
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation of secondary alcohols using solid-supported hypervalent iodine catalysts†
Frederic
Ballaschk
and
Stefan F.
Kirsch
 *
*
Organic Chemistry, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany. E-mail: sfkirsch@uni-wuppertal.de
First published on 3rd October 2019
Abstract
It is shown how secondary alcohols are oxidized to provide the corresponding ketones by use of Oxone® and solid-supported hypervalent iodine catalysts. Under experimentally simple conditions with acetonitrile at elevated temperatures, excellent conversions were achieved with low catalyst loadings (0.2–5 mol%) when employing the conjugates 5 and 6 derived from IBX and IBS. The catalysts are broadly applicable to a range of alcohol substrates. Of primary importance with respect to sustainability issues, the metal-free catalysts are easily removed from the reaction mixture through filtration, and they can be re-used in oxidation processes for multiple times, without loss of catalytic activity.
Introduction
The formation of carbonyls through alcohol oxidation is one of the most fundamental reactions in contemporary chemistry. A great plethora of methods already exist to fulfill this task, and hypervalent iodine reagents belong to the most applied oxidation agents.1–5 For example, the Dess–Martin periodiane (DMP) became the standard textbook reagent for all kinds of alcohol oxidation, after its invention in 1983.6 Its parent compound, 2-iodoxybenzoic acid (IBX, 1),7–9 is even older and was reported first in 1893 by Hartmann and Meyer (Scheme 1).10 Since then, IBX was mainly forgotten over more than half a century before it experienced a renaissance with the breakthrough reports by Frigerio and Santagostino in 1994: IBX was shown to be a moisture-stable and easy-to-handle reagent for the mild oxidation of alcohols in DMSO at room temperature.11,12 The IBX oxidation of alcohols then emerged as a standard transformation in organic synthesis with numerous applications in complex molecule synthesis.13 Nowadays IBX is considered one of the most versatile oxidizing agents, with an extraordinary array of reactivities beyond alcohol oxidation, including phenol oxidation,14–17 functional group transfer onto activated methylene compounds,18–22 and dehydrogenations.23,24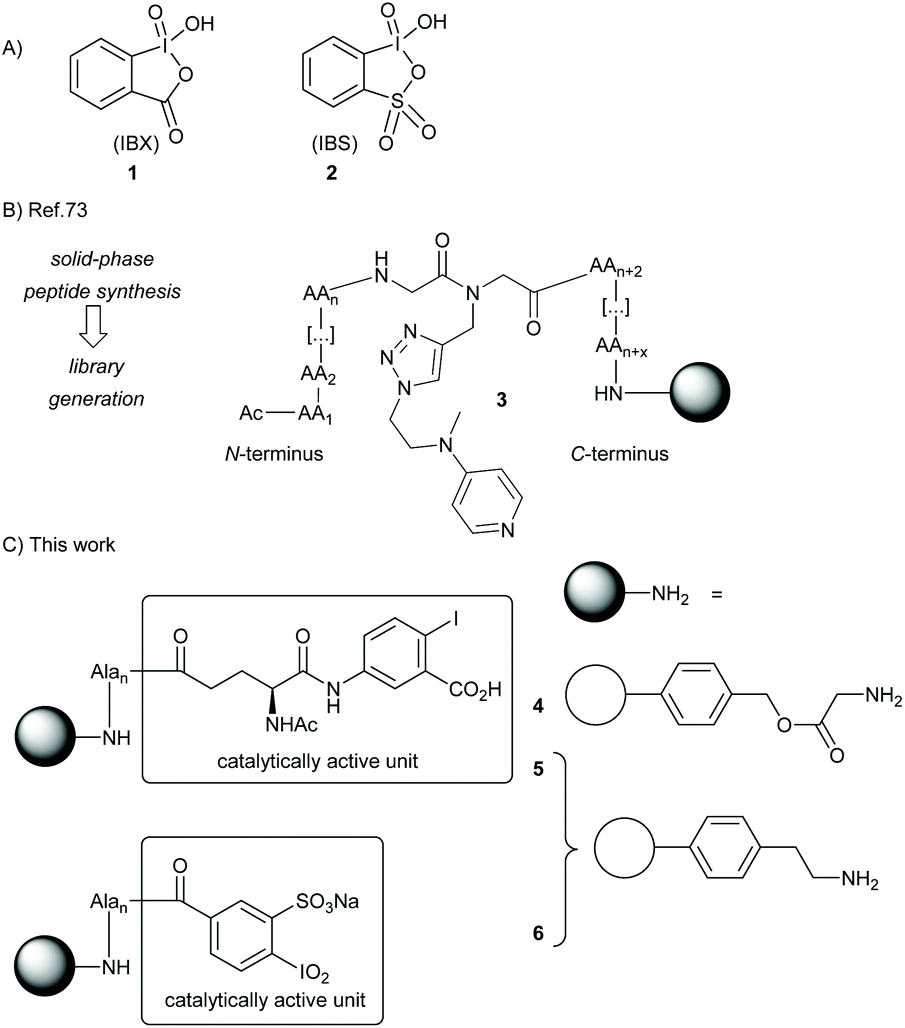 | ||
| Scheme 1 A) Structures of IBX and IBS. (B) DMAP–peptide conjugates through Fmoc-SPPS on Boc-Gly Merrifield resin. (C) Solid-supported oxidation catalysts derived from IBX and IBS. | ||
Despite the many advantages IBX offers, several issues are unattractive to practitioners: (i) similar to many other beloved oxidation agents, stoichiometric amounts of the hypervalent iodine compound are required for alcohol oxidation. This is particularly problematic since IBX is reported to be shock- and heat-sensitive,25,26 at least under certain conditions, thus weakening its routine for large-scale applications. (ii) The regeneration of the IBX species from the 2-iodosobenzoic acid (IBA) formed during the oxidative process is typically avoided, resulting in significant amounts of “organic waste”.27 (iii) DMSO is the preferred solvent for all variants of IBX oxidations,28,29 requiring sophisticated work-up protocols to remove the remainder of the hypervalent iodine reagent, for example by addition of chlorinated solvents. It is therefore reasonable to summarize that IBX and its derivatives are not the ideal reagents with respect to the principles of Green Chemistry30,31 since they are mostly used as stoichiometric and non-recyclable reagents.
