390 Search PubMed
- M. Burger, R. Glaus, V. Hubert, S. van Willigen, M. Wörle-Soares, F. Convertini, P. Lefranc, E. Nielsen and D. Günther, J. Archaeol. Sci., 2017, 82, 62–71 CrossRef CAS
- D. R. Bandura, V. I. Baranov, O. I. Ornatsky, A. Antonov, R. Kinach, X. Lou, S. Pavlov, S. Vorobiev, J. E. Dick and S. D. Tanner, Anal. Chem., 2009, 81, 6813–6822 CrossRef CAS PubMed
- O. Ornatsky, D. Bandura, V. Baranov, M. Nitz, M. A. Winnik and S. Tanner, J. Immunol. Methods, 2010, 361, 1–20 CrossRef CAS PubMed
- J. Szpunar, R. Lobinski and A. Prange, Appl. Spectrosc., 2003, 57, 102A–112A CrossRef CAS PubMed
- C. Giesen, H. A. O. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schuffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Günther and B. Bodenmiller, Nat. Methods, 2014, 11, 417–422 CrossRef CAS PubMed
- M. Moldovan, E. M. Krupp, A. E. Holliday and O. F. X. Donard, J. Anal. At. Spectrom., 2004, 19, 815–822 RSC
- M. Resano, F. Vanhaecke and M. T. C. de Loos-Vollebregt, J. Anal. At. Spectrom., 2008, 23, 1450–1475 RSC
- S. Gschwind, H. Hagendorfer, D. A. Frick and D. Günther, Anal. Chem., 2013, 85, 5875–5883 CrossRef CAS PubMed
-
B. Hattendorf and D. Günther, in Handbook of Spectroscopy, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 647–698 Search PubMed
- Y. Shao and G. Horlick, Appl. Spectrosc., 1991, 45, 143–147 CrossRef CAS
- F. Vanhaecke, C. Vandecasteele, H. Vanhoe and R. Dams, Mikrochim. Acta, 1992, 108, 41–51 CrossRef CAS
- F. Vanhaecke, R. Dams and C. Vandecastelle, J. Anal. At. Spectrom., 1993, 8, 433–438 RSC
- Z. K. Wang, B. Hattendorf and D. Günther, J. Anal. At. Spectrom., 2006, 21, 1143–1151 RSC
- I. Feldmann, W. Tittes, N. Jakubowski, D. Stuewer and U. Giessmann, J. Anal. At. Spectrom., 1994, 9, 1007–1014 RSC
- N. Jakubowski, L. Moens and F. Vanhaecke, Spectrochim. Acta, Part B, 1998, 53, 1739–1763 CrossRef
- E. R. Denoyer, S. D. Tanner and U. Voellkopf, Spectroscopy, 1999, 14, 43–54 CAS
- N. Yamada, J. Takahashi and K. i. Sakata, J. Anal. At. Spectrom., 2002, 17, 1213–1222 RSC
- J. T. Rowan and R. S. Houk, Appl. Spectrosc., 1989, 43, 976–980 CrossRef CAS
- G. C. Eiden, C. J. Barinaga and D. W. Koppenaal, Rapid Commun. Mass Spectrom., 1997, 11, 37–42 CrossRef CAS
- S. D. Tanner and V. I. Baranov, At. Spectrosc., 1999, 20, 45–46 CAS
-
P. Turner, T. Merren, J. Speakman and C. Haines, in Plasma Source Mass Spectrometry: Development and Applications, ed. G. Holland and S. D. Tanner, Special Publication of the Royal Chemical Society No. 202, Cambridge, 1997, pp. 28–34 Search PubMed
- J. P. Guzowski Jr and G. M. Hieftje, J. Anal. At. Spectrom., 2001, 16, 781–792 RSC
- GBC Scientific Equipment, OptiMass 9600 ICP-oTOFMS, 2017, www.gbcsci.com/wp-content/uploads/2017/02/01-0875-05_Optimass9600.pdf, (accessed 17.08.2018).
