
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGrafting strategies for hydroxy groups of lignin for producing materials
Armin
Eraghi Kazzaz
,
Zahra
Hosseinpour Feizi
and
Pedram
Fatehi
 *
*
Chemical Engineering Department, Lakehead University, 955 Oliver Road, Thunder Bay, Ontario P7B 5E1, Canada. E-mail: pfatehi@lakeheadu.ca
First published on 11th October 2019
Abstract
Lignin is one of the most abundant biopolymers on Earth and is considered as the primary resource of aromatic compounds. Recently, lignin has attracted attention from scientists and industrialists due to its inherent potential arising from its unique structure, which leads to its possible use in many applications. Many efforts have been made to ameliorate the reactivity and compatibility of lignin in different areas. Although methods have been proposed for endowing lignin with different properties, there continues to be a considerable demand for discovering new and effective ways of unraveling the beneficial uses of this aromatic polymer. Considering the structure of lignin, different grafting modifications can occur on the aliphatic and/or aromatic groups of lignin. To date, there has been a lack of fundamental understanding of the modification pathways of lignin for generating lignin-based products. In this review paper, we discuss comprehensively the chemical reactions that were introduced in the literature for preparing lignin with different features via modifying its phenolic and aliphatic hydroxy groups for altered uses. This review paper critically and comprehensively elaborates on the recent progress in lignin reactions as well as the challenges, advantages and disadvantages associated with the reaction procedures and the product development processes. Furthermore, the research gap in reaction strategies and product development are described throughout this study.
Introduction
Lignin is an abundant, natural polymer representing between 15 and 30 wt% of lignocellulosic biomass.1 This polymer exists in the cellular wall of cellulose fibers and provides structural support against oxidative stresses and microbial attacks for plants.2–4 Lignin, an amorphous heteropolymer, is insoluble in water and has a limited reactivity.5,6 It mainly comprises methoxylated phenylpropanoid (guaiacyl and syringyl) subunits that provide lignin with an energy density of 30% higher than that of polysaccharide polymers.1,7 Lignin can be an alternative product to petroleum feedstocks for producing different chemicals.8 The availability of lignin in the biosphere exceeds 300 billion tons, with a growth rate of around 20 billion tons every year.9 However, a small fraction of the extracted lignin is used in the formulation of adhesives,10 dispersants,11 surfactants or as antioxidants in rubbers and plastics.12–14 Thus, there is considerable room for taking greater advantage of the inherent potential of this abundant polymer in various fields.Based on the types of plants (softwood, hardwood, and non-wood), the amount of each monolignol could be different. Hardwood lignin contains the highest amount of syringyl alcohol among the three classes of lignin with a smaller amount of coniferyl alcohol monolignols. Lignin extracted from softwood resources (also called coniferous or guaiacyl lignin) merely contains coniferyl alcohol monolignols. Meanwhile, lignin from grass (i.e., non-wood lignin) contains all three monolignols while the highest amount of monolignols is uncertain.
Since methoxy groups provide steric hindrance to the aromatic hydroxy groups of monolignol, their amount in monolignol is very critical. This is due to the fact that the enzyme catalyzing monolignol units’ polymerization severely attacks the hydroxy groups, which connect monomers to generate a polymer chain. Hence, the overall cross-linking in the lignin structure decreases due to the enhancement in the steric hindrance, which lowers the ability of the aromatic part of lignin to react with other monomers. In addition, considering the internal cross-linking of the lignin structure, hardwood lignin, by virtue of having numerous units of syringyl alcohol monolignol, exhibits minimal internal cross-linking, while, lignin from grasses has more of a cross-linked structure than other lignin classes. It is worth noting that this internal cross-linked structure affects both the lignin molecule and the characteristics of lignin-based materials.15
The polymeric nature of lignin presents technical restrictions when used directly for synthesizing with other chemicals, which raises the need for its structural modification. Lignin modification and its use in alternative products has become particularly popular in biorefining processes. Biorefining can be considered as analogous to petroleum refining that is supposed to create many biodegradable, non-toxic and recyclable chemicals from the biomass.16 In the past, different modification pathways had been conducted on lignin to make it a valuable product. Based on the lignin structure, modification reactions can occur on aromatic, aliphatic, or both parts.
Lignin can be isolated from the spent pulping liquors of sulfite, kraft, organosolv, and soda processes. Among these, sulfite and kraft processes are the two dominant techniques that are commercially utilized in the pulping industry.17–19 Lignin produced from the kraft process is usually used as a fuel and burned in mills, while lignin generated in the sulfite pulping process is extracted as lignosulfonate. The solubility of kraft lignin is much lower than that of lignosulfonate due to the lack of hydrophilic groups on kraft lignin.20,21 Nonetheless, kraft lignin possesses some outstanding properties in comparison with other types of lignin, such as a higher phenolic hydroxide group content, which is raised from the cleavage of β-aryl bonds during the pulping process. Interest on lignosulfonate, on the other hand, has increased because its sulfonic acid groups are attached to its aliphatic part rendering it soluble in water and providing it with the capability of emulsifying and binding properties. There are two commercial techniques called LignoBoost and Lignoforce that utilize acidification for lignin isolation from black liquor with the lignin solid content of 50–60 wt%.22,23
Herein, the primary objective of this review is to discuss the fundamentals associated with the modification of the aromatic and aliphatic groups of lignin. Distinguishing the altered reaction pathways on lignin aromatic and aliphatic parts can possibly lead to the identification of an appropriate method for producing lignin-based products with desired properties for altered applications. Furthermore, the challenges and perspectives associated with the modification methods at both laboratory and commercial scale practices are discussed throughout this study. In addition, since reviews are available on the topics of polymerization,24,25 catalytic reactions,26,27 depolymerization,8,24,28,29 redox-neutral strategies,8 and photoredox catalysis,30 this study has excluded the discussion on the above-mentioned strategies. Lignin oxidation has also been covered briefly in this study, while more comprehensive information on the oxidation of lignin and its derivatives could be found in the literature.31–33 However, the graft modifications of lignin have not been studied comprehensively, which further begets the lack of studies on some curtail applications of lignin in industry. This study also excludes discussion on model compounds but provides comprehensive discussion on the modification of industrial lignin as the raw material.
Alternative methods for modification of phenolic structure of lignin
Phosphorylation
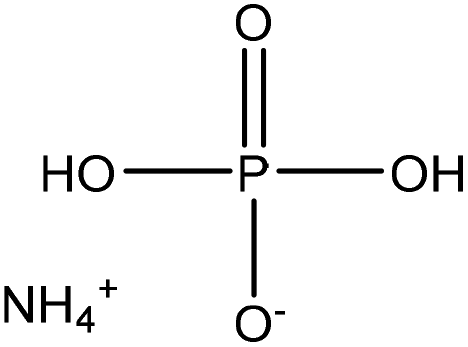
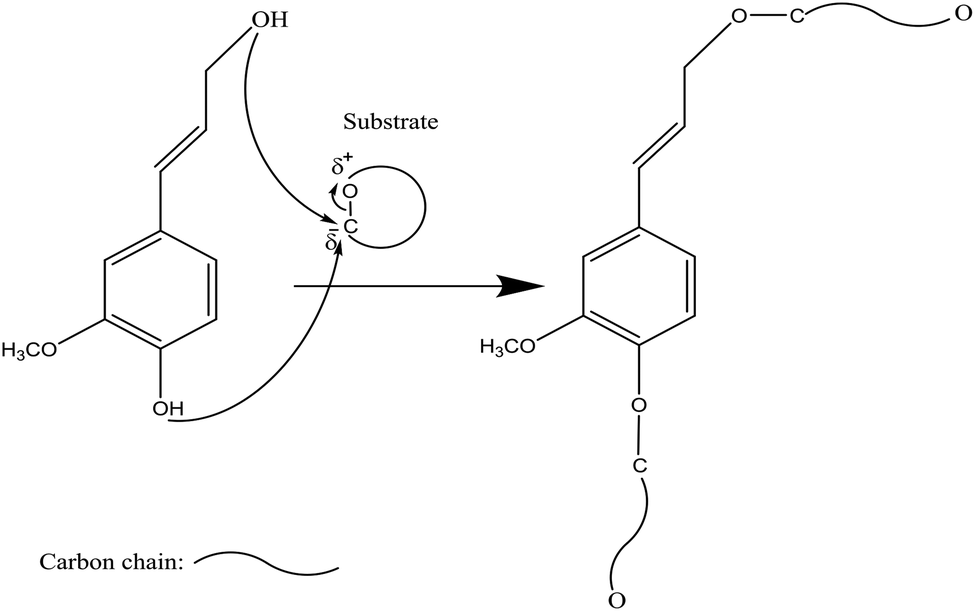
Phosphorus-containing compounds have been widely studied to develop non-toxic and environmentally friendly flame retardants to diminish the production of toxic fumes and smoke during burning and to hamper the combustibility of polymers.34–36 Phosphorylation of lignin occurs via the addition of a phosphoryl group (–PO3) to a molecule. This reaction proceeds through the SN2 reaction mechanism (Fig. 1). Generally, in an SN2 reaction, lignin's hydroxy groups, as a nucleophile, attack the carbon atom, an electrophilic center, due to the withdrawal of some electron density by the leaving-group (e.g., bromine, chlorine) from carbon, which makes the carbon partially positive. This leads the nucleophile that is the lone pair of an electron on oxygen (hydroxyl of lignin) to attack the partially positive carbon. As the nucleophilic groups of lignin generate a bond with the carbon atom, the bond among the leaving group and the carbon atom breaks, simultaneously. At the same time, the bond between carbon and the leaving group breaks, which renders the leaving-group negatively-charged. Finally, the hydroxy groups of lignin form a bond with the carbon atom to generate the product. The reaction mentioned above was reported to occur on both aromatic and aliphatic hydroxy groups of lignin.37,38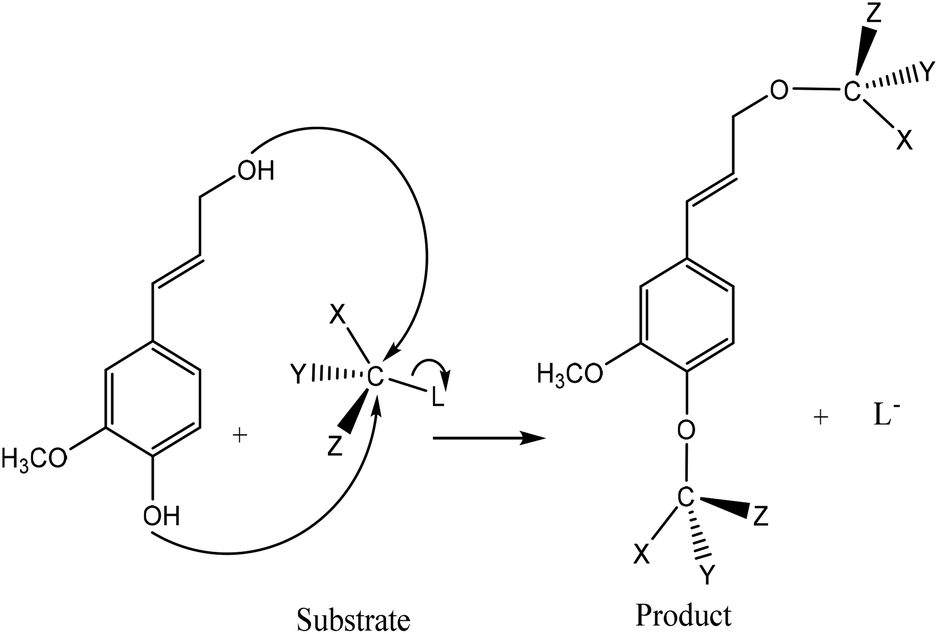 | ||
| Fig. 1 General SN2 reaction mechanism between lignin and substrate to form a new product and a leaving group (L). Substituents of the reacting carbon (X, Y, and Z) do not interfere with the reaction.39–43 | ||
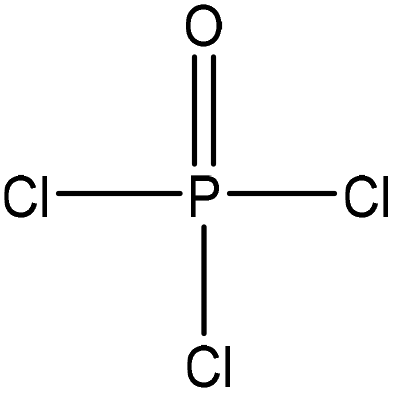
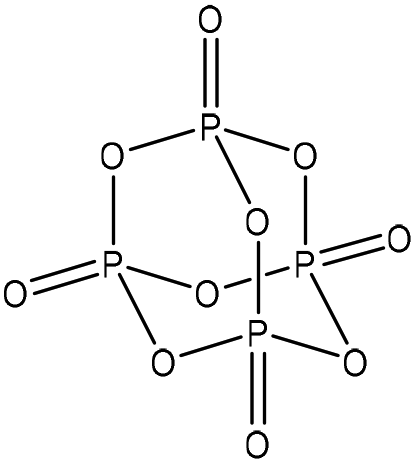
Table 1 shows some of the phosphorylation reactions conducted on lignin. Lignin phosphorylation has been carried out under different conditions using various phosphorus reagents, such as phosphorus trihalides, phosphorus oxyhalides, phosphorus thiohalides, phosphorus oxides, and phosphorus sulfides, for instance.44–47 Tetrahydrofuran,38,48 pyridine,39,49 acetonitrile,47 dimethylformamide, formaldehyde44,45 and urea37 were also used as solvents for phosphorylation in different studies. The reaction conditions were reported to occur in the time range of 1–12 h and the temperature range of 25–180 °C.38,49,50 After the reaction, lignin-based products were reported to be isolated from the reaction media using methanol, diethyl ether and ion exchange processes.38,47,49,50
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Wheat straw alkali | 12 | 95 | 3–4 |
|
Triethylamine, dimethylformamide | Methanol | Thermal stability | 92 | Flame retardant | 53 |
| Kraft | 7–8 | 20–25 | N/A |
|
Tetrahydrofuran | Water | Thermal stability | N/A | Flame retardant | 48 and 38 |
| Spruce | 1 | 80 | N/A |
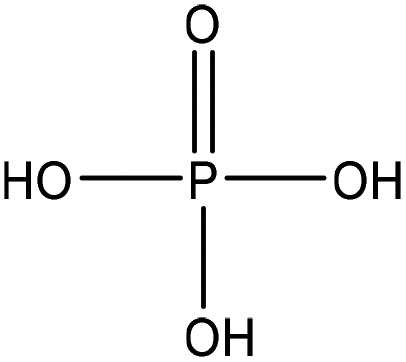
|
Urea | Water and HCl | Phosphorus content | 96 | Sorbent | 37 |
| Black liquor | 2 | 115 | N/A |
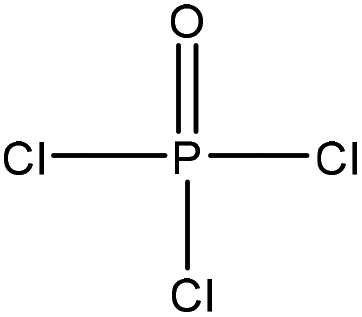
|
Pyridine | N/A | N/A | N/A | Sodium and metal ion adsorbent | 51 |
| Kraft | 12 | N/A | N/A |
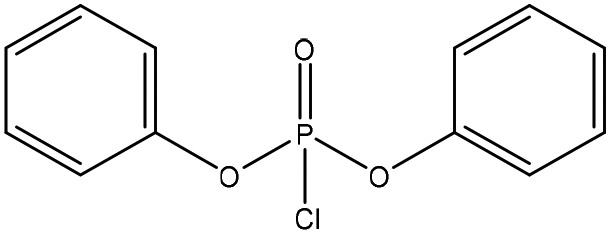
|
Pyridine | Water and DMSO | Oxidative stability | N/A | Flame retardant | 39 |
| Wheat straw alkali | 5 | 70 | 5 |

|
Dimethylformamide and formaldehyde | Water | Flame retardancy, thermal stability | N/A | Flame retardant | 44 and 45 |
| Cotton stalks | 2 | 115 | N/A |
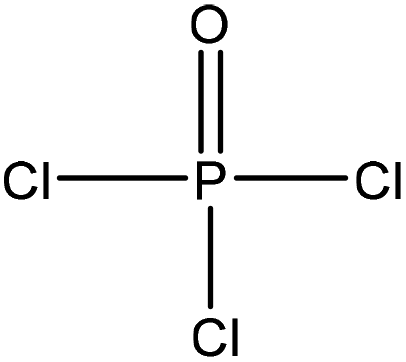
|
Pyridine | HCl | Adsorption selectivity, thermal stability | N/A | Heavy metal ion adsorbent | 51 |
| Hydrolysis | 3–6.5 | 140–180 | N/A |
|
Urea | Water | Thermal stability | 70–75 | Fire-retardant fillers for epoxy compounds | 46 |
| Alkali and an organosolv | 15 | 80 | N/A |
|
Acetonitrile | Diethyl ether | Thermal stability | N/A | Flame retardant for polybutylene succinate | 47 |
The phosphorylated group on lignin has facilitated its use as a high-performance flame-retardant additive in polyurethane, polybutylene succinate, polypropylene, epoxy and polylactic acid.39,44–47 The proposed application is attributed to the fact that the phosphorylation of aromatic compounds enhances char formation under fire conditions by acting either in the gas phase or in the condensed phase via interacting with the polymeric matrix.38,39,48 Phosphorylated lignin has also been used as a cation exchange resin49 and a sorbent of metal ions in wastewater treatment processes.37,49,51
While lignin phosphorylation has certain advantages, some drawbacks, such as long reaction times, use of toxic reagents, e.g., phosphorus oxychloride and phosphorus oxychloride, or toxic solvents, such as dimethyl formaldehyde or dioxane, exist for the phosphorylation of lignin, which may be obstacles for the development of these reaction systems at commercial scales.52
Hydroxymethylation
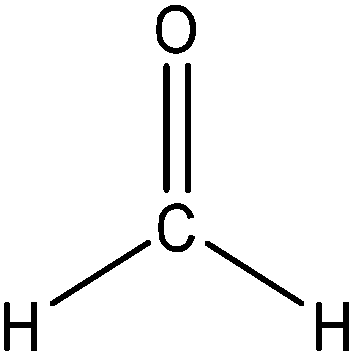
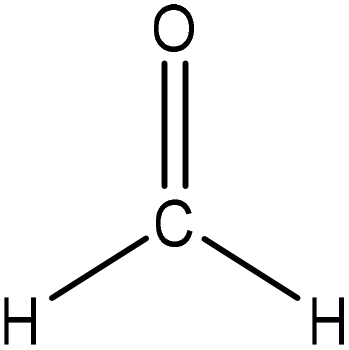
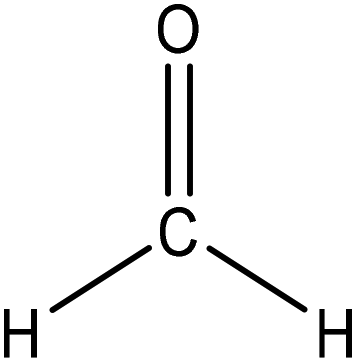
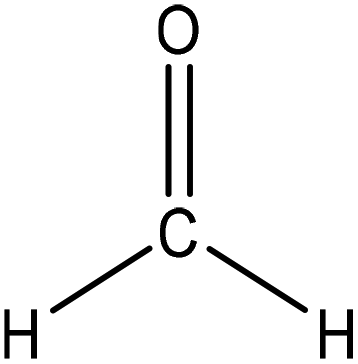
Hydroxymethylation introduces a hydroxymethyl group (–CH2OH) into lignin molecules, which is performed by electrophilic aromatic substitution (Fig. 2). Under alkaline conditions, the sigma complex of lignin loses a proton to regain its aromaticity, which makes the oxyanion become protonated. By reacting lignin and formaldehyde in hydroxymethylation, the hydroxymethyl group is introduced into the para and/or ortho position on the aromatic ring of lignin.54,55 Since this reaction is endothermic, temperature elevation would improve reaction yields.56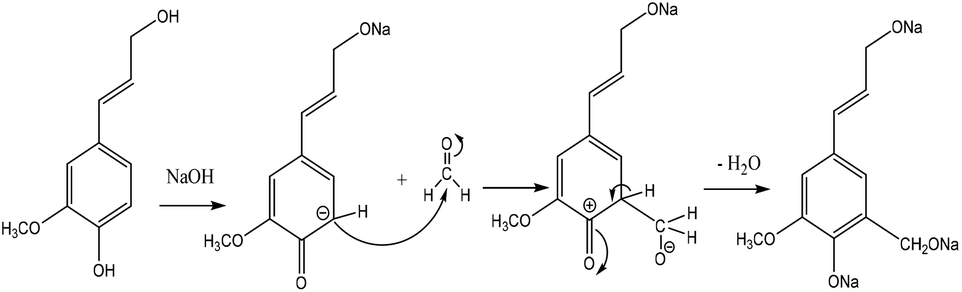 | ||
| Fig. 2 Hydroxymethylation of lignin using formaldehyde.57 | ||
Table 2 shows the results of hydroxymethylation reaction conducted on lignin in the past. In most cases, the reactions were carried out using formaldehyde in alkaline media in the temperature range of 25–90 °C for 2–8 h (or even 72 h) to produce hydroxymethylated lignin. Compared to other reagents, formaldehyde was reported to shorten the reaction time due to its high reactivity. The use of paraformaldehyde in this reaction prolongs the reaction time as it requires the release of formaldehyde monomers at a slower pace in the reaction.58 It is worth noting that formaldehyde may polymerize by itself in the hydroxymethylation reaction, which is undesirable. The hydroxymethylated lignin was reported to be separated from the reaction media by acidification and washing with acids.59 It is worth mentioning that hydroxymethylation is one of the most appealing methods used to produce lignin derivatives used for wood adhesive applications. This reaction can also be used prior to sulfonation and amination reactions to attach a methyl group to lignin for the desired reactions.20,57,61
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Solvent | Reagent | ||||||
| N/A: not available. | ||||||||||
| Organosolv | 2 | 40 | Alkaline | Water |
|
Acidification | Reactivity, thermal stability | N/A | N/A | 64 |
| Wheat straw, and grass | 3 | 90 | 10.5 | Water |
|
Acidification and centrifugation | Molecular weight | N/A | N/A | 55 and 65 |
| Kraft, and sodium lignosulfonate | 0.25–4 | 50 | Alkaline | Water |
|
N/A | Thermal stability | N/A | Phenolic resin | 66 |
| Kraft | 72 | 20–25 | 12–12.5 | Water |
|
N/A | Cross-linking ability | N/A | Adhesives and resins | 54 |
| Wheat straw, and grass | 3 | 90 | 9.7–9.9 | Water |
|
Acidification and centrifugation | Reactivity | N/A | N/A | 67 |
| Calcium lignosulfonate | 2 | 80 | 11 | Water |
|
N/A | Foaming ability, foam half-life time | N/A | N/A | 57 |
| Wheat straw | N/A | 55–90 | 10–10.5 | Water |
|
Acidification and centrifugation | Carbonyl groups, thermal degradation | N/A | As bio-protection in wood and adhesive | 68 |
| Alkali | 3 | 60–90 | 10.5–12 | Water |
|
N/A | Reactivity with resol resin, viscosity | N/A | Phenolic adhesive substitution | 69 |
| Softwood alkali | 8 | 60 | Alkaline | Water |

|
N/A | Decrease in molecular weight | N/A | Wood adhesive | 70 |
| Calcium lignosulfonate | 8 | 58 | 12–12.5 | Water |

|
N/A | N/A | N/A | Wood adhesive | 71 |
| Soda bagasse | 8 | 58 | 12–12.5 | Water |

|
N/A | N/A | N/A | Wood adhesive | 72 |
Hydroxymethylated lignin is also found to have high antioxidant activity since the phenolic OH groups are remained intact in its structure.61 Hydroxymethylated lignin has also been reported to be used in polyurethane foam production62 and as a binder in adhesive applications.63
Phenolation
Phenolation is a reaction in which the number of phenolic OH groups of lignin are increased by the addition of phenol to lignin's aliphatic chain.73,74 This reaction proceeds through SN2 (Fig. 1) or addition reaction.75 A reduction in lignin's molecular weight could be observed in some cases, which would be due to the breakage of ether bonds.76 In general, the phenolation reaction improves the flexibility, tensile strength and glass transition temperature of lignin making it preferable to be used in polyurethane films’ production.Additionally, phenolated lignin has been mostly studied when producing phenol formaldehyde resins, in which lignin is primarily phenolated to react with formaldehyde. Thus, the formaldehyde resins produced with phenolated lignin demonstrated an adequate curing time and viscosity required for panels’ production which is comparable to those of commercial resins.74
Table 3 shows the phenolation reaction carried out on lignin. Generally, lignin is mixed with phenol or cardanol, an alkyl phenol isolated from the liquid shell of cashew nut,77 mostly in water under harsh acidic conditions at a temperature range of 25–125 °C for 20 min–6 h. Phenolated lignin can be separated from the reaction mixture using filtration and/or washing with water, acetone or ether.
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Black liquor | 5 | 60 | Acidic |
|
Water | Filtration and water | Ion-exchange capacity | N/A | Making resin | 80 |
| Enzymatic hydrolysis | 2 | 110–120 | Acidic |
|
Ether | Filtration and water | Molecular weight | 100–120 | Adhesive | 73 |
| Sulfuric acid | 6 | 60 | Acidic |

|
Water | Filtration and water | Solubility and reactivity | N/A | N/A | 81 |
| Organosolv | 1 | 50–80 | N/A |
|
Water | N/A | Curing time | N/A | Resins for particleboard | 78 |
| Organosolv | 1–2 | 70–110 | Acidic |
|
Acetone and water | Filtration and water | Molecular weight and dispersity | 71–96 | Thermoset resin | 82 |
| Eucalyptus/acetosolv | 1.5 | 125 | Acidic |
|
Water | N/A | Reactivity | N/A | Resin | 83 |
| BioChoice, and a pine (softwood) kraft | 2 | 90–110 | Acidic |
|
Water | Ether, and filtration | Molecular weight decreased | 30–60 | Thermosets | 75 |
| Sulfuric acid | N/A | 20–25 | Acidic |
|
Ethanol or water | Acetone, and centrifugation | Light colored | ∼93–110 | Selective phenolation | 84 |
| Softwood kraft | N/A | 50 | Acidic |
|
Water | N/A | Tensile strength, glass transition temperature | 20–40 | Polyurethane film | 77 |
| Beech organosolv | 0.33 | 110 | Acidic |
|
Water | N/A | Strength | N/A | Wood veneer and particle board adhesion | 85 |
Additionally, phenolated lignin used in phenolic resins has been reported to have better mechanical properties than unmodified or hydroxymethylated lignin.78,79 Nonetheless, the immense amount of sulfuric acid used in lignin phenolation is not economically or environmentally attractive since it requires an expensive recovery process.
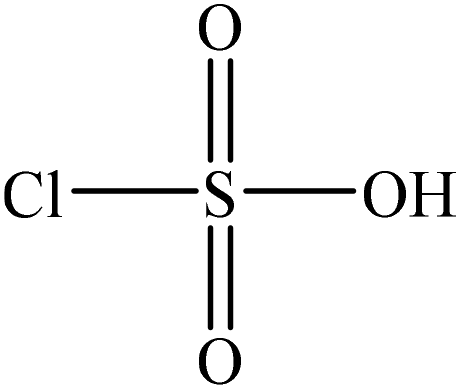
Sulfonation
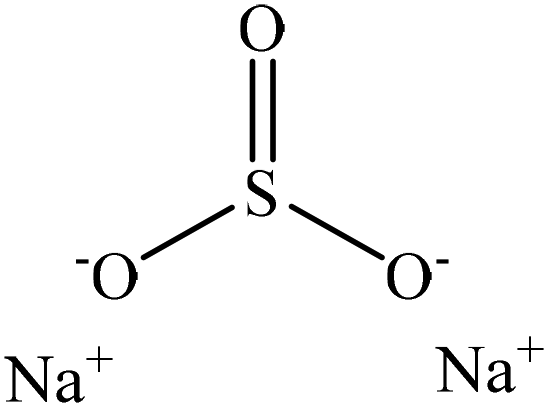
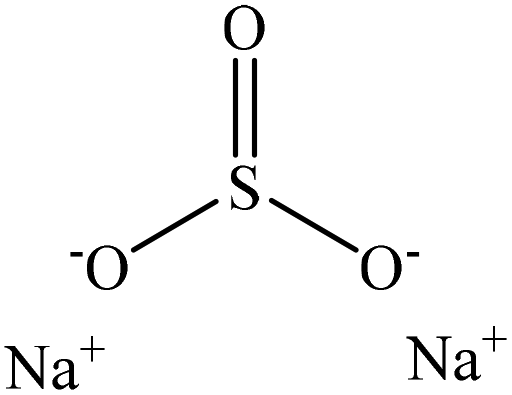
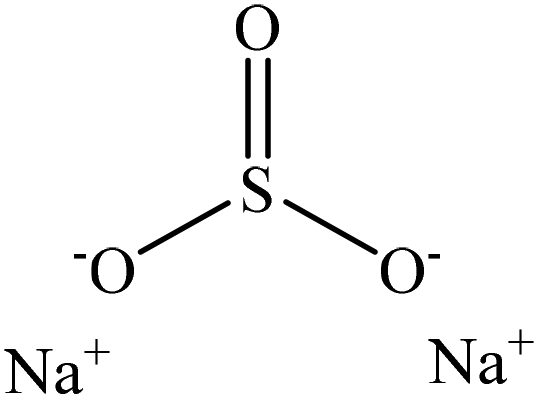
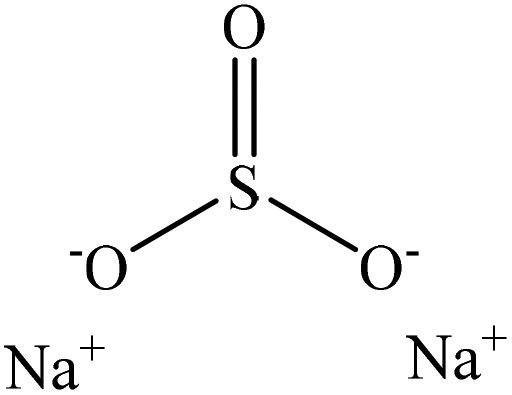
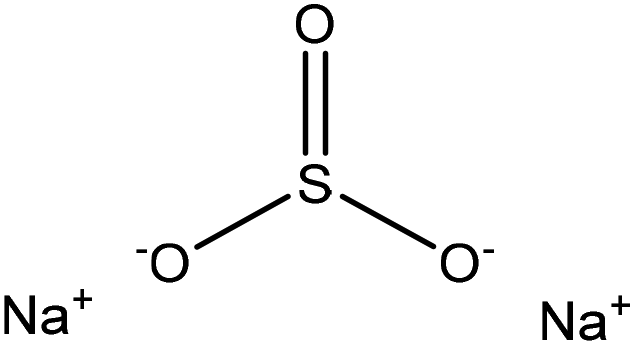
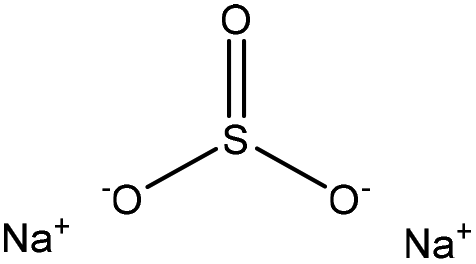
Sulfonation occurs through the substitution of a sulfonate group with lignin's aliphatic hydroxy groups through the addition reaction (Fig. 3),86 rendering lignin negatively charged. This reaction primarily occurs on the carbon of the α position.87 As an exception, sulfonation with chlorosulfonic acid occurs on the lignin's phenolic ring. Sulfonated lignin has a broad range of applications in various industries such as oil drilling,88 paper coating,89 cement and concrete production,90,91 in ion-exchange,80 and as a surfactant,93 binder, and dispersant.88,93–95 The anti-oxidant and UV absorbent properties of sulfonated lignin have also made this product attractive to be used in flame retardants and sunscreens’ production, respectively.96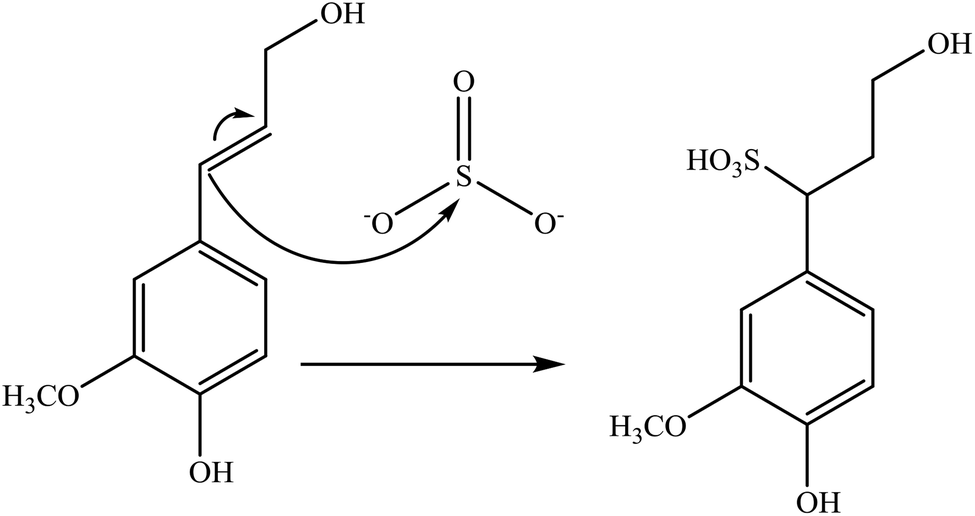 | ||
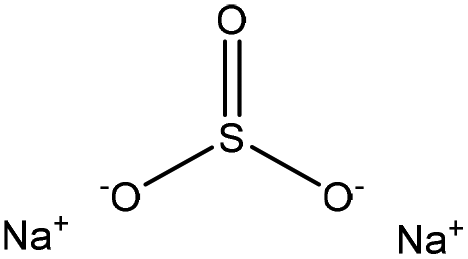
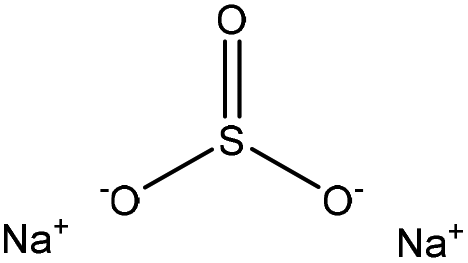
| Fig. 3 Sulfonation of lignin with sodium sulfite.97 | ||
Table 4 shows sulfonation reactions performed on lignin. The sulfonation of lignin has been reported to be conducted using either sulfur dioxide (SO2), sulfur trioxide (SO3), sulfurous acid (H2SO3), sodium metabisulfite (Na2S2O5), or bi-sulfite (M2SO3) (where M can be Ca, Na, H, Mg, K, or their combination) as a reagent. In this reaction, lignin is generally mixed with the reagent mostly in water under either acidic or alkaline conditions in a high-temperature range of 70–180 °C for 0.5 to 4 hours. Thus, the produced lignin samples could be separated and purified using filtration and a dialysis membrane.
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Esparto grass | 4 | 140 | 4–9 |
|
Water | Filtration | Solubility | N/A | Plasticizing for cement | 91 |
| Washed aqueous slurry | 2 | 140 | ∼7 |
|
Water | N/A | Solubility | N/A | Dispersant in dye | 95 |
| Kraft | 1–5 | 100–180 | Alkaline |
|
Water | Dialyzed | Molecular weight decreased | N/A | N/A | 97 |
| Corn stalks | 0.5–1 | 95 | Alkaline |
|
Water | N/A | N/A | ∼70–85 | N/A | 98 |
| Kraft | 1–2 | 70–120 | Acidic |
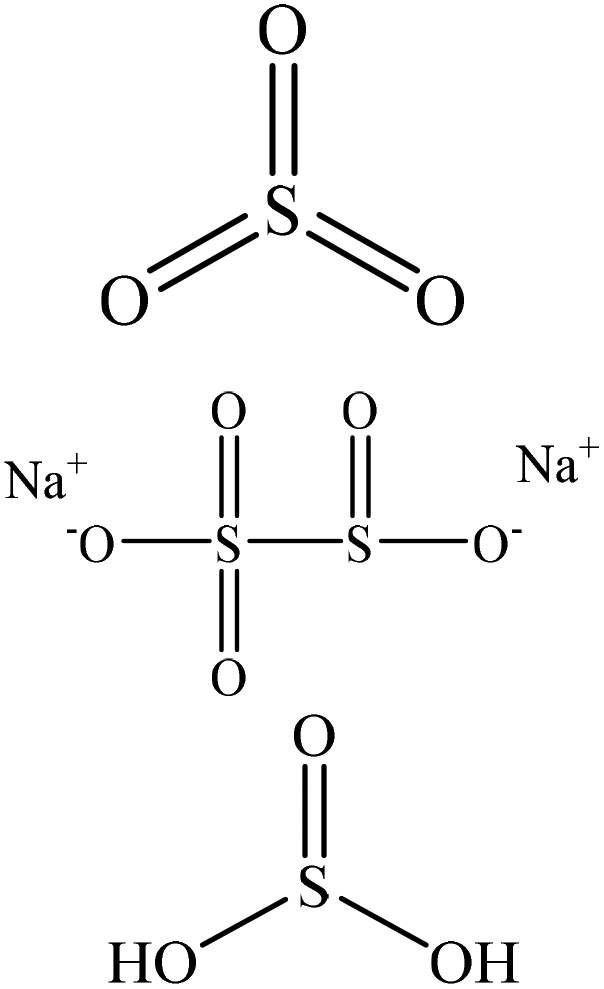
|
Water | N/A | N/A | N/A | Sulfonation of alkali pulp | 92 |
| Phenolyzed | 1.5 | 100 | Alkaline |
|
Tetrachloroethane | Filtration and water | Ion-exchange capacity | ∼33–58 | Making resin | 80 |
Sulfoalkylation
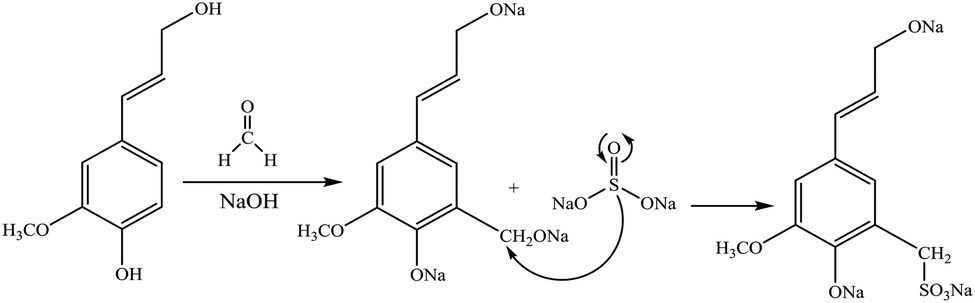 | ||
| Fig. 4 Sulfomethylation of lignin with sodium sulfite.20,57 | ||
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Enzymatic hydrolysis | 3 | 95 | Alkaline |
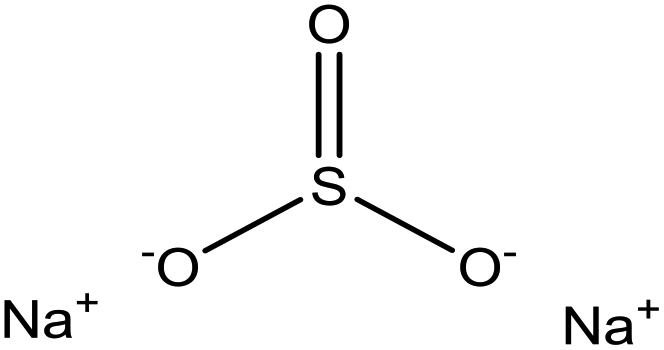
|
Formaldehyde | Filtration | Steric energy reduction | N/A | Dispersion for graphite suspension | 106 |
| Alkaline, and enzymatic hydrolysis | 1–5 | 80–130 | N/A |
|
Formaldehyde | H2SO4 | Compressive strength of concrete | ∼60–90 | Dispersant for concrete paste | 107 |
| Oxidized | 1–3 | 100 | Alkaline |
|
Water and formaldehyde | Membrane dialysis | Molecular weight, charge density | 33–38 | Flocculant for aluminum oxide suspension | 105 |
| Aminated alkaline | 2 | 90 | 10 |

|
Water | Filtration | N/A | N/A | Heavy metal ion removal | 109 |
| Oxidized softwood kraft | 0.5–4 | 60–100 | 7 |
|
Water, and formaldehyde | Membrane dialysis | Molecular weight, charge density | N/A | Dispersant for cement | 101 |
| Hardwood kraft | 1–7 | 80–140 | N/A |
|
Formaldehyde | Membrane dialysis | Molecular weight, charge density | N/A | Dispersant for cement | 20 |
| Kraft | 3 | 75 | 3–4 |
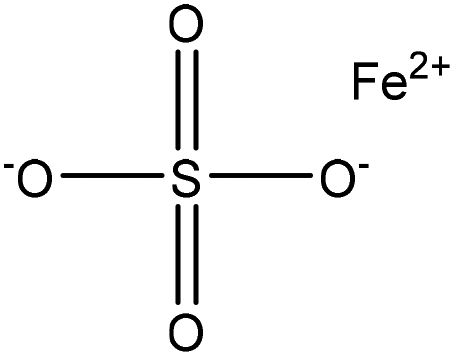
|
Formaldehyde, H2O2 | Dialyzed | Molecular weight decreased, and lighter color | N/A | N/A | 97 |
| Calcium lignosulfonate | 2 | 90 | 10 |
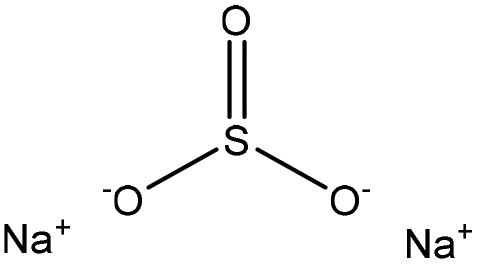
|
Formaldehyde | N/A | N/A | N/A | Dispersant for cement | 57 |
| Alkali | 5 | 90 | 10 |
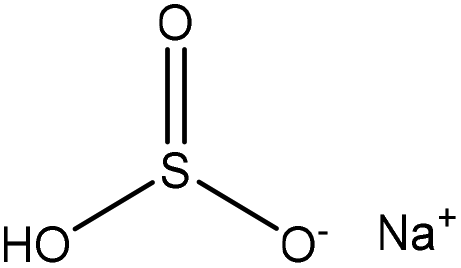
|
Water, and formaldehyde | N/A | Adsorption selectivity, wettability | N/A | Dispersant for coal–water slurry | 21 |
| Alkali-corn stalk | 2–9 | 75 | 9 |
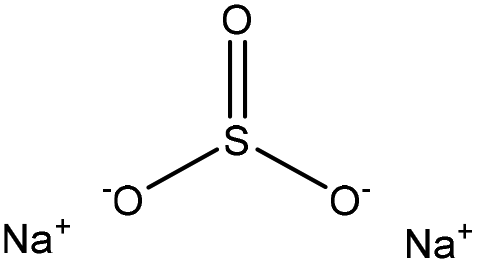
|
Formaldehyde | Filtration | N/A | N/A | Dispersant for dye | 100 |
| Hydroxymethylated alkali | 4 | 95 | 13 |
|
Formaldehyde | Neutralization and Buchner funnel | Charge density, solubility | N/A | Adsorbent | 103 |
| Wheat straw alkali | 4 | 95 | N/A |
|
Formaldehyde | Neutralization and Buchner funnel | Charge density | N/A | Dispersion for TiO2 | 104 |
However, the reactivity of this reaction is rather low and highly depends on the lignin type. In order to improve lignin reactivity toward sulfomethylation, oxidation was reported to be conducted prior to sulfomethylation.20,101
Overall, sulfomethylated lignin has been investigated to have various applications in industry, such as a dispersant for coal–water slurry21,57 and concrete paste20,101 or a flocculant for aluminum oxide suspension105 due to its augmented hydrophilicity and charge density.
However, using formaldehyde in sulfomethylation can be considered as the main drawback of this modification pathway since it is toxic, carcinogenic and mutagenic, which raises environmental concerns.
 | ||
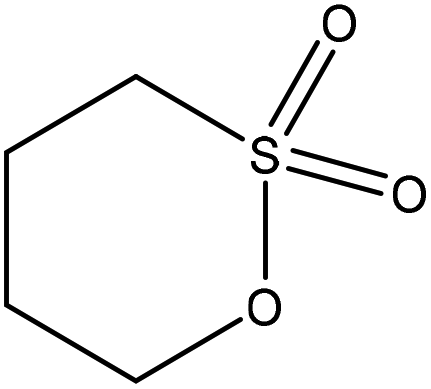
| Fig. 5 General SN2 reaction with ring-opening. Electrophilic center (carbon) is partially negatively-charged, and oxygen is partially positively-charged.109,111,112 | ||
Table 6 shows the sulfobutylation reactions conducted on lignin. In general, sulfobutylation was conducted using 1,4-butane sultone at 70 °C for 6–7 hours at pH 12. In order to separate the produced polymer from the reaction media, the ion-exchange resin and dialysis membrane have been used. Sulfobutylated lignin has been used as a dopant and a dispersant for coal–water slurry and carbendazim.109–111
| Lignin source/type | Reaction conditions | Separation | Advancement in properties | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Alkali | 7 | 70 | 12 |
|
Water | Ion-exchange resin | Molecular weight | N/A | As dopant and dispersant | 111 |
| Alkali | 6 | 65 | 12 |
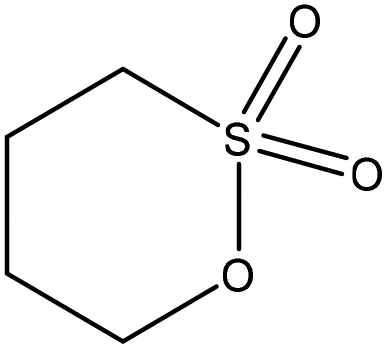
|
Water | Ion-exchange resin | Molecular weight | N/A | Dispersant for coal–water slurry | 109 |
| Eucalyptus kraft | 3 | 70 | 12 |
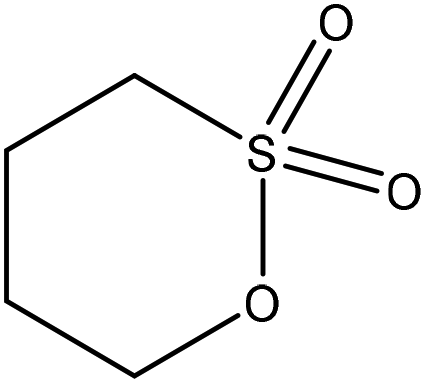
|
Water | Ethanol | Brightness | N/A | Dispersant for dye | 113 |
| Alkali | 7 | 70 | 12 |
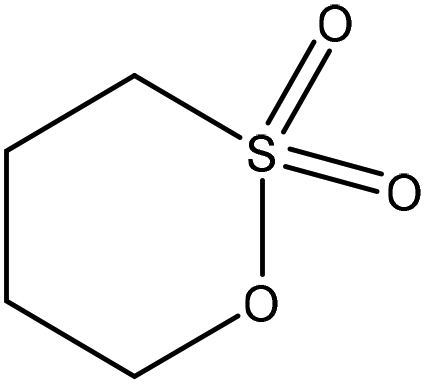
|
Water | Dialysis membrane | Molecular weight | N/A | Dispersant for carbendazim | 110 |