Several strategies were developed to tackle those issues. For example, modified IBX reagents32–37 for the use in common solvents other than DMSO and safer formulations38,39 of IBX with reduced explosive properties were described. Experimental procedures that aimed at the effective recovery of the reduced form of the hypervalent iodine compounds using biphasic protocols or fluorous techniques led to further improvements.40–44 Polymer-supported IBX reagents were also reported, with the goal to ease work-up protocols and to improve recyclability,45 including polymer-supported variants of IBX,46–50 IBX-amides,51–56 IBX-esters,57 and 2-iodylphenol ethers.58 In addition, the oxidation with catalytic amounts of IBX59–61 and particularly 2-iodoxybenzenesulfonic acid (IBS, 2)62–65 was shown to be highly efficient.66 Those catalytic methods typically rely on Oxone® as the stoichiometrically used co-oxidant,67–74 a reagent that is considered environmentally safe, non-toxic and non-explosive.75 To facilitate the recovery of the hypervalent iodine organocatalyst, fluorous IBX was recently introduced.76 However, the catalytic use of hypervalent iodine oxidants bound to a polymer support was not reported, until now. This is despite the fact that there would be obvious advantages: the iodine-based catalysts are an environmentally sustainable alternative to transition metals, relatively inexpensive and easily recoverable.77
In this paper, we present two new solid-supported hypervalent iodine reagents for the oxidation of secondary alcohols, derived from readily available starting materials: one is based on IBX, and the other is an IBS derivative. In the presence of Oxone®, the two reagents are shown to be highly effective catalysts when using a water–acetonitrile solvent mixture, with catalysts loadings as low as 1 mol%. We also demonstrate that the two metal-free organocatalysts are easily reusable, and work-up protocols consist of simple filtrations. Therefore, our hypervalent iodine compounds may become attractive ‘green catalysts’ in the ever-evolving fields of alcohol oxidations and other challenging oxidative processes.30,31,78–81
Results and discussion
We recently presented the use of solid-supported DMAP–peptide conjugates 3 for the highly regioselective acylation of polyhydroxylated small molecules (Scheme 1).82,83 We now expanded this concept to connect hypervalent iodine precursor units with peptide-modified resins. Our initial plan was to study conjugates 4 consisting of the glycine-modified Merrifield resin and a (poly)alanine linker, the N-terminus of which was connected to the iodine-containing catalyst. For reasons detailed below, we also generated the conjugates 5 and 6 where the (poly)alanine linker was attached to an aminoethyl polystyrene resin. While 5 is based on the same IBX-derived catalytically active unit used for 4, the solid-supported catalyst 6 was designed as a variant of IBS.Synthesis of the solid-supported precatalysts 4, 5 and 6
We started the synthesis of the precatalyst 4 as summarized in Scheme 2A, using 2-iodobenzoic acid as the commercially available and cheap starting material. Nitration of the aromatic core gave the 5-nitro analogue in 65% yield, as pure substance after simple recrystallization. Subsequent conversion into the tert-butyl ester furnished 7 in excellent yields. Reduction of the nitro group with palladium on charcoal and H2 pressure (8 bar) led to the desired aniline derivative 8 in 82% yield. The iodobenzoic acid variant was coupled to a protected glutamic acid with classical conditions providing the unnatural amino acid 9 in 90% yield. The methyl ester was then saponified while the tert-butyl ester remained untouched: this step was achieved with lithium hydroxide in aqueous THF in 73% yield.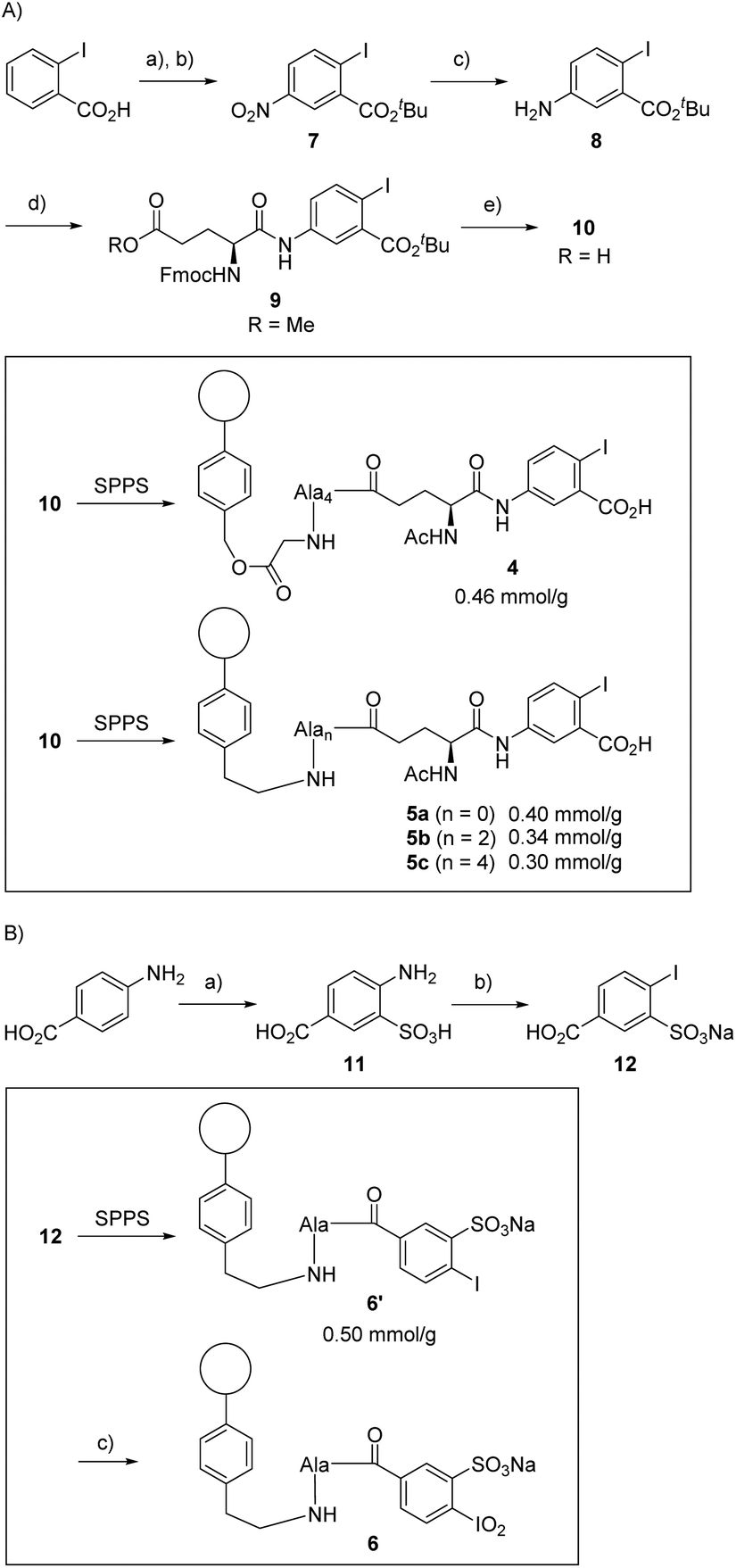 | ||
| Scheme 2 A) Synthesis of solid-supported precatalysts 4 and 5: (a) i) 0 °C, 15 min, (ii) rt, 30 min, (iii) 130 °C, 90 min, (H2SO4/HNO3), 65%; (b) Boc2O (2.5 eq.), DMAP (0.3 eq.), 70 °C, 4 h, (tBuOH), >99%; (c) H2 (8 bar), Pd/C (10 mol%), rt, 18 h, (EtOAc), 82%; (d) Fmoc-Glu(OMe)-OH (1.35 eq.), HATU (1.5 eq.), DIPEA (2 eq.), rt, 18 h, (DMF), 90%; (e) LiOH·H2O (1 eq.), 0 °C to rt, 18 h, (THF/H2O), 73%. (B) Synthesis of solid supported precatalyst 6: (a) H2SO4/SO3 (25%), (H2SO4), 69%; (b) i) NaOH (1.0 eq.), 0 °C, (ii) H2SO4 (20 eq.), 0 °C, (iii) NaNO2 (1.1 eq.), −2 °C, 60 min, (iv) urea (2.0 eq.), −2 °C, 60 min, (v) NaI (1.5 eq.), 0 °C to rt, 18 h, (water), 47%; (c) TBA-Oxone® (5 eq.), MeSO3H (5 eq.), rt, 5 h, (CH2Cl2). | ||
With the Fmoc-protected amino acid 10 in hand, the immobilization on glycin-modified Merrifield resin was accomplished using standard solid-phase peptide synthesis (see ESI† for details). Of note, alanine was chosen as the linker amino acid because of the ease of its introduction with SPPS techniques, its lack of functional groups, and its stability toward oxidative conditions (particularly in comparison to glycine). We decided to start with the conjugate 4 having four alanines as linker unit since, at the outset of our studies, we expected that the use of an elongated and flexible linker that provides a spatial separation of the catalytically active part from the polymer body will be advantageous.51 The loading of the resin was determined to be around 0.46 mmol g−1via UV-VIS measurements (see ESI†). The amino acid 10 was also linked to the aminoethyl polystyrene resin giving conjugates 5. We constructed those conjugates with several linker lengths for further tests, with 5a having no alanine unit (n = 0), 5b having two alanines (n = 2) and 5c having four alanines (n = 4). In all cases, a loading between 0.3 and 0.4 mmol g−1 was achieved.