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Fresenius. J. Anal. Chem., 2001, 370, 454–470 CrossRef CAS PubMed
- J. T. Rowan and R. S. Houk, Appl. Spectrosc., 1989, 43, 976–980 CrossRef CAS
- G. C. Eiden, C. J. Barinaga and D. W. Koppenaal, J. Anal. At. Spectrom., 1996, 11, 317–322 RSC
- D. R. Bandura, V. I. Baranov and S. D. Tanner, Fresenius. J. Anal. Chem., 2001, 370, 454–470 CrossRef CAS PubMed
- S. D. Fernández, N. Sugishama, J. R. Encinar and A. Sanz-Medel, Anal. Chem., 2012, 84, 5851–5857 CrossRef PubMed
- D. Günther, B. Hattendorf and A. Audétat, J. Anal. At. Spectrom., 2001, 16, 1085–1090 RSC
- D. P. Myers, G. Li, P. P. Mahoney and G. M. Hieftje, J. Am. Soc. Mass Spectrom., 1995, 6, 411–420 CrossRef CAS PubMed
- R. E. Sturgeon, J. W. H. Lam and A. Saint, J. Anal. At. Spectrom., 2000, 15, 607–616 RSC
-
C. A. Flory, S. C. Hansen and C. Myerholtz, Mass selective notch filter with quadrupole excision fields, US Pat., 5,672,870, 1997
- O. Borovinskaya, B. Hattendorf, M. Tanner, S. Gschwind and D. Günther, J. Anal. At. Spectrom., 2013, 28, 226–233 RSC
- D. J. Douglas and J. B. French, J. Am. Soc. Mass Spectrom., 1992, 3, 398–408 CrossRef CAS PubMed
- B. Hattendorf and D. Günther, J. Anal. At. Spectrom., 2004, 19, 600–606 RSC
- L. Hendriks, A. Gundlach-Graham, B. Hattendorf and D. Günther, J. Anal. At. Spectrom., 2017, 32, 548–561 RSC
- M. Tanner and D. Günther, Anal. Chim. Acta, 2009, 633, 19–28 CrossRef CAS PubMed
- M. Burger, G. Schwarz, A. Gundlach-Graham, D. Käser, B. Hattendorf and D. Günther, J. Anal. At. Spectrom., 2017, 32, 1946–1959 RSC
-
B. Hattendorf, PhD thesis, ETH Zürich, 2002
- H. A. O. Wang, D. Grolimund, C. Giesen, C. N. Borca, J. R. H. Shaw-Stewart, B. Bodenmiller and D. Günther, Anal. Chem., 2013, 85, 10107–10116 CrossRef CAS PubMed
- A. Gundlach-Graham, M. Burger, S. Allner, G. Schwarz, H. A. O. Wang, L. Gyr, D. Grolimund, B. Hattendorf and D. Günther, Anal. Chem., 2015, 87, 8250–8258 CrossRef CAS PubMed
- O. Borovinskaya, S. Gschwind, B. Hattendorf, M. Tanner and D. Günther, Anal. Chem., 2014, 86, 8142–8148 CrossRef CAS PubMed
- S. Gschwind, M. d. L. Aja Montes and D. Günther, Anal. Bioanal. Chem., 2015, 407, 4035–4044 CrossRef CAS PubMed
- H. P. Longerich, S. E. Jackson and D. Günther, J. Anal. At. Spectrom., 1996, 11, 899–904 RSC
- L. A. Currie, Anal. Chem., 1968, 40, 586–593 CrossRef CAS
- L. A. Currie, Pure Appl. Chem., 1995, 67, 1699–1723 CAS
- V. G. Anicich, J. Phys. Chem. Ref. Data, 1993, 22, 1469–1569 CrossRef CAS
- K. K. Murray, R. K. Boyd, M. N. Eberlin, G. J. Langley, L. Li and Y. Naito, Pure Appl. Chem., 2013, 85, 1515–1609 CAS
- K. P. Jochum and U. Nohl, Chem. Geol., 2008, 253, 50–53 CrossRef CAS
- J. W. Olesik, Appl. Spectrosc., 1997, 51, 158A–175A CrossRef CAS
- O. Borovinskaya, M. Aghaei, L. Flamigni, B. Hattendorf, M. Tanner, A. Bogaerts and D. Günther, J. Anal. At. Spectrom., 2014, 29, 262–271 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ja00275d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |