
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLytic polysaccharide monooxygenase (LPMO) mediated production of ultra-fine cellulose nanofibres from delignified softwood fibres†
Salla
Koskela
 a,
Shennan
Wang
a,
Dingfeng
Xu
a,
Xuan
Yang
b,
Kai
Li
ab,
Lars A.
Berglund
b,
Lauren S.
McKee
ab,
Vincent
Bulone
a and
Qi
Zhou
*ab
a,
Shennan
Wang
a,
Dingfeng
Xu
a,
Xuan
Yang
b,
Kai
Li
ab,
Lars A.
Berglund
b,
Lauren S.
McKee
ab,
Vincent
Bulone
a and
Qi
Zhou
*ab
aDivision of Glycoscience, Department of Chemistry, KTH Royal Institute of Technology, AlbaNova University Centre, SE-106 91 Stockholm, Sweden. E-mail: qi@kth.se; Tel: +4687909625
bWallenberg Wood Science Center, Department of Fiber and Polymer Technology, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
First published on 23rd September 2019
Abstract
The production of cellulose nanofibres (CNFs) typically requires harsh chemistry and strong mechanical fibrillation, both of which have negative environmental impacts. A possible solution is offered by lytic polysaccharide monooxygenases (LPMOs), oxidative enzymes that boost cellulose fibrillation. Although the role of LPMOs in oxidative modification of cellulosic substrates is rather well established, their use in the production of cellulose nanomaterials is not fully explored, and the effect of the carbohydrate-binding module (CBM) on nanofibrillation has not yet been reported. Herein, we studied the activity of two LPMOs, one of which was appended to a CBM, on delignified softwood fibres for green and energy-efficient production of CNFs. The CNFs were used to prepare cellulose nanopapers, and the structure and properties of both nanofibres and nanopapers were determined. Both enzymes were able to facilitate nanocellulose fibrillation and increase the colloidal stability of the produced CNFs. However, the CBM-lacking LPMO was more efficient in introducing carboxyl groups (0.53 mmol g−1) on the cellulose fibre surfaces and releasing CNFs with a thinner width (4.3 ± 1.5 nm) from delignified spruce fibres than the modular LPMO (carboxylate content of 0.38 mmol g−1 and nanofibre width of 6.7 ± 2.5 nm) through LPMO-pretreatment followed by mild homogenisation. The prepared nanopapers showed improved mechanical properties (tensile strength of 262 MPa and modulus of 16.2 GPa) compared to those obtained by conventional CNF preparation methods, demonstrating the potential of LPMOs as green alternatives for cellulose nanomaterial preparation.
Introduction
Extensive use of plastics in consumer products has caused wide-ranging environmental issues due to the long-term persistence of plastics in the global ecosystem, ingestion of microplastics by organisms, and their accumulation along food chains.1,2 Therefore, there is a pressing need to develop biodegradable materials with good properties from naturally occurring polymers, such as cellulose. In recent years, cellulose has gained popularity in sustainable materials research in the form of cellulose nanofibres (CNFs), which are three orders of magnitude smaller than intact fibre cells. Cellulose nanomaterial-enabled products are lighter, stronger, biologically compatible, and environmentally benign, which makes CNFs ideal replacements for the conventional synthetic polymer materials.3,4 To produce CNFs, cellulose fibres from natural sources need to be disintegrated into nanoparticles.5 Strong intra-fibre hydrogen bonding and a tendency to aggregate in water make cellulose fibres resistant to mechanical disintegration, and consequently strong disintegration methods that require high energy or chemical modifications of cellulose fibres are needed.6–10 Various chemical pre-treatment approaches, such as 2,2,6,6-tertamethylpiperidine-1-oxyl (TEMPO)-mediated oxidation6,8 or quaternisation via nucleophilic addition,9 are able to facilitate the fibrillation of cellulose with a kitchen blender and produce CNFs with the corresponding negative or positive surface charges and also with a uniform width of 3–4 nm, which is similar to the width of cellulose microfibrils in plants. However, there are still major environmental issues related to some chemical modifications in CNF production, particularly when periodate oxidation, TEMPO-mediated oxidation, or sulfuric acid hydrolysis is employed. The dicarboxylic acid hydrolysis method has been shown to be sustainable in cellulose nanomaterial production owing to the acid recovery.11 Enzymatic pre-treatment using glycoside hydrolases is more environmentally friendly and requires reduced energy consumption for fibrillation compared to the untreated sample.12,13 However, multiple passes (at least 30 passes) through the microfluidizer are still required in order to prepare CNFs with a thinner width of 4–12 nm.13 Thus, using carbohydrate-active enzymes to assist the production of CNFs with tailored surface properties and an ultra-fine width and significantly reduce the energy consumption for mechanical homogenisation still remains a challenge.In nature, recalcitrant wood cell wall components, including cellulose, are effectively decomposed under mild, aqueous reaction conditions mainly by fungi.14 Fungi can accomplish this by utilising an array of efficient carbohydrate-active enzymes, including lytic polysaccharide monooxygenases, LPMOs. Most fungal LPMOs are classified in the auxiliary activity family AA9 in the carbohydrate-active enzyme database (http://www.cazy.org). These recently discovered enzymes are capable of oxidative modification of recalcitrant polysaccharides, such as cellulose.15,16 On cellulose, LPMOs perform a highly specific oxidation reaction at either the C1 or C4 position, the reaction products being aldonic acids or gem-diols, respectively, of which the former contains an ionisable carboxyl group.17 When acting on low-molecular weight cellulosic substrates, C1-specific LPMOs release soluble cello-oligomers containing carboxyl groups at their reducing ends.18 On fibres, the oxidative chain cleavage does not yield soluble products, but instead carboxyl groups are formed on the fibres, mainly at the crystalline areas.19,20 The C1-localised carboxyl groups have been shown to facilitate cellulose fibrillation owing to the increased colloidal stability of the produced CNFs, in a much similar manner to that of C6-carboxyl groups derived from TEMPO-oxidation.20–24 The unique aspect of LPMO-oxidation is the chain breaks introduced by the enzyme, which might also enhance cellulose fibrillation by decreasing the crystallinity. A separate carbohydrate-binding module (CBM) possessed by some LPMOs is thought to enhance the LPMO activity by bringing the enzyme's active site into contact with the crystalline regions of cellulose,25 but little is known about the effect of the CBM on the subsequent mechanical fibrillation process, as it may also restrict the movement of the LPMO.26
In the present work, we developed an environmentally friendly and energy-efficient method to produce CNFs and cellulose nanopapers with robust mechanical properties. We used two C1-active LPMOs from the ascomycetous fungus Neurospora crassa (NcLPMO9E and NcLPMO9F) to boost the fibrillation of delignified wood fibres. NcLPMO9E contains a family 1 CBM and NcLPMO9F is non-modular. The delignified wood fibres, i.e. holocellulose, were prepared from spruce wood by using peracetic acid (PAA). Treatment with PAA selectively removes lignin while preserving the native wood fibre structure without the defects typically present in commercial wood pulp fibres, and the process is environmentally sustainable.27 The LPMOs were removed after the enzymatic reaction, and the fibres were subjected to mild and short mechanical homogenisation. The structure and morphology of the resulting nanofibres after the reaction with a CBM-containing LPMO and a non-modular LPMO were characterised and compared. The LPMO-oxidised nanofibres were further used to prepare cellulose nanopapers by vacuum filtration and their mechanical properties were studied in uniaxial tensile tests and compared to those of the unmodified holocellulose fibres.
Experimental methods
Preparation of holocellulose
The holocellulose fibres were prepared according to a previously reported method.27 Briefly, softwood spruce sticks were first soaked in water under vacuum, and subsequently treated with 4% (w/w) peracetic acid (PAA) for 45 min at 85 °C. The PAA solution was prepared by diluting 38–40% (w/w) PAA solution (Sigma-Aldrich), followed by adjusting the pH to 4.6 using NaOH before the reaction. The weight ratio of PAA and softwood was 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. Four rounds of PAA treatment were performed until the wood sticks were disintegrated into individual fibres. Most of the lignin was removed after this treatment as 1.5% (w/w) residual lignin was present in the final samples compared to the initial content of 21% (w/w). No mechanical stirring was applied during the treatment to avoid fibre damage. To increase the enzyme accessibility of holocellulose fibres, the hemicelluloses were partially removed with pressurised hot water extraction (autoclave) followed by extensive washing in hot water before LPMO treatment (Table S1, ESI†).