We next focused on the synthesis of the solid-supported precatalyst 6 (Scheme 2B), relying on our previous synthetic approach to the oxidant IBX-SO3K.37,84,85 We began with 4-aminobenzoic acid, which was selectively sulfonylated with fuming sulfuric acid to give the acid 11. Sandmeyer-type conditions furnished iodide 12. All the steps of this sequence are high-yielding without the need for a chromatographic purification, and multiple runs were successfully and reproducibly carried out in multigram scale. The carboxylic acid 12 was then coupled to the polymer resin bearing a single alanine linker to give the conjugate 6′ with a loading of 0.50 mmol g−1. We note that the direct coupling of 12 to the resin was slow and low-yielding when no alanine was involved. The polymer-supported iodoxy reagent 6 was easily obtained upon oxidation of 6′ with the Oxone® tetrabutylammonium salt (TBA-Oxone®).
Oxidations with the solid-supported precatalysts 4, 5 and 6
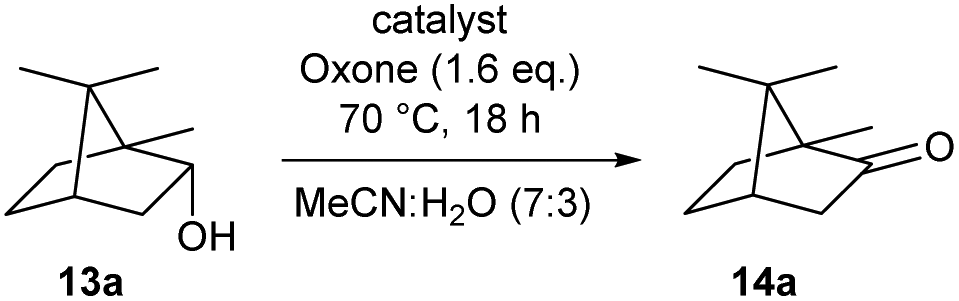
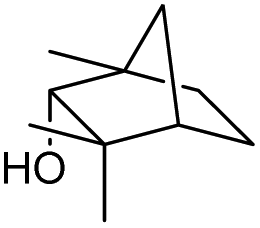
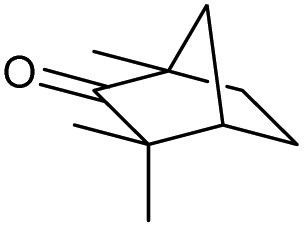
We then started to test the potential of our new catalytic system 4, choosing the oxidation of borneol 13a as a suitable benchmark reaction. Following previous reports on the catalysed oxidation with IBX and IBS with stoichiometric amounts of Oxone®, the oxidation was initially attempted with 1.6 equivalents of Oxone® at 70 °C in aqueous acetonitrile (70%), in the presence of 4 mol% of polymer bound IBX precatalyst 4. As shown in Table 1 (entry 2), borneol was completely converted into the corresponding ketone 14a under the conditions after 18 h; isolated yields of 14a were high. In a similar way, a range of secondary alcohols including 4-nonanol, menthol, 1,3-diphenylpropan-2-ol and 2,2-dimethyl-1-phenylpropan-1-ol were easily oxidized by use of catalyst 4. The formation of by-products was not detected via1H NMR or GC-FID analysis of the crude reaction mixture. Hence, the performance of the oxidation reactions was easily screened by determining the ratio of starting substrate 13 and oxidation product 14 with GC-FID in a standardized way. In the absence of precatalyst 4, 8% conversion of the starting substrate were observed after 18 h, thus indicating a minor background reaction with no synthetic use (entry 1). However, it was truly disappointing that the polymeric system 4 lacked a main asset: it was not reusable. A loss of activity was already found in the second cycle when reusing 4 after simple filtration for the borneol oxidation (entry 3). A markedly lowered conversion of 82% was observed after 18 h, under otherwise identical conditions. This trend continued, and the fifth reaction with the same batch of catalyst gave only 15% conversion, demonstrating that resin-supported catalyst 4 had no recyclability of value.|
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Cycle no.a | 13a/14ab |
| a Recycling and reuse of catalyst by filtration. b Ratio 13a/14a was determined via GC-FID (calibrated with stock solutions of 13a and 14a) after 18 h. c With 40 mol% of nBu4NHSO4, 2 h, MeCN. | |||
| 1 | — | — | 92/8 |
| 2 | 4 (4) | 1 (initial) | 0/100 |
| 3 | 4 (4) | 2 (1st reuse) | 18/82 |
| 4 | 4 (4) | 3 (2nd reuse) | 64/36 |
| 5 | 4 (4) | 4 (3rd reuse) | 82/18 |
| 6 | 4 (4) | 5 (4th reuse) | 85/15 |
| 7 | 5c (5) | 1 (initial) | 0/100 |
| 8 | 5c (5) | 2 (1st reuse) | 0/100 |
| 9 | 5c (5) | 3 (2nd reuse) | 0/100 |
| 10 | 5c (5) | 4 (3rd reuse) | 0/100 |
| 11 | 5c (5) | 5 (4th reuse) | 19/81 |
| 12c | 6 (1) | 1 (initial) | 21/79 |
| 13c | 6 (1) | 2 (1st reuse) | 14/86 |
| 14c | 6 (1) | 3 (2nd reuse) | 12/88 |
| 15c | 6 (1) | 4 (3rd reuse) | 14/86 |
| 16c | 6 (1) | 5 (4th reuse) | 19/81 |
| 17c | 6 (1) | 6 (5th reuse) | 22/78 |
The failure of 4 was attributed to the weakness of the ester bond between the Merrifield resin and the glycine linker toward hydrolysis under the reaction conditions. Indeed, control experiments showed that esters were partly hydrolysed in aqueous acetonitrile at 70 °C in the presence of Oxone® while amide bonds remained fully stable. As a result, column chromatography was required to obtain analytically pure oxidation products when employing precatalyst 4. Simple filtration, on the other hand, gave the ketones with traces of impurities that were not unequivocally identified, but may certainly stem from the partial hydrolysis of 4. The design error of 4 then led us to carefully study the systems 5 and 6 where the hypervalent iodine precursor units were connected to the aminoethyl polystyrene resin via more stable amide bonds.
Gratifyingly, it was easily possible to reuse the catalysts 5 and 6 for multiple times, after recycling of the resins through simple filtration. As shown in Table 1 (entries 7–11), the IBX-derived system 5c kept its activity over five cycles, and complete oxidation of borneol was achieved using 5 mol% of the catalyst with 1.6 equivalents of Oxone® at 70 °C in aqueous acetonitrile. Lower catalyst loadings (<2.5 mol%) were shown to provide incomplete conversions, even after elongated reaction times. The IBS variant 6 was effectively reused for five times (Table 1, entries 12–17), accomplishing a constant conversion between 78% and 88%, albeit with lowered catalyst loading (1 mol%) and reduced reaction times (2 h). As a result of the recycling tests, we decided that both systems 5 and 6 based on the aminoethyl polystyrene resin qualify well for catalyzing the oxidation of secondary hydroxy groups under the premise that they deliver easy work-up and multiple reuses.
Our optimization attempts with catalyst 5c then showed that a broad range of reaction conditions is feasible, and the oxidation proceeds equally well in water-free MeNO2, aqueous MeNO2 (30% water) and aqueous MeCN (5–30% water). However, the use of dry acetonitrile as solvent led to significantly diminished yields. Best conversions were achieved between 70 °C and 90 °C, while temperatures below 60 °C did not provide the product of oxidation. To our surprise, the linker length had almost no influence on the catalyst efficiency. As summarized in Table 2, similar conversions were achieved when using 2.5 mol% of the polymers 5a, 5b for the oxidation of nonanol 13b.
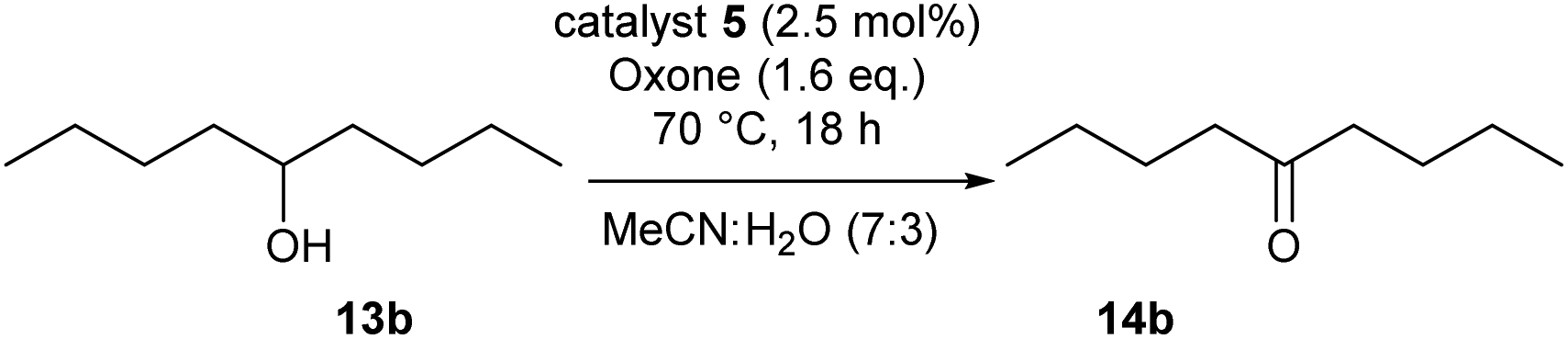
The IBS-derived catalyst system 6 turned out to be markedly more active, compared to 5: Table 3 demonstrates that 3 mol% of 6 or of its unoxidized precursor 6′ are sufficient to achieve an excellent conversion at 70 °C in aqueous acetonitrile. The required reaction times were notedly shorter than with IBX derivative 5. Under non-aqueous conditions in acetonitrile or nitromethane, the oxidation proceeded slowly, and the weak conversions were attributed to the low solubility of Oxone® (entries 3 and 4). The addition of substoichiometric amounts of nBu4NHSO4 as phase transfer catalyst resulted in a tremendous rate enhancement.62 In the presence of 0.4 equivalents of nBu4NHSO4, it was easily possible to use 6 with extremely low catalysts loadings of 1 mol% and 0.2 mol% to accomplish useful conversions (entries 8 and 9). Under the conditions, a TON up to 455 was reached with regard to each catalytic center (with a TOF of around 5.3 × 10−3 s−1).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additive | Solvent | 13b/14ba |
| a Ratio 13b/14b was determined via GC-FID (calibrated with stock solutions of 13b and 14b). b 6 h. c 2 h. d 4 h. e 24 h. | ||||
| 1b | 6′ (3) | — | MeCN–H2O (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) :3) |
5/95 |
| 2b | 6 (3) | — | MeCN–H2O (7:3) |
3/97 |
| 3c | 6 (2.5) | — | MeCN | 94/6 |
| 4c | 6 (2.5) | — | MeNO2 | 91/9 |
| 5c | 6 (2.5) | n Bu4NHSO4 (10 mol%) | MeCN | 43/57 |
| 6c | 6 (2.5) | n Bu4NHSO4 (10 mol%) | MeNO2 | 63/37 |
| 7c | 6 (2.5) | n Bu4NHSO4 (20 mol%) | MeCN | 3/97 |
| 8d | 6 (1) | n Bu4NHSO4 (40 mol%) | MeCN | 2/98 |
| 9e | 6 (0.2) | n Bu4NHSO4 (40 mol%) | MeCN | 9/91 |
| 10d | — | n Bu4NHSO4 (40 mol%) | MeCN | 94/6 |
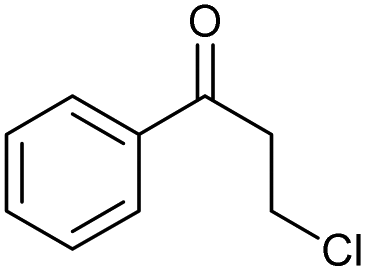
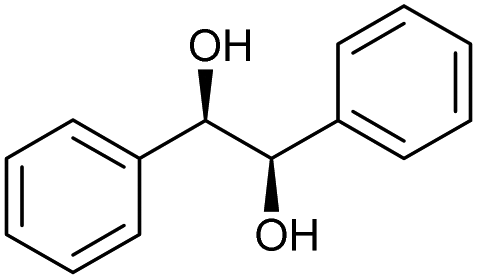
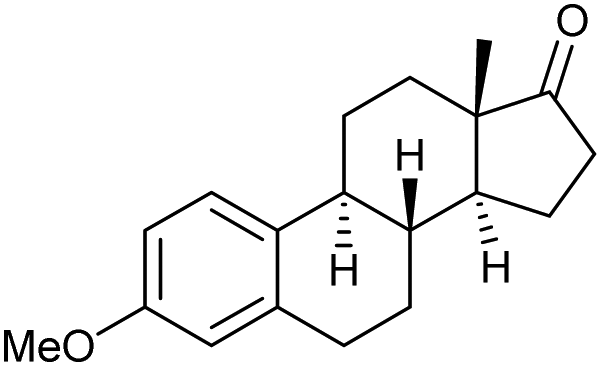
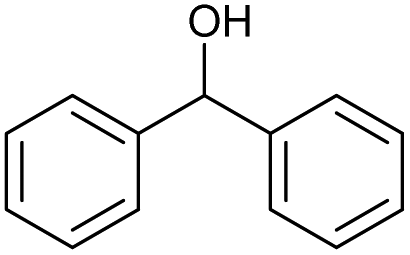
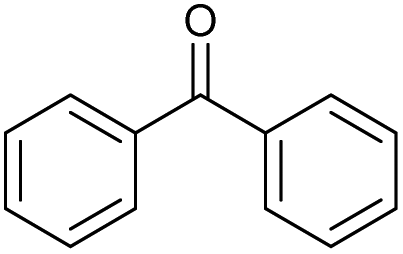
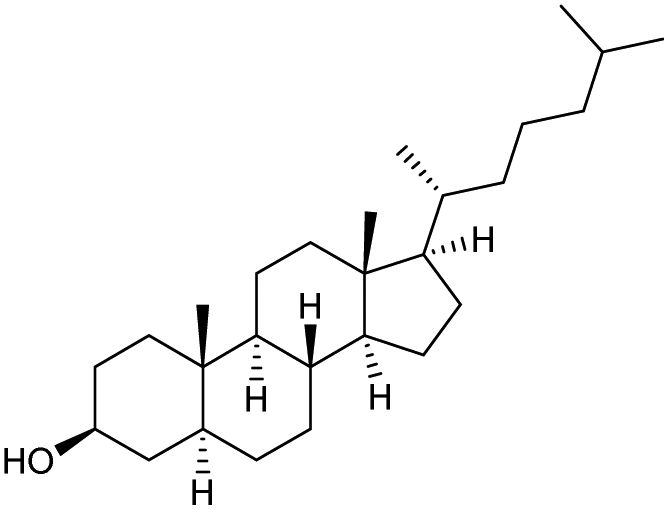
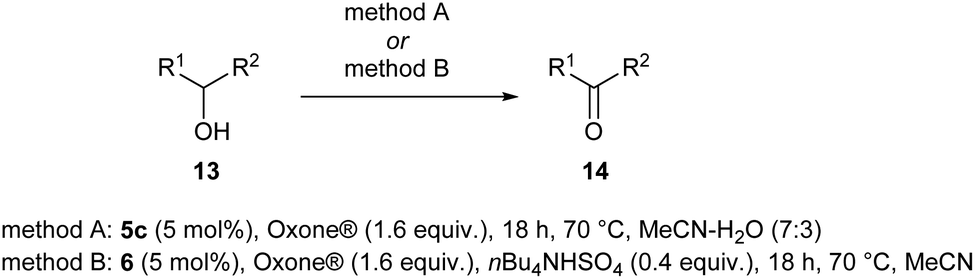
The scope of the catalyzed oxidation of secondary alcohols with Oxone® was then explored, using the solid-supported compounds 5c and 6 under standardized conditions. As summarized in Table 4, various structurally diverse secondary alcohols 13 were examined as substrates using method A [5c (5 mol%), Oxone® (1.6 equiv.), 18 h, 70 °C, MeCN–H2O (7:3)] and method B [6 (5 mol%), Oxone® (1.6 equiv.), nBu4NHSO4 (0.4 equiv.), 18 h, 70 °C, MeCN]. We point out that the addition of the phase transfer agent was only required when using non-aqueous conditions (method B); aqueous solvent mixtures (method A) do not require the additive since Oxone® is easily dissolved under the conditions. A general trend was that the performance of the IBS-derived catalyst 6 was superior. In particular, sterically demanding secondary alcohols (e.g., 13a, 13d, 13f and 13k) were easier oxidized to the corresponding ketones using method B. The reaction times until full conversion of the starting substrate were also markedly shorter with 6, compared to 5c (method A). Another advantage of catalyst 6 was that it was employed in a combination with nBu4NHSO4 under non-aqueous conditions, allowing to use other solvents than acetonitrile if required due to solubility issues of the substrate. For example, 5α-cholestan-3β-ol 13s was successfully oxidized with Oxone® and 6 in toluene under otherwise identical conditions (entry 19). The main advantage of method A (with 5c) was the simple work-up protocol consisting only of filtration to remove 5c and subsequent extraction of the aqueous layer. If the starting material was completely converted, further purification by column chromatography was typically not necessary. In the case of method B (with 6), an additional filtration over silica for the removal of the tetrabutylammonium salts was required to obtain analytically pure compounds; the phase transfer agent was not recovered.
|
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
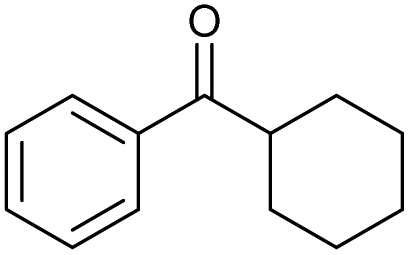
| Entry | Alcohol 13 | # | Ketone 14 | Yield [%]a,b | Entry | Alcohol 13 | # | Ketone 14 | Yield [%]a,b | ||
| A | B | A | B | ||||||||
| a Isolated yield. b n.r. = experiment was not run. c 3 h. d 24 h. e 72 h in acetone. f 90% from benzoin. g 18 h in toluene. h Determined via GC-FID (calibrated with stock solutions of 13t/14t). | |||||||||||
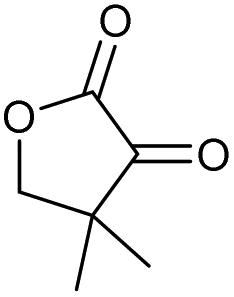
| 1 |
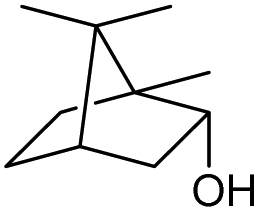
|
a |
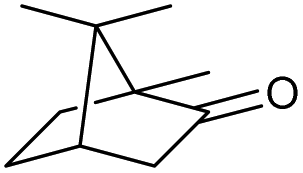
|
79 | 91c | 11 |
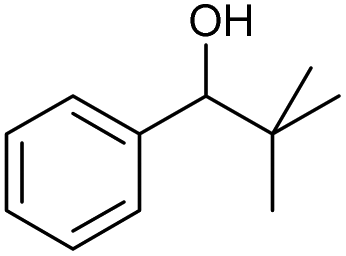
|
k |
|
76 | 98 |
| 2 |
|
b |
|
70 | 79c | 12 |
|
l |
|
82 | 84 |
| 3 |
|
c |
|
80 | 85c | 13 |
|
m |
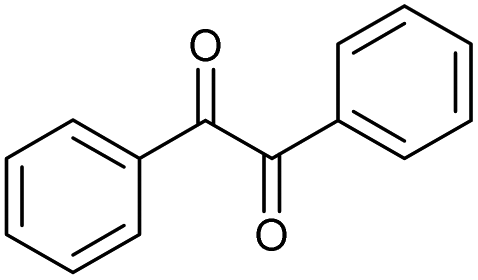
|
71 | 97f |
| 4 |
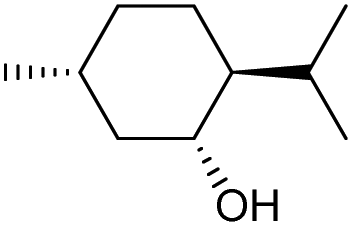
|
d |
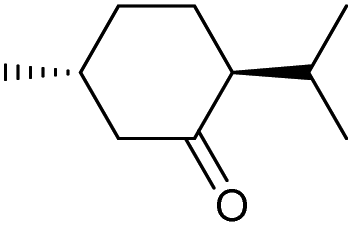
|
32 | 65d | 14 |
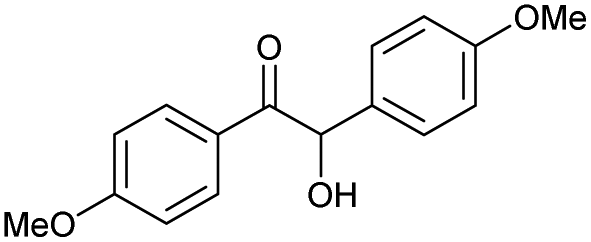
|
n |
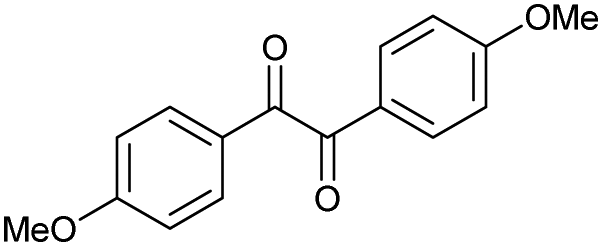
|
n.r. | 89 |
| 5 |
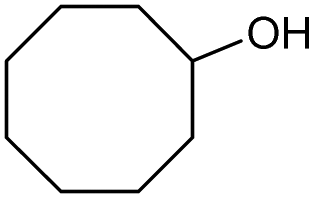
|
e |
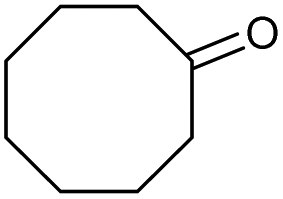
|
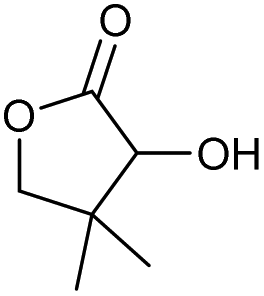
71 | 83 | 15 |
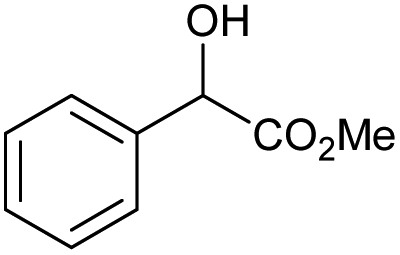
|
o |
|
0 | 66c |
| 6 |
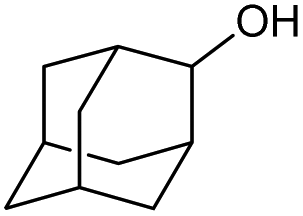
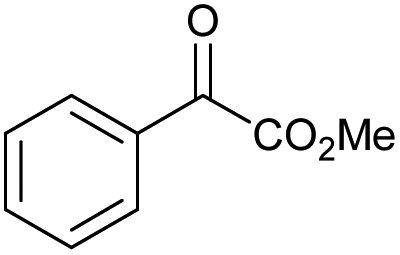
|
f |
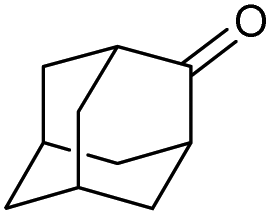
|
0 | 32e | 16 |
|
p |
|
0 | 66c |
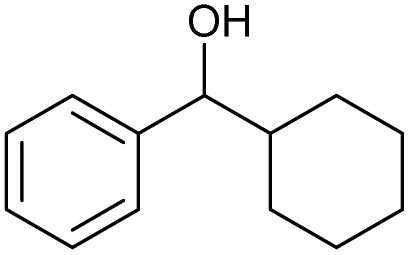
| 7 |
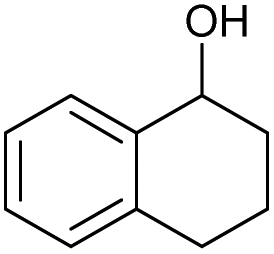
|
g |
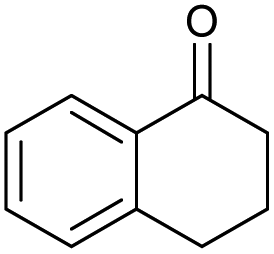
|
82 | 85 | 17 |
|
q |
|
95 | 96 |
| 8 |
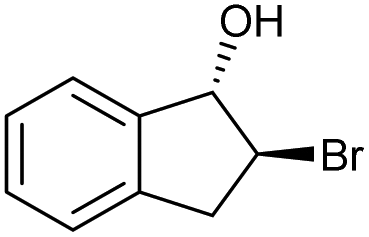
|
h |
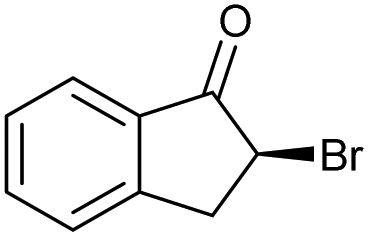
|
84 | 84 | 18 |
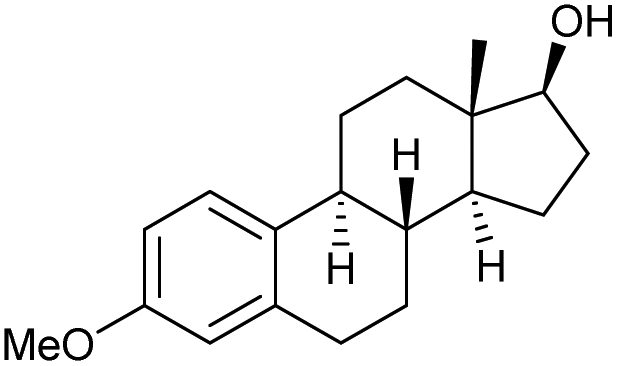
|
r |
|
0 | 61 |
| 9 |
|
i |
|
n.r. | 98c | 19 |
|
s |
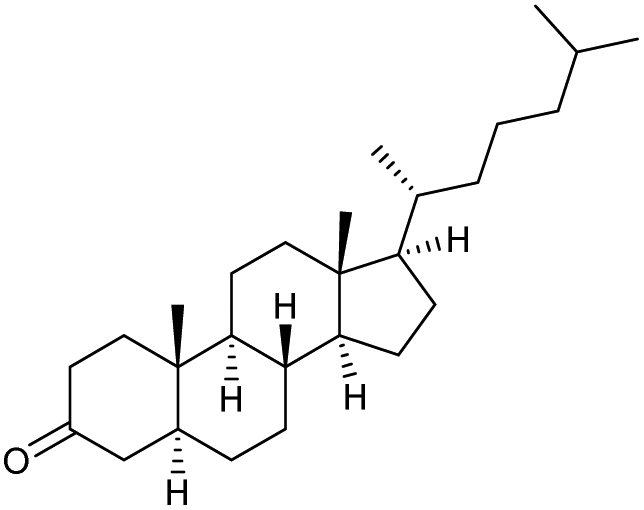
|
0 | 78g |
| 10 |
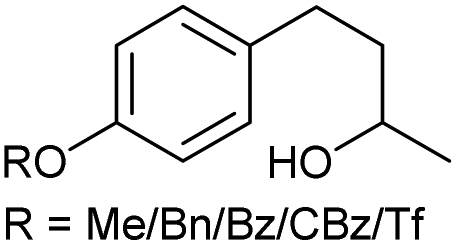
|
j |
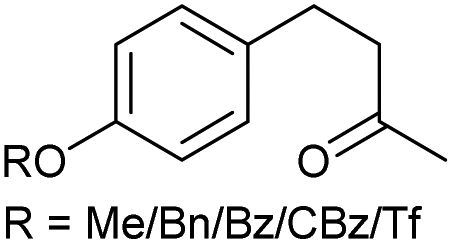
|
75/58/69/82/63 | 86/79/85/86/82 | 20 |
|
t |
|
n.r. | 75h |
We point out that primary alcohols also provide oxidation products under the conditions of method B. However, aldehydes were produced in markedly reduced yields and mixed with various products of over-oxidation, thus limiting the use of the catalytic system tremendously. In the case of primary allylic alcohols 15, it was possible to isolate the corresponding aldehydes in pure form and moderate yields, as outlined for 16a and 16b in Scheme 3. Two other limitations of our catalyst system for the oxidation of secondary alcohols were identified in the course of our studies: (1) phenols are not stable under the conditions,14,15 and (2) amines are not tolerated due to various competing condensation and oxidation reactions that were not further analyzed.
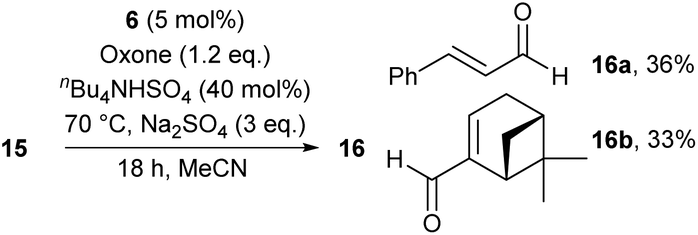 | ||
| Scheme 3 Oxidation of primary allylic alcohols 15 by use of 6. | ||
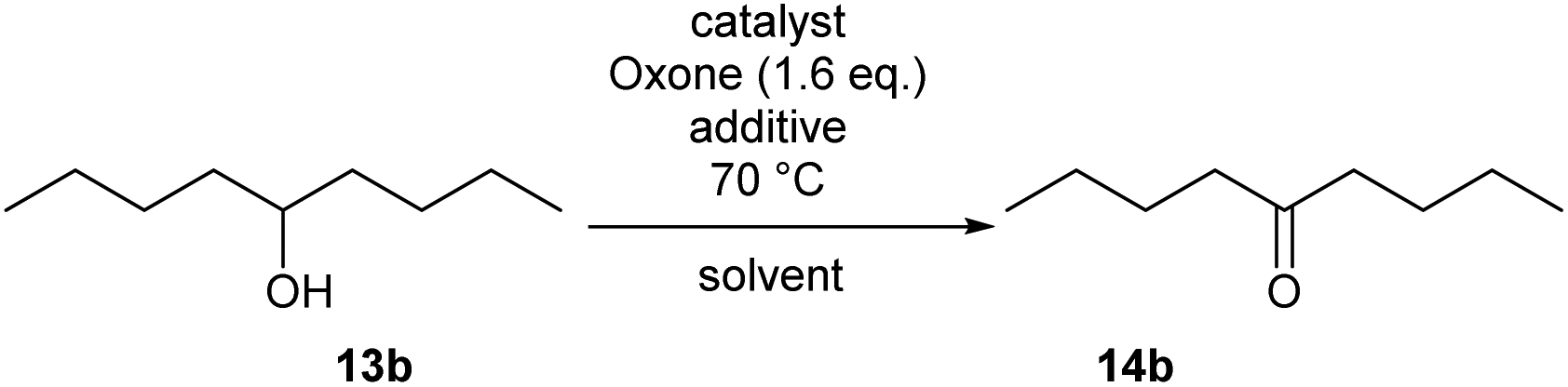
Method B using IBS-derived polymer 6 is also easily applicable to larger scales, as demonstrated in Scheme 4 for the conversion of secondary alcohol 13b into ketone 14b: 50 mmol of 13b led to the isolation of the desired ketone with 80% yield after 5 h, using only 1 mol% of catalyst 6. Of note, 95% of the supported catalyst were recovered for re-uses.
