
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methylformate from CO2: an integrated process combining catalytic hydrogenation and reactive distillation†
Martin
Scott
a,
Christian G.
Westhues
a,
Teresa
Kaiser
b,
Janine C.
Baums
a,
Andreas
Jupke
b,
Giancarlo
Franciò
 *a and
Walter
Leitner
*ac
*a and
Walter
Leitner
*ac
aInstitut für Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: francio@itmc.rwth-aachen.de; leitner@itmc.rwth-aachen.de
bAVT.FVT—Chair of Fluid Process Engineering, RWTH Aachen University, Forckenbeckstraße 51, 52074 Aachen, Germany
cMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim a. d. Ruhr, Germany
First published on 13th November 2019
Abstract
An integrated two-step process for the production of methylformate (MF) from CO2, H2 and MeOH was developed. In the first step, the hydrogenation of CO2 to a formate-amine adduct is carried out in a biphasic system comprising n-decane as the catalyst phase and MeOH as the product phase. In the second step, the resulting methanol solution containing the formate-amine adduct is subjected to reactive distillation for esterification and isolation of methylformate. The selection of the amine played an important role for devising the overall process. Whereas in the hydrogenation step basic amines work best, medium to low basic amines are preferred in the esterification step. 1,2-Dimethyl-imidazole (1,2-DMI) was identified as an effective compromise for the integration of both steps. In the hydrogenation step, a bis(diphenylphosphino)methane ligand tailored with long alkyl chains ensured effective retention of the Ru-catalyst in the non-polar phase allowing straightforward reuse of the catalyst phase. In a semi-continuous set-up, repetitive hydrogenation (8 cycles) led to a total turnover number (TTON) of 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 at an average turnover frequency (TOF) of 1400 h−1 with a cumulative catalyst leaching of only 1.4 mol% for P and 2.0 mol% for Ru. Reactive distillation was demonstrated in a continuously operated rectification unit leading to the isolation of MF at the head of the column with a purity of 91.5%.
000 at an average turnover frequency (TOF) of 1400 h−1 with a cumulative catalyst leaching of only 1.4 mol% for P and 2.0 mol% for Ru. Reactive distillation was demonstrated in a continuously operated rectification unit leading to the isolation of MF at the head of the column with a purity of 91.5%.
Introduction
The interest in the utilization of CO2 as readily available feedstock is experiencing a growing momentum in both academia and industry.1–6 Carbon dioxide can be “harvested” in high concentration at numerous point sources worldwide facilitating the implementation of carbon capture and utilization (CCU) concepts,7,8 including the use of CO2 as C1-building block for chemical synthesis as an equally attractive and challenging option.9–13 These strategies can contribute to the goal of “defossilization” of the chemical value chain, in particular when combined with energy input from renewable resources.14Catalytic hydrogenation of CO2 using transition metal complexes has evolved as a promising valorization strategy providing a range of differently functionalized products.9,15–18 In a “drop-in” approach, CO2-based products can replace chemical commodities usually produced by the refinement of fossil carbon and eventually lead to a more closed carbon cycle, which is the ultimate goal of a sustainable economy. At the same time, new products or applications may arise with the availability of “green” CO2-based intermediates.
Among the products that can be obtained by homogenously catalyzed CO2 hydrogenation, formic acid (FA) and its derivatives such as methyl formate (MF) have found particular interest for many years.19–22 While most studies focus on FA as the target product, MF provides an attractive alternative.23 In the current fossil-based industry, MF and FA are produced by carbonylation of methanol with CO, both starting materials obtained mainly from natural gas.24 Due to the principle reversibility of the reaction,25 the liquid products have been suggested as easy-to-handle CO surrogates.26,27 MF is the key intermediate for the production of FA, but also a direct source for formyl groups in many other chemical products including formate esters and formamides in commodity and specialty chemicals.5,28 Furthermore, its use as fuel component has been discussed as octane booster for spark ignition engine fuels.29–31
Considering MF as target for CO2 hydrogenation, this offers attractive features for the design of integrated reaction/separation processes. In contrast to the endergonic (ΔG° = 32.9 kJ mol−1) hydrogenation of CO2 to FA, the transformation of CO2, H2 and MeOH into MF and water is exergonic (ΔG° = −5.28 kJ mol−1) under standard conditions.20 Whereas the isolation of pure FA from the stabilizing reaction media is consequently one of the major challenges in process schemes for CO2-based FA production,20,32 the moderate polarity (ε = 8.9 at 25 °C)33 and low boiling point (T = 31.7 °C at 101.3 kPa) of MF make its isolation appear more straightforward. While the ester MF therefore offers favorable thermodynamics for the overall energy balance and potential benefits in the downstream processing, esterification imposes yet another kinetic barrier to the molecular transformation.
A very recent life cycle assessment indicates that the generation of MF from CO2 would reduce the global warming impact by 16% as compared with the fossil-based production even when using raw materials and energy from fossil sources.34 Depending on the concentration of CO2 in the feed, it is advantageous to integrate the carbon capture reference process Rectisol® (CO2 adsorption in cold methanol)35 with the carbon utilization step (up to 46% saving of the electricity demand) realizing an Integrated Carbon Capture und Utilization process (ICCU). Mandatory conditions to achieve the integration is on the one side the use of the same solvent in both processes, i.e. methanol, and on the other side to develop an efficient immobilized catalyst system.34
In the present paper, we address and fulfill these preconditions for the realization of an ICCU process to produce methylformate (MF) from CO2 and H2. We demonstrate that combining catalytic hydrogenation of CO2 with reactive distillation provides a strategy to effectively balance these seemingly contradicting requirements in a fully integrated two-step process for production of MF from CO2, H2 and MeOH. The process design depicted in Fig. 1 comprises as first step the transition metal complex-catalyzed hydrogenation of CO2 in a biphasic reaction system to generate high concentrations of an [FA·amine] adduct in methanol as the product phase. This solution is subjected directly to the second step of reactive distillation inducing the esterification to methyl formate (MF), which is isolated as the distillate at the column head while excess methanol and amine are recycled back at the bottom to the reactor. Water, the by-product of the esterification, accumulates at the bottom of the distillation column and is removed from the recycling stream when reaching a critical concentration.
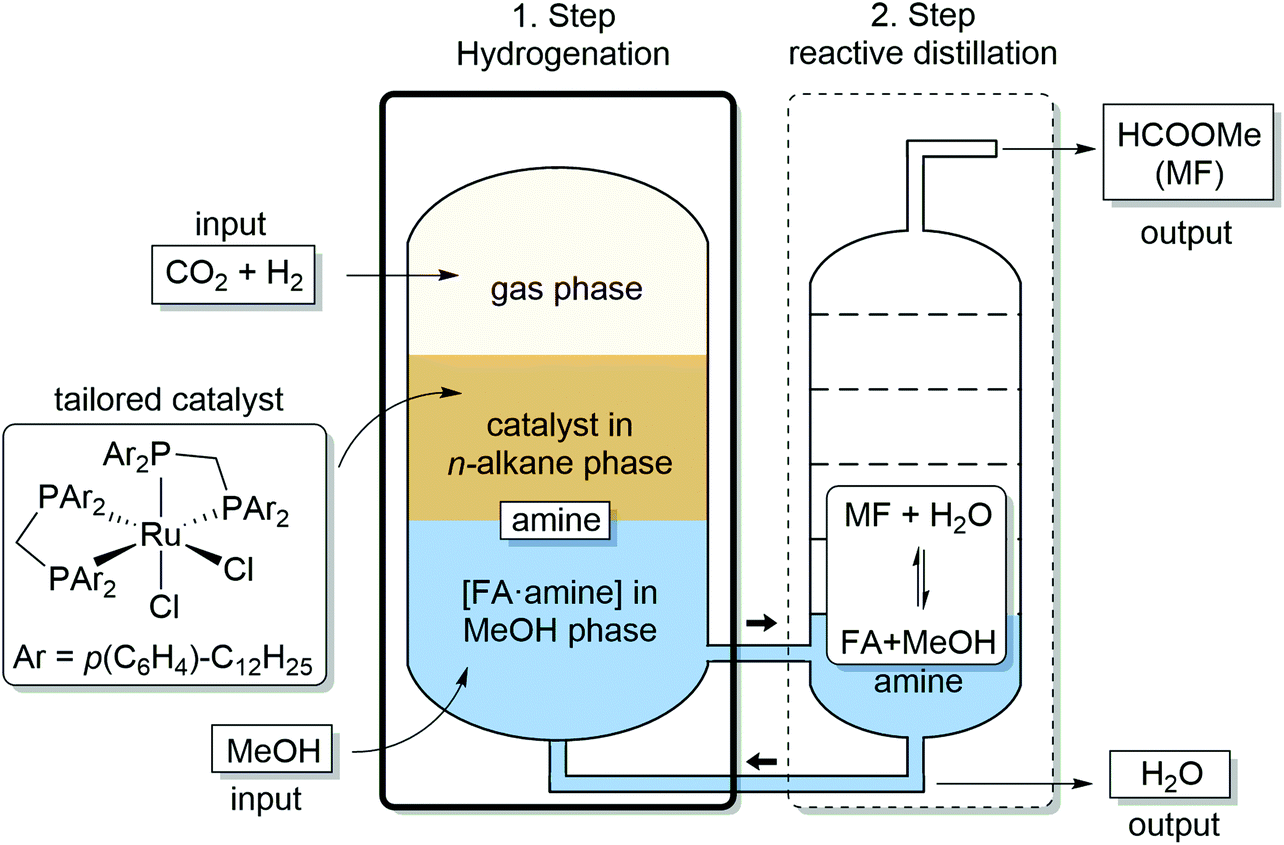 | ||
| Fig. 1 Integrated process for catalytic hydrogenation and reactive distillation for production of methyl formate (MF) from CO2, H2 and MeOH. | ||
The process shown in Fig. 1 differs from previous approaches targeting MF by achieving the overall transformation via two separate reaction steps that are integrated on a systems level. This allows to optimize reaction rates and equilibria for the two reaction steps independently, leading to higher overall efficiency (vide infra). To implement this concept, it is mandatory that the solution employed in the reactive distillation does not contain any active catalyst or metal component as this would promote the back reaction of the first step, i.e. the decomposition of FA to hydrogen and carbon dioxide,32,36 once the overpressure is released and temperature is increased. The design of the biphasic system for the first step is therefore crucial to achieve the optimized performance.
Results
The choice of methanol as recipient solvent for the formate adducts allows to perform the esterification step directly without the need of solvent replacement. On the other hand, the use of methanol significantly restricts the choice of the catalyst phase to solvents with very low polarity in order to establish a biphasic system. Long chain alkanes are suitable candidates and n-decane was selected as prototype for the catalyst phase in this study. To achieve preferential partitioning of the catalyst in the n-decane phase, the solubility properties of the catalyst must be adjusted accordingly. The complex cis-[Ru(dppm)2Cl2]37 (dppm = Ph2P-CH2-PPh2) is a powerful catalyst for CO2 hydrogenation to FA38,39 and thus was selected as lead structure for tagging with long alkyl chains at the aryl groups. The ligand synthesis starts from commercially available bromo aryl compounds bearing an octyl or a dodecyl chain in para-position. Metalation was achieved for the octyl derivative with Mg to the corresponding Grignard and of the dodecyl derivative with BuLi to the lithium reagent. Coupling of these organometallic compounds with bis(dichlorophosphino)methane yielded the new alkyl-tagged dppm derivatives L1 and L2, respectively, in good yields of 80–90% (Scheme 1). Complexation with cis-[Ru(dmso)4Cl2] resulted in the catalyst precursors cis-di(bis(bis(4-octylphenyl)phosphanyl)methane)rutheniumdichloride (1) and cis-di(bis(bis(4-dodecylphenyl)phosphanyl)methane)rutheniumdichloride (2) with good selectivities (trans complexes ≤ 7%) in high yields(80–99%) (Scheme 1).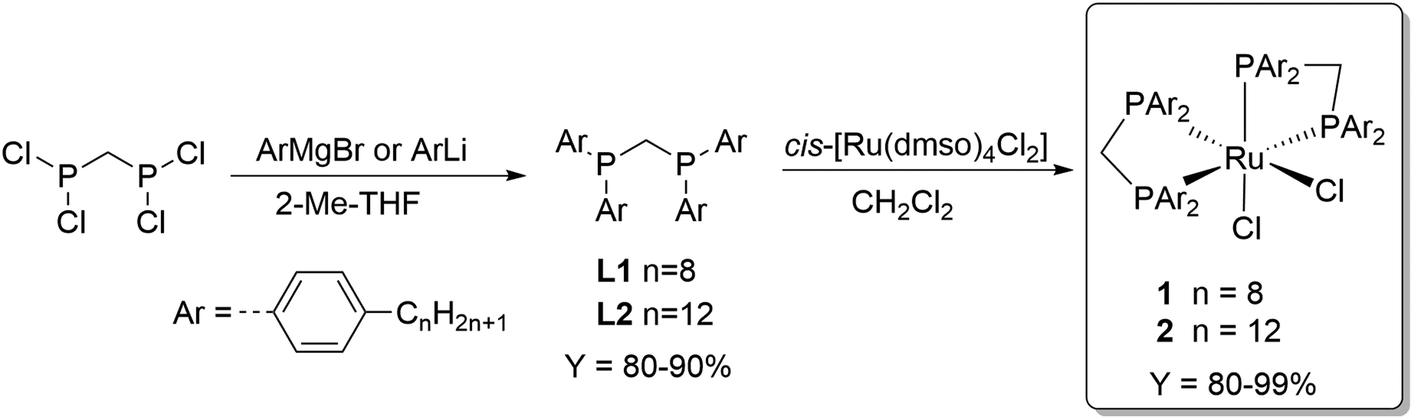 | ||
| Scheme 1 Synthesis of the alkyl-tagged complexes cis-di(bis(bis(4-octylphenyl)phosphanyl)methane)rutheniumdichloride (1) and cis-di(bis(bis(4-dodecylphenyl)phosphanyl)methane)rutheniumdichloride (2). | ||
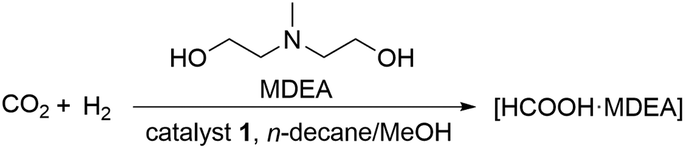
As expected, complexes 1 and 2 were found to be highly soluble in n-alkanes. For catalytic CO2 hydrogenation, the addition of amine bases to the product phase is necessary. Visual inspection of corresponding biphasic alkane/MeOH-amine mixtures confirmed that the complexes were retained with high preference in the n-decane phase (Fig. 2). Initially, methyl diethanolamine (MDEA), successfully used in biphasic organic/water systems for stabilizing FA,38,39 was chosen to validate the approach. Catalytic hydrogenation of CO2 using complex 1 was carried out under a standard set of reaction conditions and the stability of the catalyst system was tested by repetitive batch recycling of the alkane phase (Table 1). The reaction progress was monitored via the pressure drop and experiments were stopped as soon as the gas uptake was negligible (≤0.1 bar per min). A ratio of formic acid to amine χFA/MDEA of 1:1 defines the maximum conversion under these conditions and the time to reach this value as estimated from the pressure/time curves was used to derive average turnover frequencies (TOF) as lower limits for the catalyst activity.
 | ||
| Fig. 2 Observed catalyst partitioning for 2 in a typical experiment prior to addition of reaction gases. (Top: orange n-decane phase containing the catalyst; bottom: colorless MeOH phase with 1,2-dimethyl-imidazole (1,2-DMI). | ||
|
|
||||
|---|---|---|---|---|
| Cycle |
χ
FA/MDEAb [mol/mol] |
t [min] | P leachingd | Ru leachingd |
| a Reaction conditions: 10 mL window autoclave, Vn-decane = 2.5 mL, n1 = 5 μmol, nMDEA = 6.4 mmol and VMeOH/MDEA = 2.5 mL per cycle (stock solution of MDEA in MeOH was used), amine/cat (mol/mol) ≈ 1250 per cycle, pCO2 = 10 bar, pH2 = 80 bar, T = 60 °C. b Ratio determined by 1H NMR, no other side products found. c Estimated by pressure time profiles (digital manometer ±0.1 bar). d Determined by ICP-MS in aqueous matrix. e 10 mmol MDEA and 1.5 mL MeOH. f Addition of 300 μL n-decane. g Average of all 15 cycles. h Total leaching over 15 runs. | ||||
| 1e | 0.91 | 13 | 0.25% | 0.34% |
| 2 | 1.07 | 18 | 0.29% | 0.28% |
| 3 | 1.03 | 16 | 0.44% | 0.29% |
| 3 | 1.00 | 16 | 0.23% | 0.18% |
| 5 | 1.03 | 21 | 0.22% | 0.13% |
| 6f | 1.02 | 15 | 0.29% | 0.17% |
| 7 | 1.06 | 17 | 0.19% | 0.06% |
| 8 | 1.04 | 22 | 0.12% | 0.05% |
| 9 | 1.00 | 20 | 0.10% | 0.05% |
| 10 | 1.10 | 21 | 0.18% | 0.11% |
| 11f | 1.04 | 18 | 0.14% | 0.16% |
| 12 | 1.09 | 18 | 0.08% | 0.06% |
| 13 | 1.05 | 18 | 0.18% | 0.04% |
| 14 | 1.04 | 26 | 0.18% | 0.04% |
| 15 | 1.09 | 41 | 0.10% | 0.04% |
| 1.04 | 2.99% | 1.87% | ||
The first cycle was carried out with 10 mmol MDEA and 1.5 mL MeOH yielding a highly viscous product phase. Thus, more diluted solutions were employed in subsequent cycles. Rapid hydrogenation with a mean reaction time of 17 ± 4 min was observed in 13 consecutive experiments re-using the same catalyst phase, corresponding to an average TOF of ca. 4000 h−1. In run 14, catalyst deactivation started to become noticeable as indicated by longer reaction times required to reach constant pressure. A possible reason for the deactivation may originate from traces of oxygen that cannot be fully excluded by the procedure during the recycling on the small scale. ICP-MS analysis of the metal content in the product phase confirmed excellent catalyst retention leading to low losses of phosphorus (0.10–0.44%) and ruthenium (0.04–0.34%) per cycle resulting in a total leaching of P = 2.99% and Ru = 1.87%. Small amounts of n-decane were refilled to the recycled catalyst phase occasionally (Table 1, cycle 6 and 11) to compensate for losses by evaporation or cross-solubility (solubility of n-decane in MeOH = 81 g L−1 at 20 °C).40 The molecular ratio of n-decane/MeOH in the product phase measured via1H NMR never exceeded the value of 1:100, probably indicating an even reduced cross-solubility due to the formation of the polar product. In total, the catalyst phase was (re)used for 15 cycles and an average χFA/MDEA ratio of 1.04 was found in the isolated product phase corresponding to a concentration of c[FA·MDEA] ≈ 2.1 mol L−1 in methanol and a total productivity in terms of mole FA formed per mole of Ru (turnover number, TON) of ∼19000.
After successful demonstration of the hydrogenation step in combination with catalyst separation and recycling, the reactive distillation of the product solution of [FA·MDEA] in MeOH to form MF was investigated. Only small amounts of MF were observed in the distillate by 1H NMR analysis (nMF/nMeOH = 1:38) when the combined product solutions of the recycling experiment (27 g, cf. Table 1) were distilled using a 20 cm Vigreux column.41 To probe the reactive distillation in more detail, a model mixture was composed and subjected to rectification using a 45 cm long packed column and an oil bath temperature ranging from 45 °C to 75 °C over 8 hours (for detailed procedure see ESI†). In this case, a small fraction (ca. 2%) could be collected as distillate containing significantly higher amount of MF (nMF/nMeOH = 1:9, Fig. S9†). This result confirms the principle feasibility of the esterification via reactive distillation, but indicates also a slow conversion rate of [FA·MDEA] to MF and the necessity to have a higher number of separation trays to increase the production and the MF purity at the top of the column.42–44 Moreover, the residual mixture turned yellow during the distillation period of 8 h, indicating a possible degradation of the amine, a known drawback of MDEA-based scrubbers.45,46
The results from the catalytic and distillation experiments lead to the conclusion that while MDEA provides an excellent stabilizer for the hydrogenation step, this amine is not suitable for the MF formation step. In order to identify a potential amine to compromise effectively between the two steps, we screened the esterification of FA with MeOH in the presence of several amines (for complete list, data, and conditions see Table S4 and Fig. S5 and S6†). Hereby, a mixture of MeOH, FA and the selected amine was heated at 60 °C in a sealed vessel and the progress of the reaction was monitored by taking small samples at regular time intervals for 1H NMR analysis (Fig. 3).
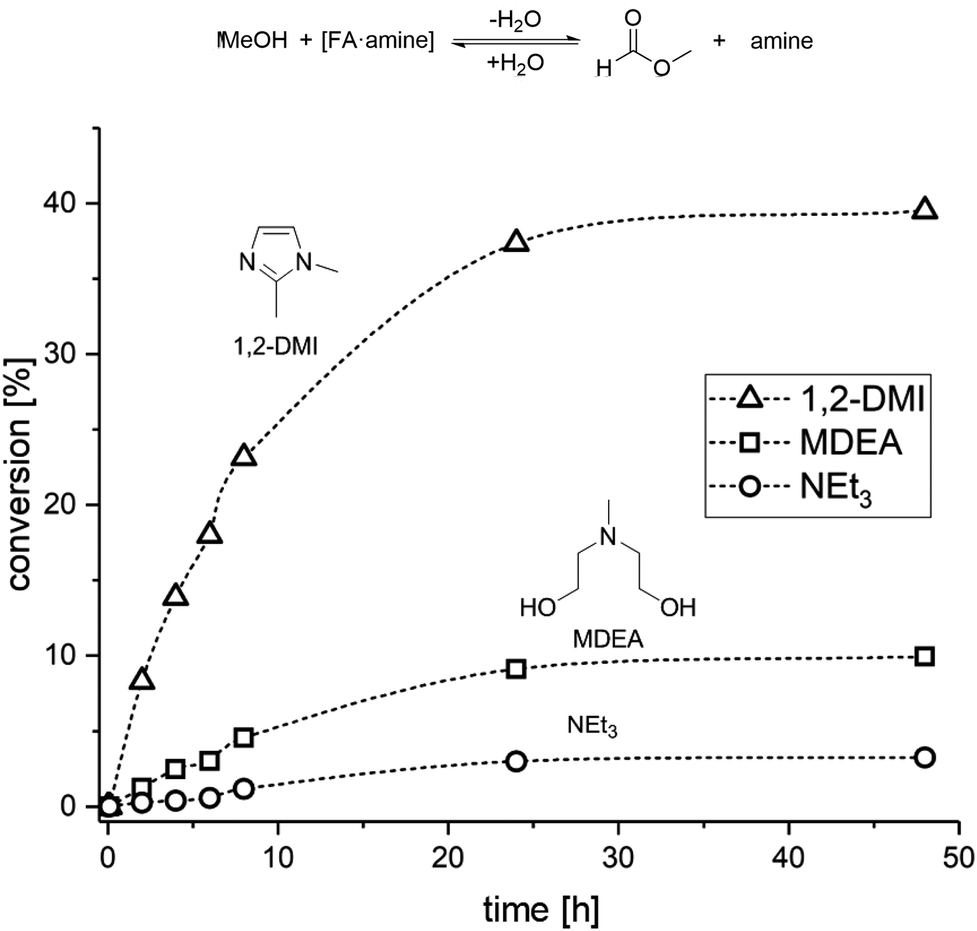 | ||
| Fig. 3 Time/conversion profiles for the esterification of FA to MF with selected amines in a closed vessel at 60 °C (VMeOH = 2 mL; nFA = 6.5 mmol; namine = 6.5 mmol). | ||
The screening showed that the esterification rate of methanol with [FA·amine] adducts increases with lower basicity of the amine. Stronger bases such as NEt3 (pKa = 10.76 at 25 °C)46 and NPr3 (pKa = 10.59 at 25 °C)47 led to very low esterification rates, while the formation of MF with the less basic MDEA (pKa = 8.57 at 25 °C)48,49 was approximately twice as fast as with NPr3 and four times faster than with NEt3 (for NPr3 see ESI, Fig. S5†). In addition to a basicity below a pKa of 8.5, further design criteria for the base can be formulated. The amine should not form an azeotrope with FA, should have a miscibility gap with water, and should be thermally stable and chemically robust under the rectification conditions. According to these features, 1,2-dimethyl-imidazole (1,2-DMI) with a pKa = 8.21 at 25 °C50 was selected as promising amine candidate. Indeed, 1,2-DMI outperformed all other tested amines yielding ∼15% MF within 5 h and 35% MF within 24 h in the screening experiments (Fig. 3).
As 1,2-DMI was identified as most promising candidate for the second reactive step, it was investigated for its performance as stabilizer in the hydrogenation of CO2 in the biphasic system n-decane/MeOH. In a first set of experiments, conditions were chosen in analogy to the study using MDEA as amine (Table 2, c1,2-DMI = 2.3 mol L−1 in MeOH). While the first reaction was completed within 70 minutes as indicated from the pressure monitoring, longer reaction times in the range of 115 ± 23 min were needed for the following four recycling experiments. In general, the reactions were roughly seven times slower (average TOF over five cycles ca. 600 h−1, Table 2) than in the presence of MDEA. Due to the lower basicity, the limiting ratio of FA to amine was also reduced to χFA/1,2-DMI = 0.58 mol/mol. Consequently, the TON under these conditions is also about 40% lower than in the MDEA system under identical conditions. While ICP-MS analysis of the product mixtures indicated a Ru-leaching in the same low range as with MDEA (0.07% to 0.33%), the P-leaching appeared to be higher, but also showed a broad scattering from batch to batch between 0.31% and 2.95%, making the results less reliable.
|
|
|||||
|---|---|---|---|---|---|
| Concentration of stock solution | cycle |
χ
FA/1,2-DMIb [mol/mol] |
t [min] | P leachingd | Ru leachingd |
| a Reaction conditions: 10 mL window autoclave, Vn-decane = 2.5 mL, n[Ru] = 3.5 μmol, nDMI = 5.7 (entries 1–5) or 14.8 (entries 7–11) mmol and VMeOH/DMI ≈ 2.5 mL per cycle (stock solution of DMI in MeOH was used), amine/cat (mol/mol) ≈ 1650 (entries 1–5) or 4200 (entries 7–11), pCO2 = 30 bar, pH2 = 60 bar, pressurized at r.t. till full saturation was reached, T = 60 °C. b Ratio determined by proton NMR, if not otherwise noted the amount of MF is ≤5% related to the amount of employed amine. c Determined by pressure time profiles, obtained with a digital manometer. d Determined by ICP-MS in an organic matrix (dioxane). e Average of the first 5 cycles. f Total of all cycles (the minor amount of formed MF is not included). | |||||
| c 1,2-DMI = 2.3 mol L−1 | 1 | 0.65 | 71 | 0.81% | 0.16% |
| 2 | 0.56 | 92 | 2.95% | 0.12% | |
| 3 | 0.51 | 110 | 0.99% | 0.33% | |
| 4 | 0.67 | 138 | 0.31% | 0.04% | |
| 5 | 0.49 | 100 | 0.44% | 0.07% | |
| 0.58 TON = 4761 | 5.50%e | 0.41%e | |||
| c 1,2-DMI = 5.9 mol L−1 | 1 | 0.34 | 227 | 0.77% | 0.28% |
| 2 | 0.34 | 186 | 4.41% | 0.16% | |
| 3 | 0.32 | 219 | 1.50% | 0.08% | |
| 4 | 0.34 | 247 | 2.24% | 0.06% | |
| 5 | 0.36 | 247 | 1.35% | 0.06% | |
| 0.35 TON = 6869 | 10.27%e | 0.64%e | |||
In a second set of experiments the loading of 1,2-DMI was substantially increased to c1,2-DMI = 5.9 mol L−1 (Table 2). Constant pressures were reached within 216 ± 31 min at a final FA to amine ratio χFA/1,2-DMI = 0.35 mol/mol corresponding to a final concentration of cFA = 2.1 mol L−1, similar to that achieved in the presence of MDEA. The total TON of ca. 6900 achieved over five cycles was even slightly higher than that reached within the same number of cycles with MDEA (cf.Table 1). Although the reaction rates and the final FA to amine ratio are significantly lower as with MDEA, the productivity of the 1,2-DMI-based catalytic system to generate formic acid is well in the range providing a promising compromise with the much more efficient subsequent esterification.
It is worth noting that during the hydrogenation reactions reported in Table 2 small amounts of MF (≤5%) were formed in agreement with the favorable kinetic of the esterification of FA to MF in the presence of 1,2-DMI. Thus, we investigated whether the amount of MF under hydrogenation conditions can be increased upon prolonging the reaction time. Accordingly, after the five experiments summarized in Table 2 at a c1,2-DMI = 2.3 mol L−1, an additional 6th cycle was carried out over 35 h reaction time. The resulting pressure-time curve shows two distinct slopes (Fig. 4). A steep decrease over the first three hours is in accordance with rapid CO2 hydrogenation. Once equilibrium to the formate adduct is reached, a much more shallow curve is observed during the following 24 h. In this second stage, the CO2 hydrogenation merely occurs to restore the amount of formate adduct transformed to MF and, thus, correlates with the much slower esterification rate. In the last ten hours of the experiment almost no pressure variation was observed indicating that the final equilibrium over both steps was reached. This experiment yielded FA at a ratio of χFA/1,2-DMI = 0.37 somehow lower than in the experiment before (Table 2, cycle 5) and MF at a ratio of χMF/1,2-DMI = 0.33, significantly higher than in all previous experiments where a ratio of up to of χMF/1,2-DMI = 0.05 was observed. Very similar results were obtained in an additional set of experiments with an extended reaction time of seven days (Table S5†). These findings demonstrate that substantial amounts of MF can be generated directly under hydrogenation conditions. However, full conversion of FA to MF cannot be reached due to equilibrium constraints in the closed vessel, leading to complex reaction mixtures. Combining the hydrogenation reaction with the reactive distillation allows to operate both steps at optimal rates and conversion levels.
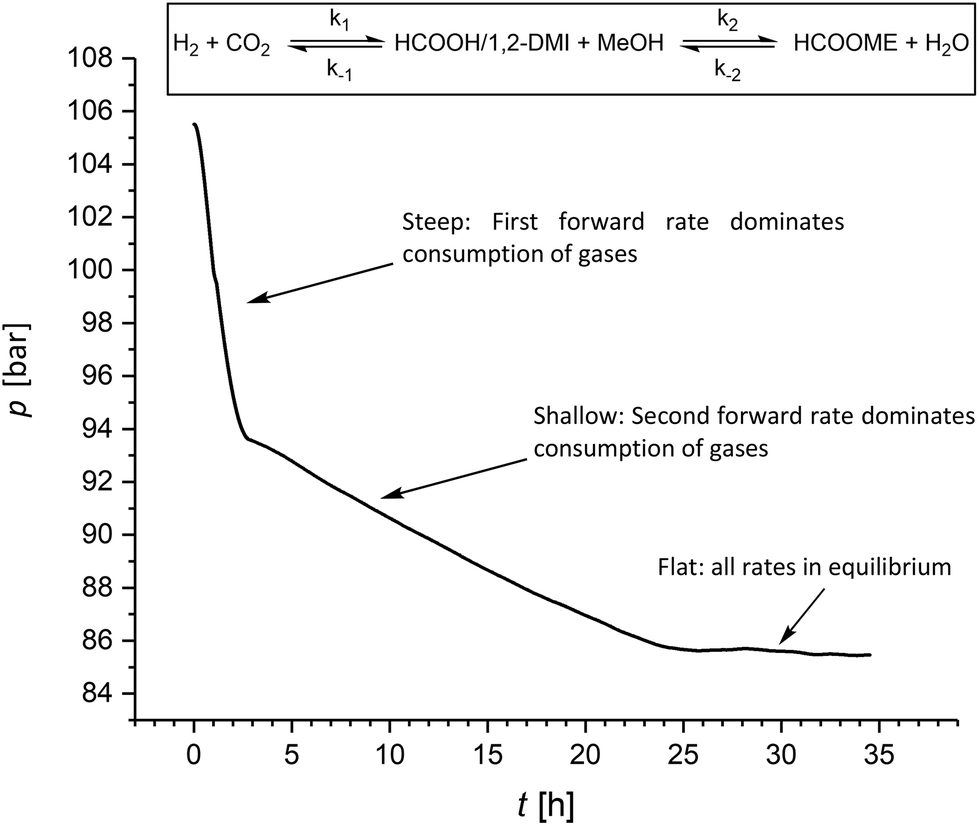 | ||
| Fig. 4 Pressure/time profile for the hydrogenation of CO2 under prolonged reaction time (conditions see Table 2; additional 6th cycle using an amine concentration of c1,2-DMI = 2.3 mol L−1). | ||
With the optimized combination of catalyst and base for the first step in hand, the robustness and potential for scale-up of the system was investigated in a semi-continuously operated set-up (Fig. 5).39
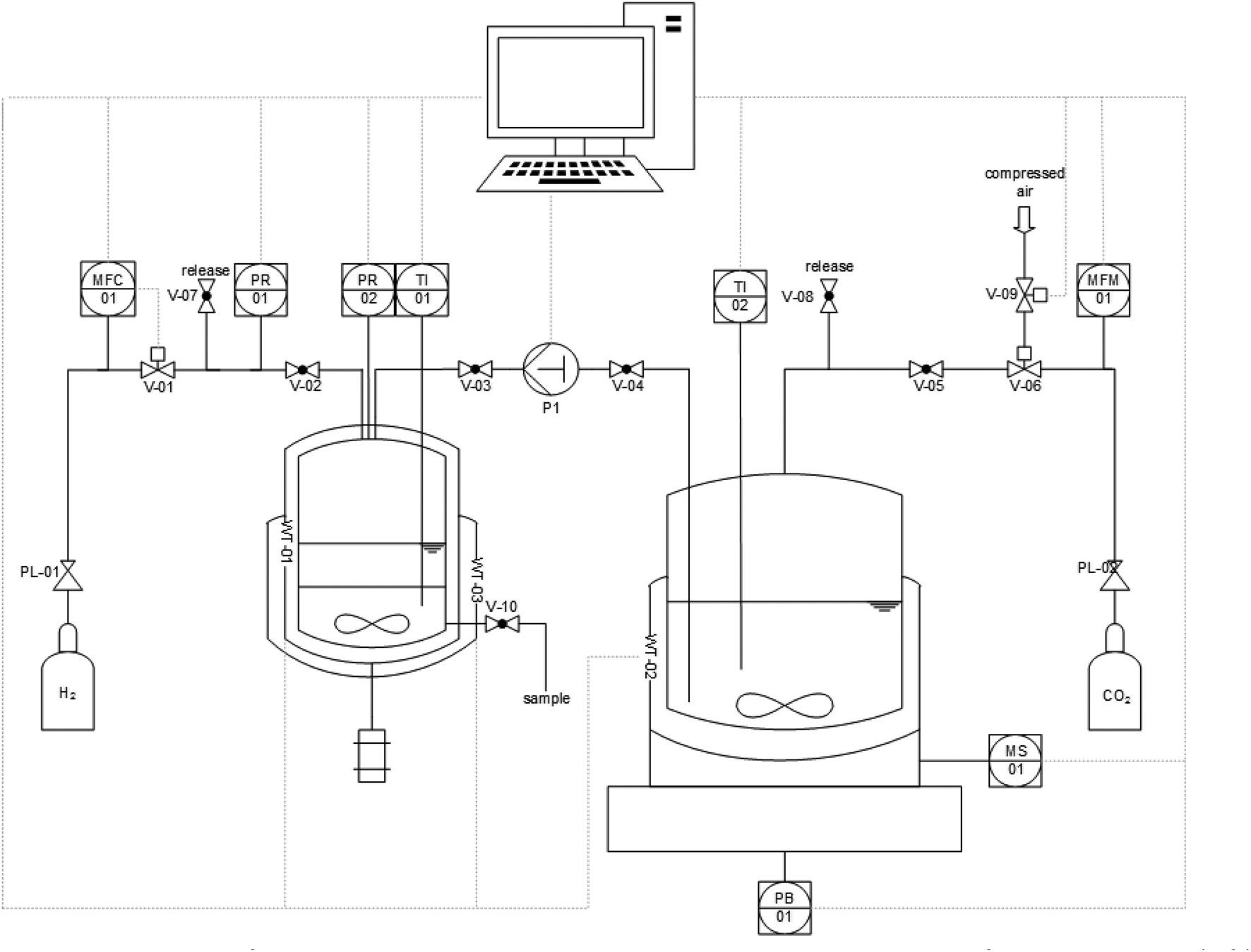 | ||
| Fig. 5 Schematic depiction of the semi-continuous operated set-up with a 100 mL autoclave for hydrogenation (left) and a 900 mL autoclave as reservoir for CO2 pre-saturated solutions of 1,2-DMI in methanol (right). | ||
The set-up comprises a 900 mL high pressure reservoir containing a CO2-saturated solution of 1,2-DMI in MeOH. This is placed on a magnetic stirring plate which is located on a balance to monitor the delivery of feed. The reservoir is connected to a 100 mL hydrogenation autoclave which is heated and cooled by a thermostat and equipped with a mechanical stirrer, a window and a sampling valve at the bottom. The reactive gases are delivered via a mass flow controller for H2 and a mass flow meter (MFM) controlled dosing valve for CO2. The graphical user interface to control the set-up was programmed using the LabView® (17.0) software. The procedure (for details see ESI† section 2.5) started by transferring a weighted amount of the feed solution of 1,2-DMI in MeOH pre-heated at 65 °C and pre-saturated with CO2 (35 bar) into the hydrogenation autoclave containing a n-decane solution of the catalyst. The reactor temperature was adjusted to 65 °C and the mixture pressurized with H2 up to 135 bar. The total pressure was kept constant by delivering H2via the MFC during the reaction. The resulting time-resolved H2 volume profile was used to monitor the reaction progress (see ESI Fig. S3 and S4†). Once the H2 flow became negligible (H2 flow ≤0.12 mLn min−1), the hydrogenation autoclave was cooled down to ∼15 °C leading to a clear phase separation within five minutes. The product phase was carefully withdrawn through the sampling valve at the bottom and the procedure started anew. The results of a series of 4 cycles are summarized in Table 3.
| Cycle | H2 Flowini. [mLn min−1] | V sub. [mL] | H2b [mmol] |
t [min] | c FA [mol L−1] | FA/1,2-DMI [mol/mol] | P leachinge [%] | Ru leachinge [%] |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: CO2 = 35 bar at rt (50 bar at 65 C); H2 up to a total pressure of 135 bar at 65 °C; T = 65 °C; 2 = 8.74 μmol; n-decane = 25 mL; c1,2-DMI = 4.2 mol L−1. b Hydrogen consumption as calculated integrating the overall H2 flow. c Time till negligible H2 flow was observed. d Average over all runs. e Determined by ICP-MS in organic matrix (dioxane). f Sum of all runs. | ||||||||
| 1 | 11.6 | 50.5 | 70.3 | 165 | 1.39 | 0.33 | 1.41 | 1.06 |
| 2 | 9.4 | 46.2 | 64.3 | 193 | 1.27 | 0.30 | 0.74 | 0.83 |
| 3 | 6.5 | 45.6 | 63.5 | 202 | 1.31 | 0.31 | 0.45 | 0.50 |
| 4 | 4.1 | 37.8 | 52.7 | 312 | 1.39 | 0.33 | 0.22 | 0.50 |
| 1.34d | 0.32d | 2.82f | 2.89f | |||||
An average final FA concentration of 1.34 mol L−1 was achieved at a ratio FA:1,2-DMI of χFA/1,2-DMI = 0.32 mol/mol, similar to what has been achieved in the small-scale experiments. A total of 180 mL of feed solution was processed corresponding to a production of 11 g FA. The prolonged reaction time in the fourth run indicates some catalyst deactivation, but a total TON of >27500 was already achieved at this point with an average TOF of 2000 h−1. The total leaching over the four runs amounted to 2.8% and 2.9% for P and Ru, respectively, underpinning the effective catalyst retention in this system.
In the envisaged integrated two-step process for MF production based on CO2 hydrogenation and subsequent esterification, the recycled methanol will contain not only 1,2-DMI but also an estimated residual water content of 6.7 mol%.51 In order to validate the compatibility of the catalyst system, a further set of hydrogenation experiments was carried out using a methanol solution of 1,2-DMI with a water content of 6.7 mol% relative to methanol (Table 4; for detailed procedure see ESI†). In the first six cycles the hydrogenation was complete within 200 min while longer times were needed for the subsequent runs (Fig. S4†). In a total of 8 cycles, 262 mL of feed solution were processed resulting in an average formic acid concentration of cFA = 1.32 mol L−1 at a χFA/1,2-DMI = 0.32 mol/mol. The productivity of 16 g FA corresponds to a TTON of 38000 generated at an average TOF of 1400 h−1. Cumulative catalyst leaching (1.4 mol% for P and 2.0 mol% for Ru) was in the same range or even slightly lower than in the experiments without added water. This may be attributed to an increased polarity of the product phase as reflected also in a more rapid phase separation. Overall, the presence of water at concentrations in this range in the recycle stream has no negative effect on the catalyst performance and may even be beneficial for the separation of the product phase.
| Entry | Cycle | H2 Flowini. [mLn min−1] | V sub. [mL] | H2b [mmol] |
t [min] | c FA [mol L−1] | FA/1,2-DMI [mol/mol] | P leachinge [%] | Ru leachinge [%] |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: CO2 = 35 bar at rt (50 bar at 65 C); H2 up to a total pressure of 135 bar at 65 °C; T = 65 °C; 2 = 8.74 μmol; n-decane = 25 mL; 1,2-DMIMeOH = 4.2 M; H2O = 6.7 mol% with respect to MeOH. b Hydrogen consumption as calculated integrating the overall H2 flow. c Time till negligible H2 flow was observed. d Average over all runs. e Determined by ICP-MS in an organic matrix (dioxane). f Sum of all runs. | |||||||||
| 1 | 1 | 11.5 | 42.5 | 59.2 | 193 | 1.38 | 0.33 | 0.72 | 1.14 |
| 2 | 2 | 11.9 | 40.6 | 56.5 | 191 | 1.42 | 0.34 | 0.25 | 0.38 |
| 3 | 3 | 18.8 | 47.8 | 66.5 | 165 | 1.25 | 0.30 | 0.15 | 0.17 |
| 4 | 4 | 12.3 | 36.0 | 50.1 | 188 | 1.30 | 0.31 | 0.13 | 0.17 |
| 5 | 5 | 6.6 | 25.4 | 35.4 | 211 | 1.31 | 0.32 | 0.07 | 0.04 |
| 6 | 6 | 6.1 | 25.4 | 35.5 | 210 | 1.15 | 0.28 | 0.03 | 0.04 |
| 7 | 7 | 4.3 | 32.5 | 32.7 | 295 | 1.26 | 0.30 | 0.04 | 0.04 |
| 8 | 8 | 0.3 | 12.1 | 16.9 | 426 | 1.45 | 0.35 | 0.04 | 0.04 |
| 9 | 1.32d | 0.32d | 1.43f | 2.02f | |||||
After successful demonstration of the n-decane/MeOH/1,2-DMI system for the FA production and catalyst separation, the esterification step from the MeOH solution was examined in a continuously operated reactive distillation. The feed for the reactive distillation was composed to represent a typical product phase obtained in hydrogenation experiments (MeOH as solvent, 1,2-DMI = 0.67 mol L−1, FA = 0.235 mol L−1, χFA/1,2-DMI = 0.35 mol/mol). The physico-chemical data for the components are listed in Table S6.† The rectification setup consists of a reboiler containing a total volume of 675 mL liquid phase, a DN50 bubble-cap tray column with 10 trays, and a condenser with an integrated reflux divider (Fig. 6). Every second tray of the column is equipped with a sampling point. The model mixture can be fed continuously into the bottom of the column where a manually operated electrical heating device is installed. The temperature is detected at the bottom as well as at the head of the column and the condenser is cooled with water at T = 7 °C. The pressure is monitored via a digital manometer and was kept at ambient pressure throughout the distillation process.
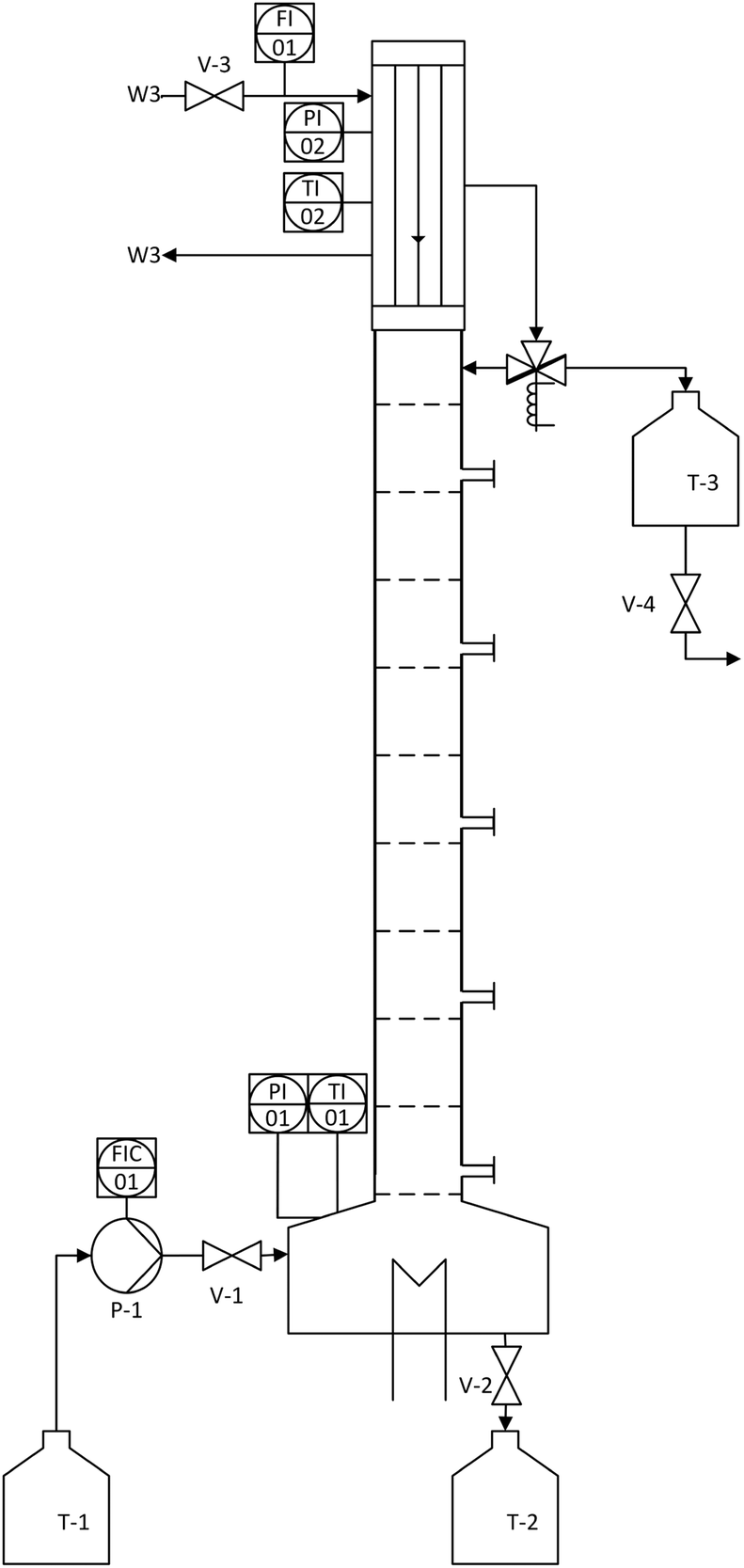 | ||
| Fig. 6 Schematic depiction of the rectification setup used for the reactive distillation of the MeOH/FA/1,2-DMI mixture. | ||
The rectification column was operated for a total time of 7 h with a reflux ratio (split ratio) of 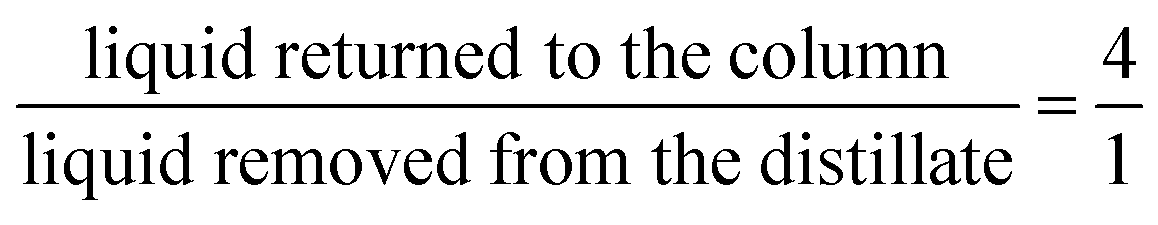 . After a heating phase of 115 minutes, continuous delivery of the feed solution directly into the bottom of the distillation column was started with a flowrate of 1–2 mL min−1 to assure a constant filling level of the reboiler. The temperature at the bottom of the rectification varied over the experimental time between 68.0 °C–79.3 °C. First samples were taken after 140 min and after that, samples were taken along the column length every 60 min and analyzed by 1H-NMR spectroscopy. The highest MF purity of the distillate was detected after 740 min and the measured composition at each sampled tray is plotted in Fig. 7. Although steady state conditions were not yet reached within this period, the results are significant and show a clear trend. The high boilers 1,2-DMI and FA completely remained in the reboiler. Although a color change of the liquid in the reboiler was notable, no degradation of 1,2-DMI was observed in the distillation residue and no side products were detected by 1H-NMR. Along the column, the percentage of MeOH diminished with the percentage of MF continuously increasing representing the progress of the esterification (Fig. 7). As expected, the highest MF purity was found in the distillate with 91.5 mol%. Water, which could not be detected with the used analytical method, is expected to accumulate at the bottom of the column. This stands in agreement with the calculation of the ternary system MF-MeOH-H2O with a Non-Random-Two-Liquid (NRTL) model, which predicts no azeotropes for the ternary composition. Due to the conversion of FA to MF, the ratio of FA to 1,2-DMI at the bottom of the column changed over time from the initial value of χFA/1,2-DMI = 0.35 mol/mol to χFA/1,2-DMI = 0.14 mol/mol at the end of this experiment accounting for a total FA conversion of XFA = 58.8%. Under the assumption that FA decomposition to CO is negligible at these low temperatures, this conversion corresponds to a total production of 0.767 kg MF per 1.00 kg FA entering the column under continuous operation.
. After a heating phase of 115 minutes, continuous delivery of the feed solution directly into the bottom of the distillation column was started with a flowrate of 1–2 mL min−1 to assure a constant filling level of the reboiler. The temperature at the bottom of the rectification varied over the experimental time between 68.0 °C–79.3 °C. First samples were taken after 140 min and after that, samples were taken along the column length every 60 min and analyzed by 1H-NMR spectroscopy. The highest MF purity of the distillate was detected after 740 min and the measured composition at each sampled tray is plotted in Fig. 7. Although steady state conditions were not yet reached within this period, the results are significant and show a clear trend. The high boilers 1,2-DMI and FA completely remained in the reboiler. Although a color change of the liquid in the reboiler was notable, no degradation of 1,2-DMI was observed in the distillation residue and no side products were detected by 1H-NMR. Along the column, the percentage of MeOH diminished with the percentage of MF continuously increasing representing the progress of the esterification (Fig. 7). As expected, the highest MF purity was found in the distillate with 91.5 mol%. Water, which could not be detected with the used analytical method, is expected to accumulate at the bottom of the column. This stands in agreement with the calculation of the ternary system MF-MeOH-H2O with a Non-Random-Two-Liquid (NRTL) model, which predicts no azeotropes for the ternary composition. Due to the conversion of FA to MF, the ratio of FA to 1,2-DMI at the bottom of the column changed over time from the initial value of χFA/1,2-DMI = 0.35 mol/mol to χFA/1,2-DMI = 0.14 mol/mol at the end of this experiment accounting for a total FA conversion of XFA = 58.8%. Under the assumption that FA decomposition to CO is negligible at these low temperatures, this conversion corresponds to a total production of 0.767 kg MF per 1.00 kg FA entering the column under continuous operation.
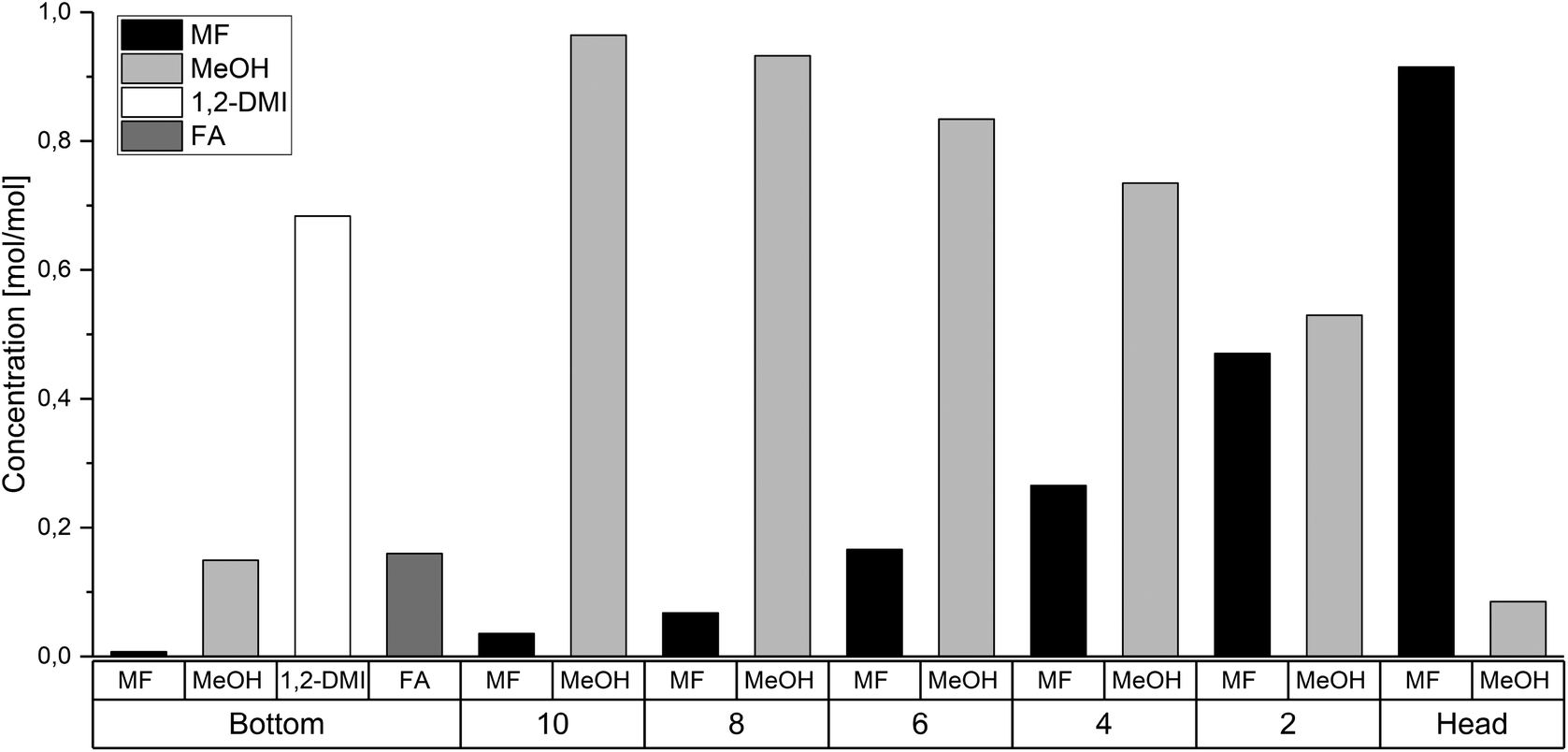 | ||
| Fig. 7 Molar ratios of MF, MeOH, 1,2-DMI and FA in the reboiler, at the trays and in the column head of the reactive distillation column after 740 min. | ||
Summary and conclusion
An integrated two-step process was developed for the production of methylformate (MF) from carbon dioxide, hydrogen and methanol. The first step comprises the hydrogenation of CO2 to formic acid (FA) in the presence of an amine and has been realized using a biphasic system with methanol as the product phase and n-decane as the catalyst phase allowing efficient immobilization of alkyl-tagged ruthenium phosphine complexes. The second step comprises the esterification of formic acid and methanol via reactive distillation directly from the product mixture of the first step. The basicity of the employed amine was identified as a key design parameter. Whereas high basicity of the amine is beneficial in general for the hydrogenation step, medium to low basicity is required for high reaction rates in the esterification reaction. Commercially available 1,2-dimethyl imidazole (1,2-DMI) was identified to represent a good compromise for both steps allowing the realization of a fully integrated process. Repetitive batch experiments demonstrated the suitability of the biphasic system to generate a suitable feed for the second step (average cFA = 1.32 mol L−1 at a χFA/1,2-DMI ≈ 0.32 mol/mol, TTON in the range of 38000, average TOF 1400 h−1) with excellent catalyst retention (cumulative leaching over 8 cycles 1.4 mol% for P and 2.0 mol% for Ru). Methylformate production from the methanol solution containing the FA/1,2-DMI adduct was shown to be possible when directly subjected to a reactive distillation step, yielding MF as the distillate with good purity of 91.5% even under non-optimized rectification conditions. With the identification of a suitable reaction system compromising both the hydrogenation and reactive distillation step, a block diagram for a fully integrated continuous process can be devised (Fig. 8).
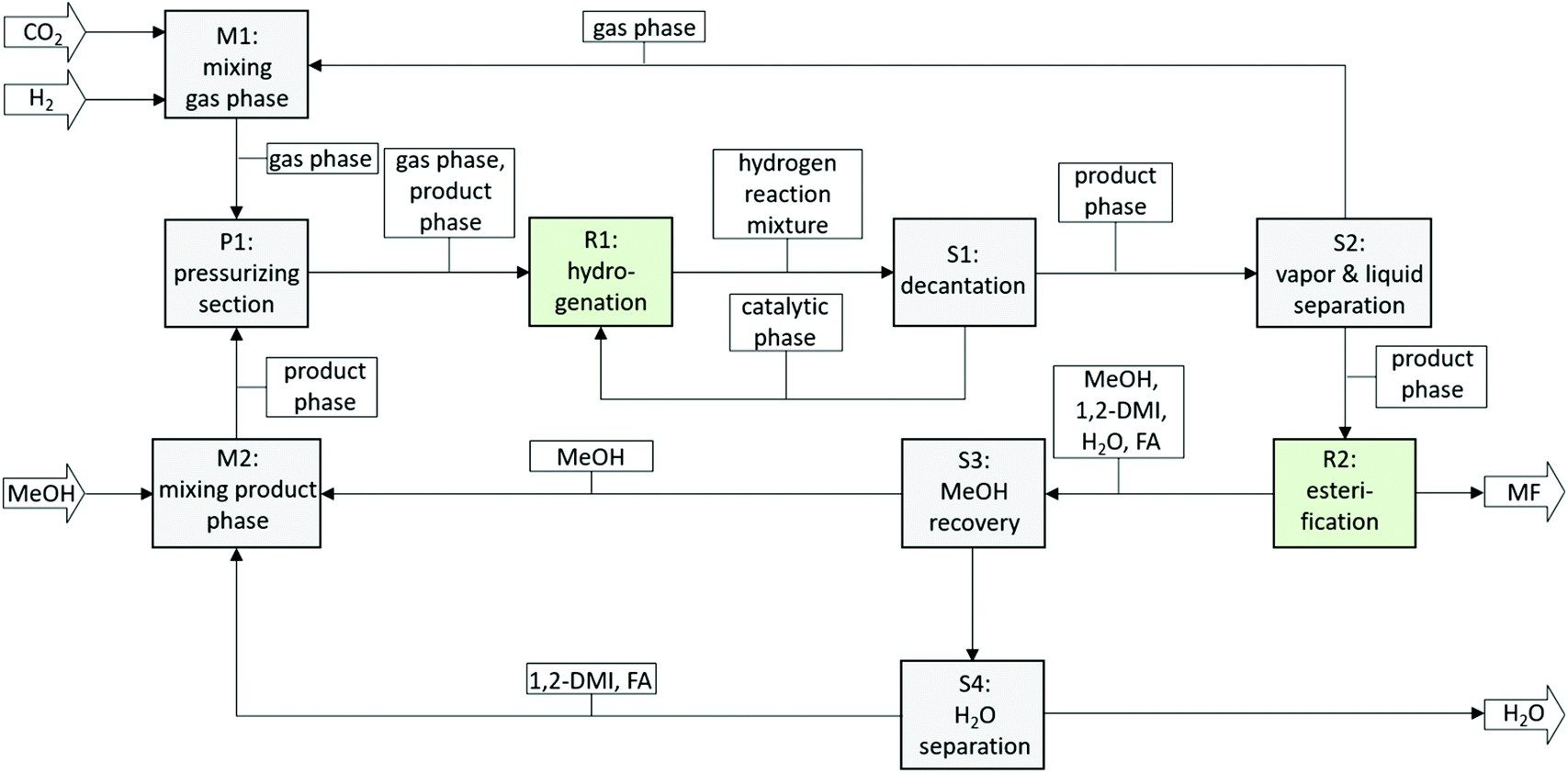 | ||
| Fig. 8 Block flow diagram of a continuous process for the production of MF from CO2. Ri = Reaction units, Si = separation units, Mi = mixing units. | ||
The process concept to produce methyl formate schematically shown in Fig. 1 and summarized as block diagram in Fig. 8 offers an attractive alternative to the isolation of formic acid from CO2 hydrogenation mixtures, which proves to be notoriously difficult.52 With the increasing availability of methanol53 from biomass,54,55 recycled waste,56 or even CO2 and hydrogen,57,58 both reactive components of the methyl formate process can be produced from renewable electricity and resources. Given the broad portfolio of methylformate in direct applications or as intermediate, the CO2-based production outlined in this paper holds therefore significant potential within the so-called “Power-to-X” concept.59,60
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been carried out within the project “Carbon-2-Chem” (03EK3042C) was funded by the German Federal Ministry of Education and Research (BMBF). Additional financial support by the Cluster of Excellence 2186 “The Fuel Science Center” funded by the Deutsche Forschungsgemein-schaft (DFG) is gratefully acknowledged. Open Access funding provided by the Max Planck Society. The authors thank Julia Nießen for carrying out the ICP-MS measurements.References
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuehn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2017, 118, 372–433 CrossRef PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, Green Chem., 2016, 18, 5831–5838 RSC.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- J. Klankermayer and W. Leitner, Science, 2015, 350, 629–630 CrossRef CAS.