:100. Four rounds of PAA treatment were performed until the wood sticks were disintegrated into individual fibres. Most of the lignin was removed after this treatment as 1.5% (w/w) residual lignin was present in the final samples compared to the initial content of 21% (w/w). No mechanical stirring was applied during the treatment to avoid fibre damage. To increase the enzyme accessibility of holocellulose fibres, the hemicelluloses were partially removed with pressurised hot water extraction (autoclave) followed by extensive washing in hot water before LPMO treatment (Table S1, ESI†).
LPMO selection and sequence analysis
We selected LPMOs that have been previously shown to possess C1-oxidation ability, i.e. NcLPMO9E and NcLPMO9F from the ascomycetous fungus N. crassa.28,29 The amino acid sequences of the LPMOs were obtained from UniProt (protein-IDs: Q1K4Q1; Q7RWN7). Native signal peptides were predicted with SignalIP software 4.1 (http://www.cbs.dtu.dk/services/SignalP/). The theoretical molecular masses of the mature recombinant LPMOs were calculated from the amino acid sequences without the signal peptides with ExPASy ProtParam (http://web.expasy.org/prot-param/). The calculated theoretical molecular masses of NcLPMO9E and NcLPMO9F are 32 kDa and 24 kDa, respectively.Gene synthesis and cloning
Genes encoding the LPMOs were commercially synthesised and codon optimised by GenScript, USA. Codon-optimised gene sequences for NcLPMO9E and NcLPMO9F are shown in Fig. S1 and S2, ESI,† respectively. The synthetic genes encoded native secretion signals at the N-terminus and (His)6 tags at the C-terminus of the corresponding proteins. The genes were inserted into the EcoRI-NotI position of the P. pastoris expression vector pPICZB, in which the c-myc epitope and polyhistidine tag were omitted. The constructs were transformed into E. coli TOP10 cells (Thermo Fisher Scientific) by electroporation, and the transformants were cultivated in low salt Luria–Bertani (LB) broth containing 25 μg mL−1 zeocin (Invitrogen). The plasmid DNA was extracted using a GeneJet Plasmid Miniprep kit (Thermo Scientific). The constructs were confirmed by restriction pattern analysis (EcoRI and NotI – Thermo Scientific) (Fig. S3, ESI†), and the positive clones were sequenced by Eurofins Genomics, Germany. Constructs with verified sequences were linearised using PmeI and approximately 10 μg of DNA was used to transform P. pastoris X33 cells by electroporation. The clones were selected on YPDS agar containing 100 μg mL−1 zeocin. Integration of the heterologous genes into P. pastoris genomic DNA was confirmed by colony PCR utilising AOX1 primers and a standard protocol (Fig. S4, ESI†).Production and purification of the LPMOs
The LPMOs were produced by cultivating the P. pastoris transformants in 250 mL flasks containing 30 mL of the growth medium. The cultivation conditions were 30 °C and 230 rpm. After overnight cultivation in BMGY, the cells were suspended into BMMY for a final cell density (OD600) of ∼1.0. BMGY and BMMY contained 10 g L−1 yeast extract, 20 g L−1 peptone, 1.34% YNB, 4 × 10−5% biotin and 100 mM potassium phosphate buffer at pH 6.0. BMGY also contained 1% glycerol. The cultures were supplemented daily with methanol and CuSO4 at final concentrations of 2% (v/v) and 0.1 mM, respectively. The recombinant protein production was followed daily with SDS-PAGE and western-blot analyses as shown in Fig. S5 and S6, ESI.†The His-tagged LPMOs were purified by immobilised metal-ion affinity chromatography (IMAC) using an Äkta purifier FPLC system (GE Healthcare). The pH of the culture supernatant was adjusted to 7.4 before loading on a 5 mL IMAC FF HiTrap column (Bio-Rad), which was previously charged with a metal (Cu2+) and equilibrated in 50 mM NaCl – 5 mM imidazole – 20 mM sodium phosphate, pH 7.4 (for NcLPMO9E) or 50 mM NaCl – 20 mM sodium phosphate, pH 7.4 (for NcLPMO9F). Gradient elution was performed using 500 mM imidazole and 500 mM NaCl. The pooled proteins were gel filtered on a Biogel P-6 (Bio-Rad) column to 20 mM sodium phosphate, pH 7.4. Finally, the proteins were concentrated in 5 kDa cut-off Vivaspin® 20 centrifugal concentrators (GE Healthcare). Protein concentrations were determined using a Pierce™ BCA protein assay kit (Thermo Scientific). The molecular masses of LPMOs were estimated on SDS-PAGE. The samples were diluted in sample buffer containing 200 mM β-mercaptoethanol, 0.05% bromophenol blue, 5% SDS, and 50% glycerol in 225 mM Tris-HCl pH 6.8, and heated at 95 °C for 5 min before loading on Mini-protean® TGX™ 4–20% gels (Bio-Rad). PageRuler Prestained protein ladder (Thermo Scientific) was used as a molecular mass marker, and the gels were stained using a PageBlue™ protein staining solution (Thermo Scientific).
SDS-PAGE separated proteins were transferred using a Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad) to 0.45 μm nitrocellulose membranes (Bio-Rad). Mouse anti-His antibodies were used as primary antibodies and anti-mouse IgG antibodies conjugated to HRP were used as secondary antibodies (Invitrogen). The membranes were washed in TBS buffer that contained 2.42 g L−1 Tris-base, 8 g L−1 NaCl and 0.1% (v/v) Tween®20, pH 7.4. Visualisation was performed using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare) according to the manufacturer's instructions. The membranes were photographed using a Fujifilm LAS1000 camera running Image Reader LAS-1000 software version 2.6.
LPMO activity assay
The peroxidase activity of the purified LPMOs was determined spectrophotometrically (Cary 50 UV-VIS) against the soluble synthetic substrate 2,6-dimethoxyphenol (Sigma Aldrich). Formation of the reaction product (coerulignone) was monitored at a wavelength of 469 nm (ε469 nm 53200 cm−1 M−1). The assay contained appropriate amounts of the enzymes, 1 mM 2,6-DMP, 100 μM H2O2 and 50 mM sodium phosphate buffer pH 6.5 in a final volume of 1 mL, as described by Breslmayr et al.30 The reaction temperature was 25 °C. Three technical replicates were used for all assays. To determine their stability, the LPMOs were incubated for 0, 6, 24, 48, 72, and 120 h at 25 °C, after which their residual activity was assayed at 25 °C as described above.
LPMO oxidation of holocellulose and fibrillation
Holocellulose fibres (300 mg) were gently dispersed in 50 mM sodium phosphate buffer, pH 6.5 and soaked overnight. For the LPMO reactions, the fibres were diluted to a final concentration of 0.4% (w/v). The LPMO reactions contained 122 mg g−1 NcLPMO9E or 88 mg g−1 NcLPMO9F at an approximate molar ratio of 4 μmol g−1 of enzyme to cellulose, and 10 μM L-ascorbic acid as the electron donor for the enzymes. Reactions without LPMOs were conducted as controls. The reactions were performed at 25 ± 2 °C under magnetic stirring for 2 days. The reactions were stopped by separating the fibres from the suspension. To remove the enzymes, the fibres were dispersed in 10 μM NaOH pH 9.0 and incubated overnight. NaOH was removed by extensive washing with ultrapure water in a vacuum filtration unit equipped with a Durapore® 0.65 μM DVPP membrane (Merck). The LPMO-treated and the control holocellulose fibres were suspended in 500 mL distilled water and homogenised for 2 min at 25000 rpm with an IKA T 25 digital ULTRA-TURRAX©, corresponding to 0.02 kW h.
Preparation of nanopapers
100 mg of the homogenised fibres was gently suspended in ultrapure water at a final concentration of 0.05% (w/w). Water was removed from the suspensions with a vacuum-filtration setup equipped with a filter funnel (Φ = 70 mm) and filter membrane (0.22 μm, DVPP). The wet films obtained from filtration were carefully peeled off and placed between two stainless steel meshes. The films were dried under pressure at 50 °C for 24 h.Characterisation
The chemical composition of the LPMO-treated and unmodified holocellulose fibres was determined by carbohydrate analysis. 100 mg of the fibres were hydrolysed with sulphuric acid, and the monosaccharides were quantified using a Dionex ICS-3000 chromatography system (Thermo Fisher Scientific, USA). The amount of glucose in glucomannan was estimated based on the glucose and mannose contents using a 1:3 ratio of glucose:mannose and subsequently subtracted out when calculating the cellulose content. Soluble sugars released from the holocellulose fibres by NcLPMO9E and NcLPMO9F treatment were determined by the phenol–sulphuric acid method. Samples of 100 μL were taken from the supernatants of the LPMO reactions and mixed with 100 μl of 5% phenol and 500 μl of 98% sulphuric acid. The absorbance was measured after 10 min at 490 nm, and the soluble sugars were quantified as glucose equivalent units against a standard curve of glucose.31 The extent of the fibrillation was examined under an inverted light microscope (Nikon Eclipse Ti-S, USA) running imaging software Nis-Elements version 4.60. The solid content of the homogenised holocellulose fibres was adjusted to 0.1% (w/w) before observation. Transmission electron microscopy (TEM) observations were conducted using a Hitachi Model HT7700 transmission electron microscope operated in high-contrast mode at 100 kV. The specimens were prepared by depositing a drop of the dilute water suspension of cellulose nanofibres on a freshly glow-discharged, carbon-coated grid and stained with 1% uranyl acetate as a negative stain. Atomic force microscopy (AFM) analysis was performed on a MultiMode 8 Atomic Force Microscope (Digital Instruments, Inc., USA) in the ScanAsyst® mode. The X-ray diffraction (XRD) patterns were recorded using a Philips X'Pert Pro diffractometer (model PW 3040/60) in the reflection mode (5–35° 2θ angular range, steps of 0.05°). CuKα radiation (λ = 1.5418 Å) was generated at 45 kV and 40 mA and monochromatised using a 20 μm Ni-filter. Diffractograms were recorded from rotating specimens using a position sensitive detector. The crystallinity index (CI) of cellulose was calculated from the ratio between the intensity of the 200 peak (I200) and the intensity of the minimum (IAM) between the 200 and 110 peaks.32 The zeta (ζ) potentials of the holocellulose fibres were measured using a Zetasizer nano ZS instrument (Malvern, Worcestershire, UK) at 25 °C following the Smoluchowski method. For the measurements, the fibres were suspended in distilled water at a final concentration of 0.05% (w/v) and disintegrated by sonication (amplitude 30%, duration 30 s, pulse 50%). The amounts of the carboxyl groups introduced by the LPMOs were determined by conductometric titration. 100 g suspensions containing 50 mg of the holocellulose fibres were adjusted to a pH value of 2.5 with 0.1 M HCL. The suspensions were titrated with 0.01 M NaOH and the conductivity was monitored with a conductometric station (SevenCompact, Mettler-Toledo). The titration curve showed the typical presence of strong and weak acid groups. The amount of strong acid corresponded to the added HCl, and that of the weak acid corresponded to the carboxyl content. Fourier transform infrared spectroscopy (FTIR) was performed by using a PerkinElmer Spectrum 2000 instrument equipped with an MKII Golden Gate Single Reflection ATR system from Specac Ltd, UK. The ATR crystal was an MKII heated diamond 45° ATR top plate. The measurement was performed in a spectral range of 600–4000 cm−1 with a resolution of 4 cm−1.