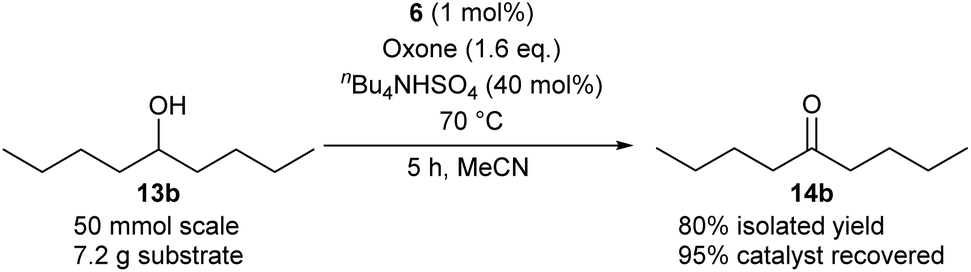 | ||
| Scheme 4 Oxidation of 13b on a 50 mmol-scale by use of 6. | ||
Conclusions
We have presented two new catalytic systems 5c (based on IBX) and 6 (based on IBS) that are outstanding tools for the oxidation of secondary alcohols with Oxone® under ketone formation. The catalysts, derived from hypervalent iodine reagents linked to a polymer support, are highly active, and a broad scope is demonstrated for both catalysts. However, the catalytic oxidation with 6 proceeded more rapidly and cleanly. After completion of the reaction, the catalysts were recovered through simple filtration, and it was possible to reuse them for multiple times, thus rendering them highly attractive for the objectives of Green Chemistry: recoverable and prepared from readily available starting materials consisting of abundant chemical elements.Our future studies will make use of the catalyst systems for chemoselective oxidations, by varying the linker units. We will also show in due course how the immobilized catalysts are of use in flow chemical oxidation processes.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- V. V. Zhdankin, ARKIVOC, 2009, 1–62 Search PubMed.
- V. V. Zhdankin, J. Org. Chem., 2011, 76, 1185–1197 CrossRef CAS PubMed.
- A. Varvoglis, Tetrahedron, 1997, 53, 1179–1255 CrossRef CAS.
- M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086–2099 RSC.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- A. Duschek and S. F. Kirsch, Angew. Chem., Int. Ed., 2011, 50, 1524–1552 CrossRef CAS PubMed.
- T. Wirth, Angew. Chem., Int. Ed., 2001, 40, 2812–2814 CrossRef CAS PubMed.
- V. Satam, A. Harad, R. Rajule and H. Pati, Tetrahedron, 2010, 66, 7659–7706 CrossRef CAS.
- C. Hartmann and V. Meyer, Ber. Dtsch. Chem. Ges., 1893, 26, 1727–1732 CrossRef.
- M. Frigerio and M. Santagostino, Tetrahedron Lett., 1994, 35, 8019–8022 CrossRef CAS.
- M. Frigerio, M. Santagostino, S. Sputore and G. Palmisano, J. Org. Chem., 1995, 60, 7272–7276 CrossRef CAS.
- J. L. F. Silva and B. Olofsson, Nat. Prod. Rep., 2011, 28, 1722–1754 RSC.
- L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235–2261 CrossRef.
- S. Quideau, L. Pouységu and D. Deffieux, Synlett, 2008, 467–495 CrossRef CAS.
- D. Magdziak, A. A. Rodriguez, R. W. Van De Water and T. R. R. Pettus, Org. Lett., 2002, 4, 285–288 CrossRef CAS PubMed.
- S. Quideau, L. Pouysegu, A. Ozanne and J. Gagnepain, Molecules, 2005, 10, 201–216 CrossRef CAS PubMed.
- S. F. Kirsch, J. Org. Chem., 2005, 70, 10210–10212 CrossRef CAS PubMed.
- A. Duschek and S. F. Kirsch, Chem. – Eur. J., 2009, 15, 10713–10717 CrossRef CAS PubMed.
- Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436–4454 CrossRef CAS PubMed.
- B. Crone and S. F. Kirsch, Chem. Commun., 2006, 764–766 RSC.
- T. Harschneck, S. Hummel, S. F. Kirsch and P. Klahn, Chem. – Eur. J., 2012, 18, 1187–1193 CrossRef CAS PubMed.
- K. C. Nicolaou, Y. L. Zhong and P. S. Baran, J. Am. Chem. Soc., 2000, 122, 7596–7597 CrossRef CAS.
- K. C. Nicolaou, T. Montagnon and P. S. Baran, Angew. Chem., Int. Ed., 2002, 41, 993–996 CrossRef CAS.
- M. Frigerio, M. Santagostino and S. Sputore, J. Org. Chem., 1999, 64, 4537–4538 CrossRef CAS.
- J. B. Plumb and D. J. Harper, Chem. Eng. News, 1990, 3 CAS.
- T. K. Achar, S. Maiti and P. Mal, RSC Adv., 2014, 4, 12834–12839 RSC.
- Z. Liu, Z.-C. Chen and Q.-G. Zheng, Org. Lett., 2003, 5, 3321–3323 CrossRef CAS PubMed.
- J. D. More and N. S. Finney, Org. Lett., 2002, 4, 3001–3003 CrossRef CAS PubMed.
- C. Parmeggiani, C. Matassini and F. Cardona, Green Chem., 2017, 19, 2030–2050 RSC.
- F. Cavani and J. Henrique Teles, ChemSusChem, 2009, 2, 508–534 CrossRef CAS PubMed.
- A. P. Thottumkara and T. K. Vinod, Tetrahedron Lett., 2002, 43, 569–572 CrossRef CAS.
- R. D. Richardson, J. M. Zayed, S. Altermann, D. Smith and T. Wirth, Angew. Chem., Int. Ed., 2007, 46, 6529–6532 CrossRef CAS PubMed.
- L.-Q. Cui, Z.-L. Dong, K. Liu and C. Zhang, Org. Lett., 2011, 13, 6488–6491 CrossRef CAS PubMed.
- S. Seth, S. Jhulki and J. N. Moorthy, Eur. J. Org. Chem., 2013, 2445–2452 CrossRef CAS.
- J. N. Moorthy, K. Senapati, K. N. Parida, S. Jhulki, K. Sooraj and N. N. Nair, J. Org. Chem., 2011, 76, 9593–9601 CrossRef CAS PubMed.
- A. Bredenkamp, F. Mohr and S. F. Kirsch, Synthesis, 2015, 47, 1937–1943 CrossRef CAS.
