
Cellulose conversion into lactic acid over supported HPA catalysts†

Abstract
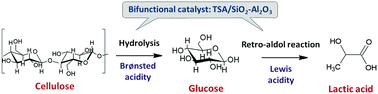
Lactic acid is one of the most important high added value chemicals with various applications in diverse fields, e.g. food industry, pharmaceuticals, and plastics. Recently, it has been gaining even more attention as the starting material for the synthesis of bio-based chemicals such as acrylic acid, 2,3-pentanedione and acetaldehyde. The current industrial process for lactic acid synthesis is based on enzymatic fermentation of carbohydrates (e.g. glucose and sucrose), which is a sensitive process in terms of feed quality/purity, requires strict control of the reaction conditions and produces a considerable amount of waste. Therefore, increasing research effort is being placed on the development of alternative green and sustainable chemocatalytic processes. Lactic acid can be synthesized from glucose via a retro-aldol reaction pathway, which is favored under basic conditions, or via catalysts with pronounced Lewis acidity. On the other hand, glucose is produced from cellulose hydrolysis, which requires Brønsted acidity. In view of developing a one-pot chemocatalytic process for the synthesis of lactic acid from cellulose, the present work investigates the effect of oxides (SiO2, SiO2-Al2O3, Nb2O5, Nb2O5-SiO2, Nb2O5-Al2O3), heteropolyacids (HPAs) (TSA, PTA) and supported HPAs on the oxides as bifunctional catalysts with varying ratios of Lewis to Brønsted acid sites on cellulose conversion into lactic acid. According to the experimental results, the type of acidity played a key role in the reaction pathway and consequently in the product distribution. Among the supported catalysts tested, TSA/SiO2-Al2O3 with the highest Lewis to Brønsted acidity ratio led to the highest lactic acid selectivity (38.4%) and yield (23.5%), at 61.2% cellulose conversion. For this catalyst the effect of reaction conditions, catalyst concentration, cellulose crystallinity and the presence of other biomass components (hemicellulose and lignin) was further evaluated. The stability and reuse of TSA/SiO2-Al2O3 were confirmed for at least 3 reaction cycles, whereas the reaction pathway of cellulose conversion was confirmed and validated via a power law kinetic modeling scheme.
- This article is part of the themed collection: International Symposium on Green Chemistry 2019
 Please wait while we load your content...
Please wait while we load your content...