The nanopaper surfaces and their cross-sections after freeze-fracture were analysed by field-emission scanning electron microscopy (FE-SEM) (Hitachi S-4800). The samples were attached with carbon tape onto metal stubs and then coated with platinum/palladium using a sputter coater (Cressington 208HR). The mechanical properties of the nanopapers were tested under uniaxial tension with a Universal testing machine (Instron 5944, USA). The prepared nanopapers were cut into strips of 3 mm in width and conditioned at a relative humidity of 50% for 2 d. At least ten specimens were tested from each sample and the results are reported as an average of at least five specimens. The test was performed by using a strain rate of 2 mm min−1 and a gauge length of 20 mm. The modulus was determined from the slope of the initial low strain region of the stress–strain curve. Toughness, defined as work to fracture, was calculated as the area under the stress–strain curve.
Results and discussion
Activity of NcLPMO9E and NcLPMO9F
The apparent molecular weights of the recombinant enzymes, 43 kDa and 25 kDa for NcLPMO9E and NcLPMO9F (Fig. 1), respectively, corresponded well to those reported in a previous study where the enzymes were produced from P. pastoris using a similar protocol,33 but without the C-terminal purification tag (His)6 utilised in this study. In order to confirm the activity of the LPMOs, we utilised a spectrophotometric assay that detects coloured oxidation products of the synthetic substrate 2,6-dimethoxyphenol (2,6-DMP) from an LPMO-catalysed peroxidase reaction.30 The peroxidase reaction is dependent on the presence of catalytic copper on the enzyme's active sites, and as it can only be demonstrated by LPMOs that are correctly processed and folded by the host, it could be used to confirm the activity of the recombinant LPMOs. Indeed, both of the N. crassa LPMOs were active on 2,6-DMP at pH 6.5 and 25 °C, and these conditions were also selected for later experiments using a more complex, natural cellulosic substrate. Under these conditions, the activity towards the synthetic substrate 2,6-DMP was observed to be higher for the NcLPMO9E than the NcLPMO9F, the highest observed specific activities being 105 nkat mmol−1 and 25 nkat mmol−1, respectively (Fig. 2). The observed differences in the activities between the two enzymes corresponded to previous results utilising synthetic substrates.33 While the peroxidase activity on the soluble synthetic substrate cannot be used to predict the activity on the native insoluble cellulosic substrate,28,29,34 the assay is a convenient method to characterise the stability of the enzymes under certain reaction conditions to estimate the appropriate reaction time. The results showed that the modular NcLPMO9E demonstrated good stability at 25 °C, retaining 40% and 20% of its original activity after 48 h and 120 h, respectively. The non-modular NcLPMO9F was less stable under the same conditions, as the activity was observed to be lost after 48 h of incubation.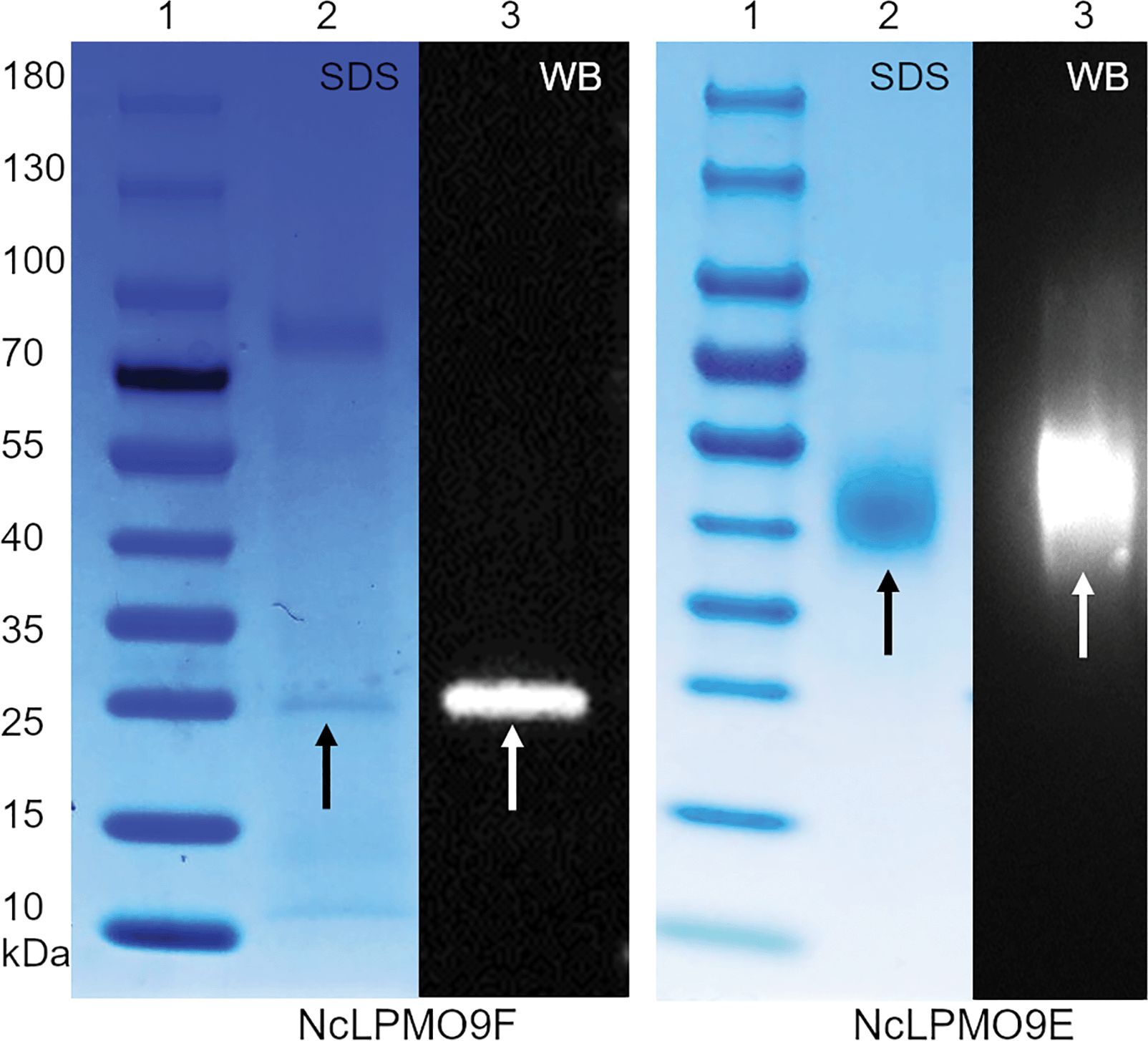 | ||
| Fig. 1 SDS-PAGE and western blot analyses of the recombinantly produced N. crassa LPMOs NcLPMO9F and NcLPMO9E. In each case, lane 1 shows a size marker protein ladder, and lanes 2 and 3 contain the produced LPMOs. The bands indicated by arrows show the heterologously expressed proteins. SDS = SDS-PAGE and WB = western blot. | ||
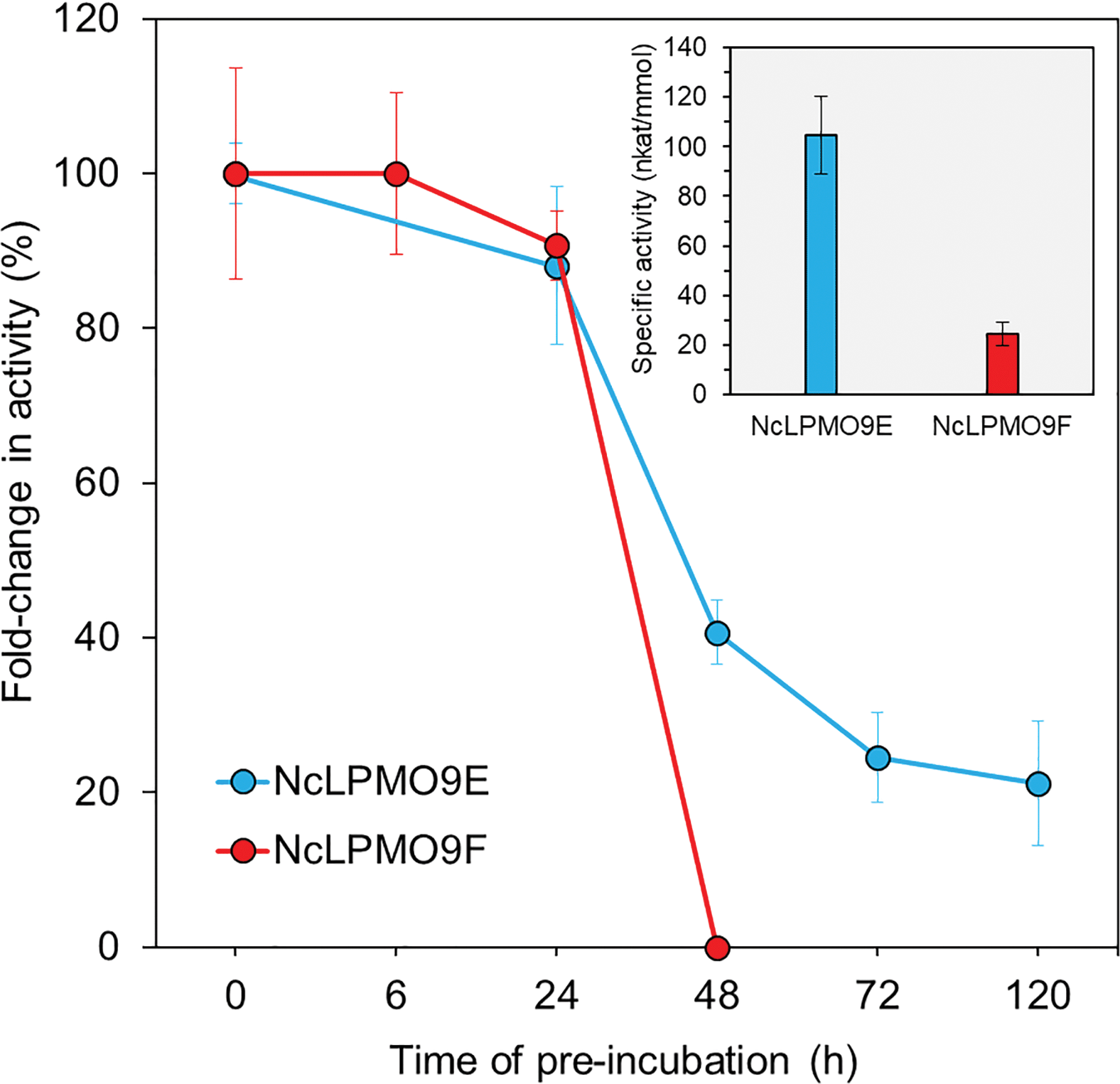 | ||
| Fig. 2 Specific activity and stability of the LPMOs measured using a 2,6-DMP assay at pH 6.5. The stability was measured after pre-incubation at 25 °C for increasing lengths of time. | ||
LPMO-assisted holocellulose fibrillation
Spruce holocellulose was incubated with NcLPMO9E and NcLPMO9F for 48 h before brief mechanical homogenisation. The enhanced fibrillation resulting from the LPMO treatment was clearly visible in the fibre suspensions after the mechanical treatment (Fig. 3a–c). This was further examined by using inverted light microscopy (Fig. 3d–f) and TEM (Fig. 3g and h). Microscopy analysis revealed extensive fibrillation down to the nanoscale resulting from the LPMO treatments, while the control non-enzymatically treated fibres remained relatively intact and could not be analysed by TEM. The nanofibres obtained after the LPMO treatments were well individualised with some aggregations due to drying during sample preparation for TEM observation. Of the two studied enzymes, the CBM-lacking NcLPMO9F yielded smaller nanofibres with an average width of approximately 5 nm (Fig. 3h), while the nanofibres resulting from the CBM-containing NcLPMO9E-treatment had an average width of around 9 nm (Fig. 3g). This was also confirmed by AFM analysis as shown in Fig. S7, ESI.