- I. M. Kumanyaev, M. A. Lapitskaya, L. L. Vasiljeva and K. K. Pivnitsky, Mendeleev Commun., 2012, 22, 129–131 CrossRef CAS.
- A. Ozanne, L. Pouységu, D. Depernet, B. François and S. Quideau, Org. Lett., 2003, 5, 2903–2906 CrossRef CAS PubMed.
- V. Tesevic and J. A. Gladysz, Green Chem., 2005, 7, 833–836 RSC.
- H. Tohma, A. Maruyama, A. Maeda, T. Maegawa, T. Dohi, M. Shiro, T. Morita and Y. Kita, Angew. Chem., Int. Ed., 2004, 43, 3595–3598 CrossRef CAS PubMed.
- A. Kirschning, M. S. Yusubov, R. Y. Yusubova, K.-W. Chi and J. Y. Park, Beilstein J. Org. Chem., 2007, 3, 19 Search PubMed.
- M. S. Yusubov, R. Y. Yusubova, T. V. Funk, K.-W. Chi, A. Kirschning and V. V. Zhdankin, Synthesis, 2010, 3681–3685 CrossRef CAS.
- A. Yoshimura, C. T. Banek, M. S. Yusubov, V. N. Nemykin and V. V. Zhdankin, J. Org. Chem., 2011, 76, 3812–3819 CrossRef CAS PubMed.
- C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275–3300 CrossRef CAS.
- M. Mülbaier and A. Giannis, Angew. Chem., Int. Ed., 2001, 40, 4393–4394 CrossRef.
- N. N. Reed, M. Delgado, K. Hereford, B. Clapham and K. D. Janda, Bioorg. Med. Chem. Lett., 2002, 12, 2047–2049 CrossRef CAS PubMed.
- G. Sorg, A. Mengel, G. Jung and J. Rademann, Angew. Chem., Int. Ed., 2001, 40, 4395–4397 CrossRef CAS PubMed.
- G. Sorg, B. Thern, O. Mader, J. Rademann and G. Jung, J. Pept. Sci., 2005, 11, 142–152 CrossRef CAS PubMed.
- Z. Lei, C. Denecker, S. Jegasothy, D. C. Sherrington, N. K. H. Slater and A. J. Sutherland, Tetrahedron Lett., 2003, 44, 1635–1637 CrossRef CAS.
- W.-J. Chung, D.-K. Kim and Y.-S. Lee, Synlett, 2005, 2175–2178 CAS.
- U. Ladziata, J. Willging and V. V. Zhdankin, Org. Lett., 2006, 8, 167–170 CrossRef CAS PubMed.
- H.-S. Jang, Y.-H. Kim, Y.-O. Kim, S.-M. Lee, J. W. Kim, W.-J. Chung and Y.-S. Lee, J. Ind. Eng. Chem., 2014, 20, 29–36 CrossRef CAS.
- B. M. Bizzarri, I. Abdalghani, L. Botta, A. R. Taddei, S. Nisi, M. Ferrante, M. Passacantando, M. Crucianelli and R. Saladino, Nanomaterials, 2018, 8, 516 CrossRef PubMed.
- W.-J. Chung, D.-K. Kim and Y.-S. Lee, Tetrahedron Lett., 2003, 44, 9251–9254 CrossRef CAS.
- D.-K. Kim, W.-J. Chung and Y.-S. Lee, Synlett, 2005, 279–282 CAS.
- H.-S. Jang, W.-J. Chung and Y.-S. Lee, Tetrahedron Lett., 2007, 48, 3731–3734 CrossRef CAS.
- R. R. Karimov, Z.-G. M. Kazhkenov, M. J. Modjewski, E. M. Peterson and V. V. Zhdankin, J. Org. Chem., 2007, 72, 8149–8151 CrossRef CAS PubMed.
- P. C. B. Page, L. F. Appleby, B. R. Buckley, S. M. Allin and M. J. McKenzie, Synlett, 2007, 1565–1568 CrossRef CAS.
- R. D. Richardson and T. Wirth, Angew. Chem., Int. Ed., 2006, 45, 4402–4404 CrossRef CAS PubMed.
- F. V. Singh and T. Wirth, Chem. – Asian J., 2014, 9, 950–971 CrossRef CAS PubMed.
- M. Uyanik, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 251–262 CrossRef CAS PubMed.
- M. Uyanik, R. Fukatsu and K. Ishihara, Org. Lett., 2009, 11, 3470–3473 CrossRef CAS PubMed.
- M. Uyanik, T. Mutsuga and K. Ishihara, Molecules, 2012, 17, 8604–8616 CrossRef CAS PubMed.
- L.-Q. Cui, K. Liu and C. Zhang, Org. Biomol. Chem., 2011, 9, 2258–2265 RSC.
- T. Dohi and Y. Kita, Chem. Commun., 2009, 2073–2085 RSC.
- S. K. Alla, P. Sadhu and T. Punniyamurthy, J. Org. Chem., 2014, 79, 7502–7511 CrossRef CAS PubMed.
- R. Bikshapathi, P. S. Prathima and V. J. Rao, New J. Chem., 2016, 40, 10300–10304 RSC.
- F. Drouet, G. Masson and J. Zhu, Org. Lett., 2013, 15, 2854–2857 CrossRef CAS PubMed.
- J. N. Moorthy and K. N. Parida, J. Org. Chem., 2014, 79, 11431–11439 CrossRef CAS PubMed.
- A. Schulze and A. Giannis, Synthesis, 2006, 257–260 CAS.
- A. P. Thottumkara, M. S. Bowsher and T. K. Vinod, Org. Lett., 2005, 7, 2933–2936 CrossRef CAS PubMed.
- A. Yoshimura, K. R. Middleton, M. W. Luedtke, C. Zhu and V. V. Zhdankin, J. Org. Chem., 2012, 77, 11399–11404 CrossRef CAS PubMed.
- A. Yoshimura, K. R. Middleton, A. D. Todora, B. J. Kastern, S. R. Koski, A. V. Maskaev and V. V. Zhdankin, Org. Lett., 2013, 15, 4010–4013 CrossRef CAS PubMed.
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed.
- T. Miura, K. Nakashima, N. Tada and A. Itoh, Chem. Commun., 2011, 47, 1875–1877 RSC.
- M. S. Yusubov and V. V. Zhdankin, Resour.-Effic. Technol., 2015, 1, 49–67 Search PubMed.
- S. Hazra, M. Deb and A. J. Elias, Green Chem., 2017, 19, 5548–5552 RSC.
- K.-J. Liu, S. Jiang, L.-H. Lu, L.-L. Tang, S.-S. Tang, H.-S. Tang, Z. Tang, W.-M. He and X. Xu, Green Chem., 2018, 20, 3038–3043 RSC.
- S. Hazra, A. K. Kushawaha, D. Yadav, P. Dolui, M. Deb and A. J. Elias, Green Chem., 2019, 21, 1929–1934 RSC.
- W. Schilling, D. Riemer, Y. Zhang, N. Hatami and S. Das, ACS Catal., 2018, 8, 5425–5430 CrossRef CAS.
- F. Huber and S. F. Kirsch, Chem. – Eur. J., 2016, 22, 5914–5918 CrossRef CAS PubMed.
- M. L. Tong, F. Huber, E. S. T. Kaptouom, T. Cellnik and S. F. Kirsch, Chem. Commun., 2017, 53, 3086–3089 RSC.
- K. D. Umland, C. Mayer and S. F. Kirsch, Synlett, 2014, 813–816 CAS.
- P. Klahn, H. Erhardt, A. Kotthaus and S. F. Kirsch, Angew. Chem., Int. Ed., 2014, 53, 7913–7917 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data and copies of 1H, 13C NMR-spectra. See DOI: 10.1039/c9gc02605c |
| This journal is © The Royal Society of Chemistry 2019 |