- C. Chauvier and T. Cantat, ACS Catal., 2017, 7, 2107–2115 CrossRef CAS.
- A. Tortajada, F. Julia-Hernandez, M. Boerjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed.
- X.-F. Liu, X.-Y. Li and L.-N. He, Eur. J. Org. Chem., 2019, 2437–2447 CrossRef CAS.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2017, 118, 434–504 CrossRef PubMed.
- C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254–6257 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- K. Thenert, K. Beydoun, J. Wiesenthal, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12266–12269 CrossRef CAS PubMed.
- Y. Inoue, H. Izumida, Y. Sasaki and H. Hashimoto, Chem. Lett., 1976, 5, 863–864 CrossRef.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207–2221 CrossRef CAS.
- G. Jenner, Appl. Catal., A, 1995, 121, 25–43 CrossRef CAS.
- (a) M. Pazicky, T. Schaub, R. Paciello, A. G. Altenhoff and D. M. Fries, DE102012014159A1, 2013; (b) N. Westhues, M. Belleflamme and J. Klankermayer, ChemCatChem, 2019 DOI:10.1002/cctc.201900627.
- J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2016, DOI:10.1002/14356007.a12_013.pub3.
- S. R. Akuri, C. Dhoke, K. Rakesh, S. Hegde, S. A. Nair, R. Deshpande and P. Manikandan, Catal. Lett., 2017, 147, 1285–1293 CrossRef CAS.
- E. Karakhanov, A. Maksimov, Y. Kardasheva, E. Runova, R. Zakharov, M. Terenina, C. Kenneally and V. Arredondo, Catal. Sci. Technol., 2014, 4, 540–547 RSC.
- M. S. Yalfani, G. Lolli, A. Wolf, L. Mleczko, T. E. Mueller and W. Leitner, Green Chem., 2013, 15, 1146–1149 RSC.
- J. S. Lee, J. C. Kim and Y. G. Kim, Appl. Catal., 1990, 57, 1–30 CrossRef CAS.
- R. Kumar, A. Katoch, A. Singhal and S. Kumar, Energy Fuels, 2018, 32, 12936–12948 CrossRef CAS.
- J. M. Ngugi, P. N. Kioni and J. K. Tanui, J. Clean Energy Technol., 2018, 6, 74–82 CrossRef CAS.
- B. Akih-Kumgeh and J. M. Bergthorson, Energy Fuels, 2010, 24, 396–403 CrossRef CAS.
- T. Schaub and R. A. Paciello, Angew. Chem., Int. Ed., 2011, 50, 7278–7282 CrossRef CAS PubMed.
- E. Plichta, M. Salomon, S. Slane and M. Uchiyama, J. Solution Chem., 1987, 16, 225–235 CrossRef CAS.
- C. M. Jens, L. Müller, K. Leonhard and A. Bardow, ACS Sustainable Chem. Eng., 2019, 7, 12270–12280 CAS.
- M. Gatti, E. Martelli, F. Marechal and S. Consonni, Appl. Therm. Eng., 2014, 70, 1123–1140 CrossRef CAS.
- R. Kuhlmann, K. U. Künnemann, L. Hinderink, A. Behr and A. J. Vorholt, ACS Sustainable Chem. Eng., 2019, 7, 4924–4931 CrossRef CAS.
- R. Mason, D. W. Meek and G. R. Scollary, Inorg. Chim. Acta, 1976, 16, L11–L12 CrossRef CAS.
- M. Scott, B. Blas Molinos, C. Westhues, G. Franciò and W. Leitner, ChemSusChem, 2017, 10, 1085–1093 CrossRef CAS PubMed.
- W. Leitner, G. Franciò, M. Scott, C. Westhues, J. Langanke, M. Lansing, C. Hussong and E. Erdkamp, Chem. Ing. Tech., 2018, 90, 1504–1512 CrossRef CAS.
- R. W. Kiser, G. D. Johnson and M. D. Shetlar, J. Chem. Eng. Data, 1961, 6, 338–341 CrossRef CAS.
- Although MF and methanol do not form an azeotrope, the boiling-point diagram of these compounds only offers a narrow window (see ESI Fig. S13 and S14†), making their separation challenging on a laboratory scale.
- A. T. Sundberg, P. Uusi-Kyyny, M. Pakkanen and V. Alopaeus, J. Chem. Eng. Data, 2011, 56, 2634–2640 CrossRef CAS.
- J. Polak and B. C. Y. Lu, J. Chem. Thermodyn., 1972, 4, 469–476 CrossRef CAS.
- N. Kozub, H. Schuberth and E. Leibnitz, J. Prakt. Chem., 1962, 17, 282–292 CrossRef CAS.
- A. Chakma and A. Meisen, Can. J. Chem. Eng., 1997, 75, 861–871 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, DOI:10.1002/14356007.a02_001.
- A. Tagiuri, M. Mohamedali and A. Henni, J. Chem. Eng. Data, 2015, 61, 247–254 CrossRef.
- A. Hartono, M. Saeed, I. Kim and H. F. Svendsen, Energy Procedia, 2014, 63, 1122–1128 CrossRef CAS.
- B. Lenarcik and P. Ojczenasz, J. Heterocycl. Chem., 2002, 39, 287–290 CrossRef CAS.
- Complete water removal by distillation, or other means, is an extremely energy intensive process and would dramatically affect the energy efficiency of the whole process. The residual amount of water considered here derives from theoritical simulations through the Aspen software and will be reported in detail somewhere else.
- C. M. Jens, M. Scott, B. Liebergesell, C. G. Westhues, P. Schaefer, G. Franciò, K. Leonhard, W. Leitner and A. Bardow, Adv. Synth. Catal., 2019, 361, 307–316 CrossRef CAS.
- Methanol: The Basic Chemical and Energy Feedstock of the Future, ed. M. Bertau, H. Offermanns, L. Plass, F. Schmidt and H.-J. Wernicke, Springer, Amsterdam, 2013, DOI:10.1007/978-3-642-39709-7.
- D. Gullu and A. Demirbas, Energy Convers. Manage., 2001, 42, 1349–1356 CrossRef CAS.
- N. S. Shamsul, S. K. Kamarudin, N. A. Rahman and N. T. Kofli, Renewable Sustainable Energy Rev., 2014, 33, 578–588 CrossRef CAS.
- https://enerkem.com/ .
- R. M. Navarro, M. A. Pena and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS.
- https://www.carbonrecycling.is/george-olah .
- A. Sternberg and A. Bardow, Energy Environ. Sci., 2015, 8, 389–400 RSC.
- J. Klankermayer and W. Leitner, Philos. Trans. R. Soc., A, 2016, 374, 20150315 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03006a |
| This journal is © The Royal Society of Chemistry 2019 |