† The NcLPMO9F oxidised nanofibres had a width ranging from 2 to 8 nm and an average value of 4.3 ± 1.5 nm, while NcLPMO9E oxidised nanofibres had a width from 3 to 14 nm and an average of 6.7 ± 2.5 nm, respectively. The width measured by TEM was slightly higher due to the negative staining. The length of both nanofibres was longer than 1 μm and the fibre ends were not apparent in the TEM images due to aggregation. The widths of these nanofibres are significantly smaller compared to what has been previously observed for kraft pulp treated with other LPMOs.20,22 The widths are also smaller than that of the nanofibres prepared by using a pre-treatment with glycoside hydrolases followed by much stronger mechanical disintegration using a microfluidizer (10–40 nm).12,35 These results suggest that the N. crassa LPMOs made the holocellulose more susceptible to mild mechanical homogenisation (25000 rpm, 2 min) and facilitated the production of CNFs. The yield of the nanofibres was assessed by mass recovery compared to the control sample. The yields of nanofibres from the non-modular NcLPMO9F and CBM-containing NcLPMO9E treated holocellulose were 81.8 ± 5.0% and 65.4 ± 0.5%, respectively. The analysis of soluble sugar in the supernatant confirmed that the lower nanofibre yield obtained with the CBM-containing NcLPMO9E correlated with a higher amount of released glucose equivalents (25.2 ± 1.2 mg g−1 of cellulose), compared to the non-modular NcLPMO9F, which released 10.7 ± 0.9 mg g−1 glucose equivalents. The different mass recoveries and varying amounts of soluble sugars most likely resulted from different modes of action of the modular and non-modular enzymes. A similar effect of the CBM-module on the sugar release has been recently reported by Chalak et al.36
 | ||
| Fig. 3 Photographs of the fibre suspensions (0.1%, w/v) and inverted light microscopy images of the holocellulose fibres illustrate the changes in fibre dimensions after treatment with the LPMOs followed by mild homogenisation. TEM images reveal the morphology of the produced CNFs. Control without enzyme-treatment (a and d) and holocellulose treated with NcLPMO9E (b, e and g) and NcLPMO9F (c, f and h), respectively. | ||
To assess the predominant mechanism by which the N. crassa LPMOs were able to enhance the mechanical fibrillation of holocellulose, total sugar analysis, X-ray diffraction (XRD) and ζ-potential measurements were carried out to determine the sugar compositions, crystallinities and surface charges of the LPMO-oxidised fibres, respectively. Before the LPMO reaction, the holocellulose was autoclaved and the hemicelluloses (59 wt% of xylan and 22 wt% of glucomannan) were partially removed (Table S1, ESI†). The purpose was to expose the cellulose surface and avoid hemicellulose precipitation on cellulose after the delignification step using PAA, and increase the LPMO accessibility. The hemicellulose content did not change significantly after the LPMO treatment (Table S1, ESI†), which is different from the TEMPO-mediated oxidation method where the hemicelluloses are partially removed after the oxidation step.37 The glucose content of the LPMO-treated holocellulose decreased significantly due to the difficulties in hydrolysis of aglycones after the formation of aldonic acids at the reducing ends.
The results from XRD revealed that both LPMOs induced a moderate reduction by 2% in the cellulose crystallinity compared to the control sample (Fig. 4), indicating that the LPMOs mainly targeted the crystalline regions of cellulose.38 However, no significant difference in crystallinity was found between the holocellulose nanofibres oxidised by the CBM-containing NcLPMO9E and those oxidised by the non-modular NcLPMO9F. The surface charges obtained from the ζ-potential measurements showed a 1.5-fold increase in both enzyme-treated holocellulose fibres compared to the non-enzymatically treated fibres (Fig. 5). The obtained ζ-potential values (−37.6 ± 3.1 mV for NcLPMO9E and −37.7 ± 2.4 mV for NcLPMO9F) suggested stable colloidal suspensions, which were speculated to result from the presence of deprotonated C1-carboxyl groups on the fibre surfaces.19 The ζ-potential of the control holocellulose fibres (−25.4 ± 1.5 mV) corresponded well to previous results obtained with the same type of fibres.27 Conductometric titration on the fibres also revealed that the LPMOs increased the carboxyl content by 27% and 88% compared to the control. The obtained carboxyl contents were 0.38 and 0.53 mmol g−1 for NcLPMO9E and NcLPMO9F, respectively (Fig. 5). The carboxyl content was observed to be 38% higher when the fibres were treated with the CBM-lacking NcLPMO9F, compared to the modular LPMO. The difference in carboxyl content could result from more restricted movement of the CBM-containing NcLPMO9E on the cellulose surface. However, as inferred from the ζ-potential, the particle charge density appeared to be similar for the nanofibres produced by both enzymes. This is probably due to the fact that the width of the nanofibres produced by NcLPMO9E was almost twofold compared to that of the nanofibres produced by NcLPMO9F, as measured by TEM (Fig. 3). These results indicate that the fibrillation was facilitated by better dispersion of the fibres resulting from the repulsion between the LPMO-generated C1 carboxyl groups on fibre surfaces, rather than the disruption in the ordered crystalline areas of the fibres by the LPMOs. The carboxyl content obtained by using the CBM-lacking NcLPMO9F was 0.53 mmol g−1, similar to that of CNFs generated by partial carboxymethylation of fibres followed by fibrillation, which has a charge of 515 μequiv. g−1.7 This value is five times higher than the previously reported value when LPMO was used together with endoglucanase and xylanase on hardwood kraft pulp.22 Interestingly, TEMPO-mediated oxidation of spruce was also found to be more efficient with higher carboxylate contents and higher stability of the nanofibre dispersion than the oxidation of eucalyptus.37
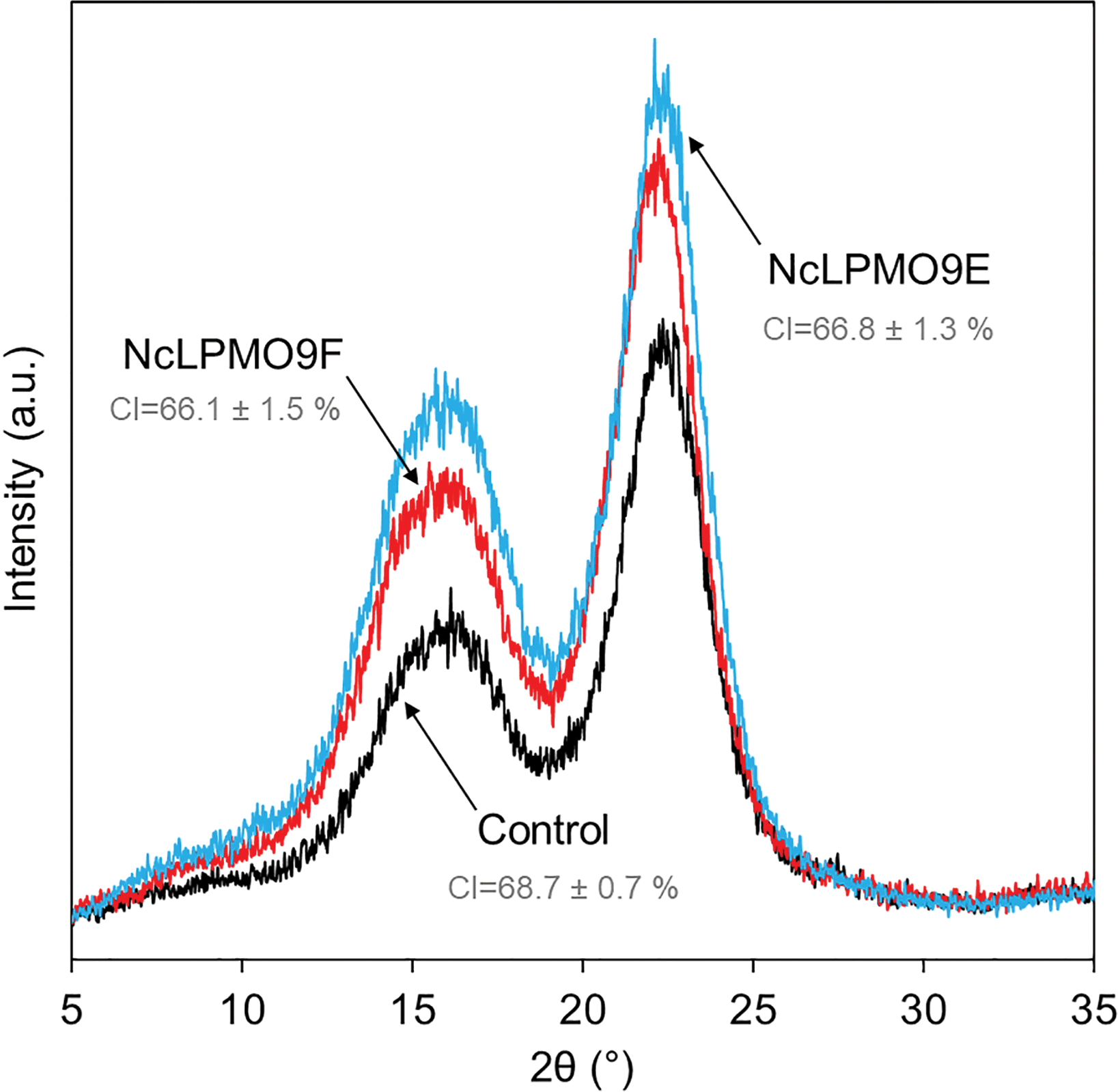 | ||
| Fig. 4 X-ray diffraction (XRD) patterns of the LPMO-oxidised holocellulose and the non-oxidised control fibres. CI = crystallinity index. | ||
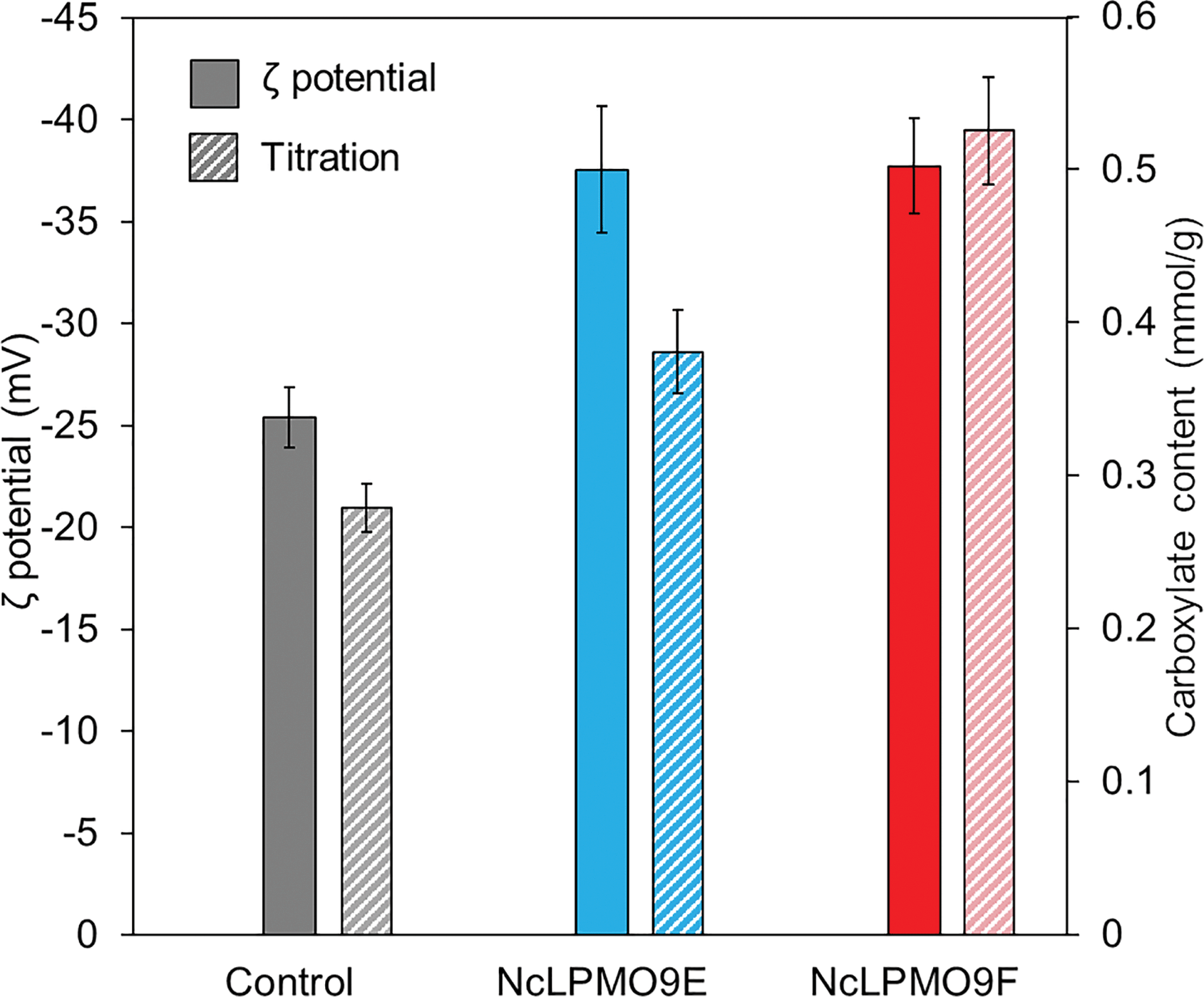 | ||
| Fig. 5 ζ-Potentials and carboxylate contents from conductometric titration of holocellulose suspensions (0.05%, w/v) treated with two N. crassa LPMOs, NcLPMO9E and NcLPMO9F, as indicated. The ‘control’ sample represents holocellulose that was not treated with LPMO. The increased ζ-potential following LPMO treatment indicates the presence of negative charges of the deprotonated carboxyl group on the fibre surface resulting from the LPMO-reaction. | ||
The presence of the LPMO-induced C1-carboxyl groups in the nanofibres was also confirmed by FTIR. The FTIR spectra showed distinct changes in the region associated with carboxyl groups derived from the LPMO treatment (Fig. 6). The observed peaks at 1733 cm−1 were attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of carbonyl groups from the partially acetylated galactoglucomannans in the holocellulose. A new peak at approximately 1595 cm−1 was observed for the LPMO-oxidised holocellulose, which corresponds to the asymmetric stretching vibrations of the deprotonated carboxyl groups from the aldonic acids at the reducing ends. The new peak was more distinct for the NcLPMO9F-oxidised nanofibres, which correlates with the conductometric titration results.
O stretching frequency of carbonyl groups from the partially acetylated galactoglucomannans in the holocellulose. A new peak at approximately 1595 cm−1 was observed for the LPMO-oxidised holocellulose, which corresponds to the asymmetric stretching vibrations of the deprotonated carboxyl groups from the aldonic acids at the reducing ends. The new peak was more distinct for the NcLPMO9F-oxidised nanofibres, which correlates with the conductometric titration results.
 | ||
| Fig. 6 FTIR spectra of holocellulose fibres treated with two C1-active LPMOs, NcLPMO9E and NcLPMO9F. The curves have been normalised at 1028 cm−1. The peaks appeared at 1733 and 1595 cm−1 corresponded to –CO and –COONa, respectively. | ||
Structure and mechanical properties of the nanopapers
The nanopapers were prepared from water suspensions of the nanofibres by vacuum filtration akin to the papermaking procedure. As shown in Fig. 7c and e, a fibrous nanofibril network structure with a random-in-plane orientation was present on the surfaces of the nanopaper prepared from the LPMO-oxidised nanofibres, while large cellulose fibril aggregates were observed in the control nanopaper from holocellulose without the LPMO treatment (Fig. 7a). A uniform layered structure was apparent in the cross-sections of the nanopapers prepared from the LPMO-oxidised nanofibres, resulting from nanofibre deposition during vacuum filtration (Fig. 7d and f). The cross-section of the control nanopaper also showed a layered structure, but the layers were heterogeneous with large aggregates and voids (Fig. 7b). This is due to the fact that the holocellulose was only partially fibrillated by mild homogenisation without LPMO treatment. Fig. 8 shows typical stress–strain curves under uniaxial tension for the holocellulose nanopapers, and the physical and mechanical properties of the nanopapers are summarised in Table S2, ESI.† The mechanical performance of the nanopapers was substantially improved when the nanofibres were prepared using the LPMOs, particularly with NcLPMO9F which did not contain a CBM. The tensile strengths were 257.0 ± 6.2 MPa and 262.2 ± 10.1 MPa for NcLPMO9E and NcLPMO9F, which are 73% and 76% higher than that of the control (148.7 ± 4.2 MPa), respectively. The strain-to-failure values of the nanopapers prepared from the LPMO-oxidised CNFs were also higher (4.2 ± 0.7% and 3.7 ± 0.4%) compared to the control paper, which failed at 2.6 ± 0.3%. The improved mechanical properties derived from the LPMO oxidation were attributed to a better fibrillation and production of nanofibres with a width below 10 nm, which led to enhanced entanglement of the nanofibre network and allowed inter-fibril slippage during uniaxial tension before fibril breakage. This resulted in higher strain-to-failure and higher tensile strength after strain hardening, and thus higher toughness, i.e. work of fracture, for the nanopapers. This is consistent with the scaling law of the mechanical properties of cellulose nanopapers: the smaller, the stronger and the tougher, as reported by Zhu et al.39 The tensile strength (262 MPa) and modulus (16.3 GPa) of the nanopaper obtained with NcLPMO9F-oxidised CNFs were also higher than those of the MFC films prepared by enzymatic pre-treatment using glycoside hydrolases, which had a tensile strength of 214 MPa and a modulus of 13.2 GPa,35 owing to a much thinner width of LPMO-oxidised CNFs compared to the MFC nanofibres prepared using cellulases. | ||
| Fig. 7 FE-SEM micrographs of the nanopapers prepared from the control holocellulose (a, b), NcLPMO9E-oxidised holocellulose nanofibres (c and d), and NcLPMO9F-oxidised holocellulose nanofibres (e and f). The left and right columns show the nanopaper surfaces and cross-sections created with freeze-fracture, respectively. | ||
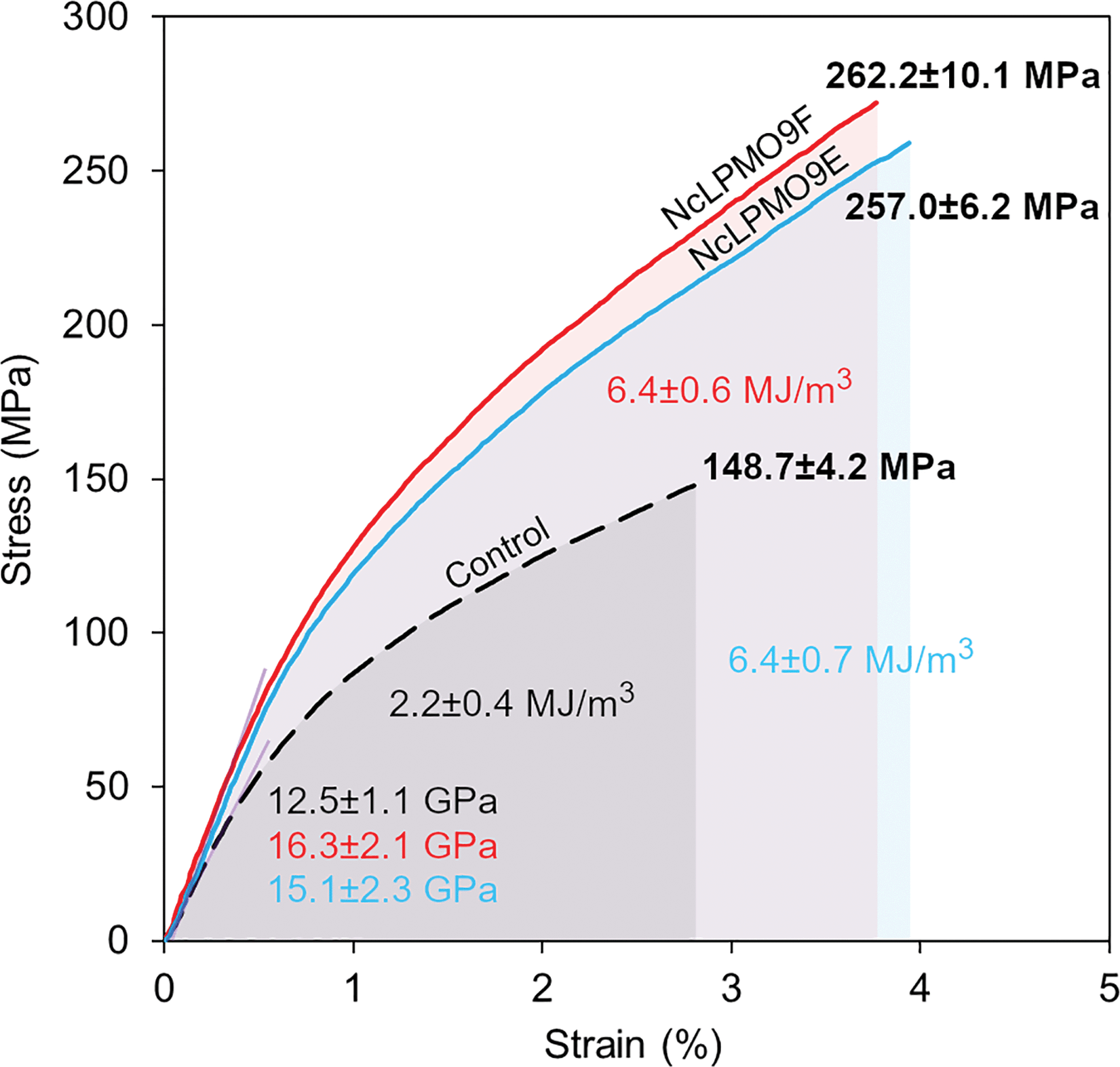 | ||
| Fig. 8 Representative stress–strain curves for nanopapers prepared from the holocellulose fibres. Control fibres without enzyme treatment (black), NcLPMO9E-treated fibres (blue), and NcLPMO9F-treated fibres (red). | ||
Impact of the carbohydrate-binding module
The CBM-lacking NcLPMO9F efficiently introduced carboxyl groups on the cellulose fibre surfaces, resulting in better fibrillation and production of nanofibres with a thinner width compared to the CBM-containing NcLPMO9E. This also resulted in better mechanical properties of the prepared nanopapers. While the surface charge densities (ζ-potentials) of the prepared nanofibres were similar after oxidation with both C1-active LPMOs from N. crassa, the carboxyl contents were significantly different, as the CBM-lacking NcLPMO9F was able to generate 38% more carboxyl croups (0.53 mmol g−1) than the CBM-containing NcLPMO9E (0.38 mmol g−1). This difference in activity is most likely due to enhanced movement of the CBM-lacking enzyme on the cellulose surfaces, leading to a less localised oxidation pattern during the reaction on cellulose. Indeed, it has been previously shown that a CBM might increase the residence time of LPMOs on the substrate, confining the oxidation to a more specified area,26 indicating that an LPMO without an appended CBM might be free to perform oxidation in a more disperse manner over the entire cellulose surface. It was also found that the nanofibre yield was higher when the fibres were treated with the non-modular NcLPMO9F, while the CBM-containing NcLPMO9E was able to release more soluble sugars from the holocellulose substrate. This is most likely due to the localisation of the CBM-containing enzyme and subsequently higher amount of proximate cleavages of the cellulose chains enabling the release of shorter products compared to the freely moving enzyme. Similar ζ-potentials and different carboxylate contents after the LPMO oxidation might be due to the fact that the ζ-potential reflects the surface charge density rather than the total charges, as a higher total charge in nanofibres is often related to a thinner width and a larger surface area. Such a result was also observed in TEMPO oxidation of cellulose from different origins, where the selective oxidation of C6 primary hydroxyls yielded nanofibres with similar ζ-potentials regardless of the total carboxylate contents.40Conclusions
In summary, we have successfully produced CNFs from delignified softwood fibres by a completely green and energy-efficient method utilising oxidative enzymes, C1-active LPMOs from N. crassa. Lignin was first removed from the spruce fibres with PAA, followed by enzymatic oxidation of cellulose with the LPMOs. After these pre-treatments, CNFs with a thin width (<10 nm) could be prepared by a rather mild mechanical homogenisation. Thinner nanofibres (width of 4.3 ± 1.5 nm) with a higher carboxylate content (0.53 mmol g−1) were obtained from spruce holocellulose by using a CBM-lacking LPMO, NcLPMO9F, compared to the modular LPMO, NcLPMO9E, owing to the free movement of the CBM-lacking enzyme and more homogeneous oxidation over the fibre surface. The non-modular NcLPMO9F also enabled the production of higher yields of nanofibres, 82% from the original fibre mass. These results suggest that LPMOs without CBMs might be better suited for cellulose nanofibrillation purposes than CBM-containing LPMOs. The LPMO-oxidised nanofibres were further used to prepare cellulose nanopaper films. The films showed a high tensile strength of 262 MPa and Young's modulus of 16.2 GPa under uniaxial tension, higher than those for nanofibres from enzymatic treatment with cellulases and TEMPO-mediated oxidation. These results demonstrate the unique utility of LPMOs in cellulose nanomaterial preparation, reducing the use of toxic chemicals and high energy for nanofibrillation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Swedish Research Council VR (2015-05030), the Swedish Foundation for International Cooperation in Research and Higher Education STINT (CH2017-7275) and Wallenberg Wood Science Centre (WWSC) for supporting this work. S. Wang thanks the China Scholarship Council (CSC) for supporting his PhD study.Notes and references
- M. Smith, D. C. Love, C. M. Rochman and R. A. Neff, Curr. Environ. Health Rep., 2018, 5, 375–386 CrossRef CAS PubMed.
- A. A. D. Machado, W. Kloas, C. Zarfl, S. Hempel and M. C. Rillig, Global Change Biol., 2018, 24, 1405–1416 CrossRef PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- O. Nechyporchuk, M. N. Belgacem and J. Bras, Ind. Crops Prod., 2016, 93, 2–25 CrossRef CAS.
- T. Saito and A. Isogai, Biomacromolecules, 2004, 5, 1983–1989 CrossRef CAS PubMed.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnas, Langmuir, 2008, 24, 784–795 CrossRef PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- A. H. Pei, N. Butchosa, L. A. Berglund and Q. Zhou, Soft Matter, 2013, 9, 2047–2055 RSC.
- Q. Q. Wang, J. Y. Zhu, R. Gleisner, T. A. Kuster, U. Baxa and S. E. McNeil, Cellulose, 2012, 19, 1631–1643 CrossRef CAS.
- L. H. Chen, J. Y. Zhu, C. Baez, P. Kitin and T. Elder, Green Chem., 2016, 18, 3835–3843 RSC.
- M. Henriksson, G. Henriksson, L. A. Berglund and T. Lindström, Eur. Polym. J., 2007, 43, 3434–3441 CrossRef CAS.
- W. X. Wang, M. D. Mozuch, R. C. Sabo, P. Kersten, J. Y. Zhu and Y. C. Jin, Cellulose, 2015, 22, 351–361 CrossRef CAS.
- K. Hildén and M. R. Mäkelä, in Reference Module in Life Sciences, Elsevier, 2018, DOI:10.1016/B978-0-12-809633-8.12424-0.
- J. A. Langston, T. Shaghasi, E. Abbate, F. Xu, E. Vlasenko and M. D. Sweeney, Appl. Environ. Microbiol., 2011, 77, 7007–7015 CrossRef CAS PubMed.
- A. V. Monclaro and E. X. Ferreira, Int. J. Biol. Macromol., 2017, 102, 771–778 CrossRef CAS PubMed.
- M. Dimarogona, E. Topakas and P. Christakopoulos, Comput. Struct. Biotechnol. J., 2012, 2, e201209015 CrossRef PubMed.
- B. Westereng, J. W. Agger, S. J. Horn, G. Vaaje-Kolstad, F. L. Aachmann, Y. H. Stenstrøm and V. G. H. Eijsink, J. Chromatogr. A, 2013, 1271, 144–152 CrossRef CAS PubMed.
- M. Eibinger, T. Ganner, P. Bubner, S. Rošker, D. Kracher, D. Haltrich, R. Ludwig, H. Plank and B. Nidetzky, J. Biol. Chem., 2014, 289, 35929–35938 CrossRef CAS PubMed.
- A. Villares, C. Moreau, C. Bennati-Granier, S. Garajova, L. Foucat, X. Falourd, B. Saake, J. G. Berrin and B. Cathala, Sci. Rep., 2017, 7, 40262 CrossRef CAS PubMed.
- S. Ladevèze, M. Haon, A. Villares, B. Cathala, S. Grisel, I. Herpoël-Gimbert, B. Henrissat and J. G. Berrin, Biotechnol. Biofuels, 2017, 10, 215 CrossRef PubMed.
- J. Hu, D. Tian, S. Renneckar and J. N. Saddler, Sci. Rep., 2018, 8, 3195 CrossRef PubMed.
- C. Moreau, S. Tapin-Lingua, S. Grisel, I. Gimbert, S. Le Gall, V. Meyer, M. Petit-Conil, J. G. Berrin, B. Cathala and A. Villares, Biotechnol. Biofuels, 2019, 12, 156 CrossRef PubMed.
- C. Valls, F. I. J. Pastor, M. B. Roncero, T. Vidal, P. Diaz, J. Martínez and S. V. Valenzuela, Biotechnol. Biofuels, 2019, 12, 161 CrossRef PubMed.
- L. I. Crouch, A. Labourel, P. H. Walton, G. J. Davies and H. J. Gilbert, J. Biol. Chem., 2016, 291, 7439–7449 CrossRef CAS PubMed.
- G. Courtade, Z. Forsberg, E. B. Heggset, V. G. H. Eijsink and F. L. Aachmann, J. Biol. Chem., 2018, 293, 13006–13015 CrossRef CAS PubMed.
- X. Yang, F. Berthold and L. A. Berglund, Biomacromolecules, 2018, 19, 3020–3029 CrossRef CAS PubMed.
- C. M. Phillips, W. T. Beeson, J. H. Cate and M. A. Marletta, ACS Chem. Biol., 2011, 6, 1399–1406 CrossRef CAS PubMed.
- V. V. Vu, W. T. Beeson, C. M. Phillips, J. H. D. Cate and M. A. Marletta, J. Am. Chem. Soc., 2014, 136, 562–565 CrossRef CAS PubMed.
- E. Breslmayr, M. Hanžek, A. Hanrahan, C. Leitner, R. Kittl, B. Šantek, C. Oostenbrink and R. Ludwig, Biotechnol. Biofuels, 2018, 11, 79 CrossRef PubMed.
- M. DuBois, K. A. Gilles, J. K. Hamilton, P. A. Rebers and F. Smith, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- H. Tang, N. Butchosa and Q. Zhou, Adv. Mater., 2015, 27, 2070–2076 CrossRef CAS PubMed.
- R. Kittl, D. Kracher, D. Burgstaller, D. Haltrich and R. Ludwig, Biotechnol. Biofuels, 2012, 5, 79 CrossRef CAS PubMed.
- A. Várnai, K. Umezawa, M. Yoshida and V. G. H. Eijsink, Appl. Environ. Microbiol., 2018, 84, e00156–e00118 CrossRef PubMed.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed.
- A. Chalak, A. Villares, C. Moreau, M. Haon, S. Grisel, A. d'Orlando, I. Herpoël-Gimbert, A. Labourel, B. Cathala and J.-G. Berrin, Biotechnol. Biofuels, 2019, 12, 206 CrossRef PubMed.
- G. Rodionova, T. Saito, M. Lenes, O. Eriksen, O. Gregersen, R. Kuramae and A. Isogai, J. Polym. Environ., 2013, 21, 207–214 CrossRef CAS.
- K. E. H. Frandsen and L. Lo Leggio, Iucrj, 2016, 3, 448–467 CrossRef CAS PubMed.
- H. L. Zhu, S. Z. Zhu, Z. Jia, S. Parvinian, Y. Y. Li, O. Vaaland, L. B. Hu and T. Li, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8971–8976 CrossRef CAS PubMed.
- Y. Okita, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1696–1700 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sugar analysis, mechanical properties data, gene sequences, restriction pattern analysis, colony PCR, SDS-PAGE and western-blot. See DOI: 10.1039/c9gc02808k |
| This journal is © The Royal Society of Chemistry 2019 |