| Alkali | 7 | 50 | 12 |

|
Water | Filtration and dialysis | Hydrodynamic size | N/A | Aggregation-induced emission | 114 |
| Kraft | 1–6 | 50–90 | Alkaline |
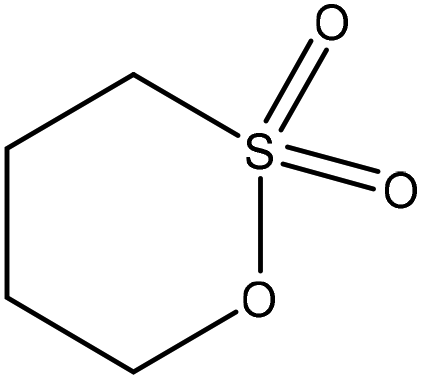
|
Water | Dialyzed | Molecular weight decreased, and lighter color | N/A | N/A | 97 |
Sulfobutylation seems to be a more favorable reaction over sulfomethylation because (1) sulfobutylation needs lower temperature and pressure (Tables 5 and 6), which results in using less expensive and simpler equipment, (2) the use of toxic formaldehyde in sulfomethylation is another downside of this reaction,109 and (3) sulfomethylation can only occur on the phenolic part of lignin, while sulfobutylation can occur on both aliphatic and aromatic parts. However, the 1,4-butane sultone reagent used in sulfobutylation is substantially more expensive than sulfomethylation reagents (Na2SO3, Na2S2O5), which may make this modification process expensive.
Carboxyalkylation
Table 7 shows the lignin carboxymethylation reactions performed in the literature. The carboxymethylation reaction has been carried out by mixing lignin with NaOH followed by sodium chloroacetate or monochloroacetic acid in the time and temperature range of 1–6 h and 30–90 °C, respectively.40,115–119 Alternative pathways were considered for purifying the products, such as acidification,118 membrane dialysis,40 and washing with ethanol.116,119,120
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Organosolv | 3.5 | 55 | N/A |
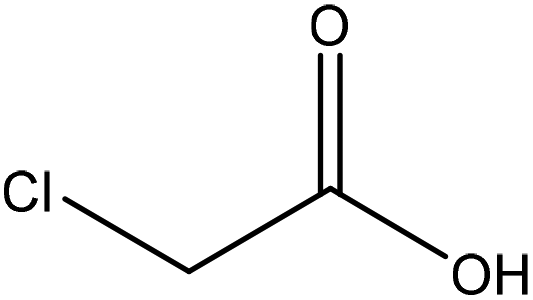
|
Ethanol | Filtration | N/A | N/A | Stabilizer in ceramic industries | 116 |
| Harwood kraft | 1–6 | 30–70 | Alkaline |
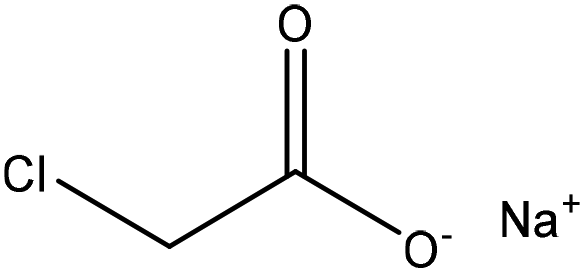
|
Water | Membrane dialysis | Charge density, solubility, molecular weight | N/A | Dispersant for clay suspension | 40 |
| Kraft | 3.5 | 55 | N/A |
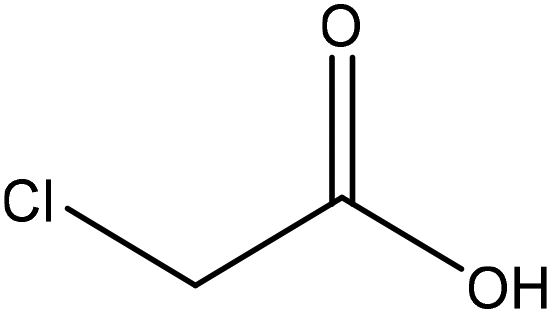
|
Ethanol | Filtration and ethanol | Surface tension | N/A | Stabilizer for oil-in-water emulsions | 117 |
| Kraft and organosolv | 3.5 | 55 | N/A |
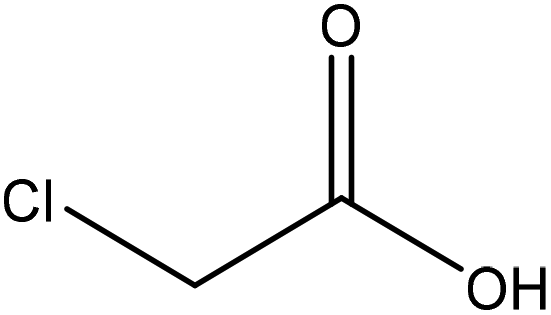
|
Ethanol | Filtration and HCl | Decrease in heat capacity, and surface tension | 90 | Stabilizer of crude bitumen | 121 |
| Wheat straw alkali | 1–3 | 50–90 | N/A |
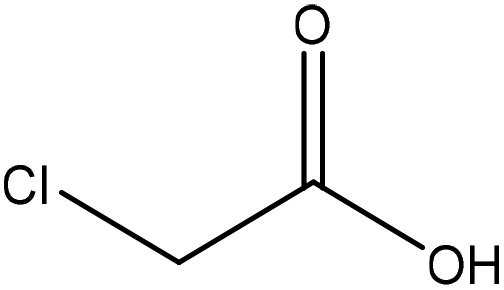
|
Water | HCl | N/A | ∼80 | Dispersant | 122 |
| Kraft | N/A | N/A | Alkaline |
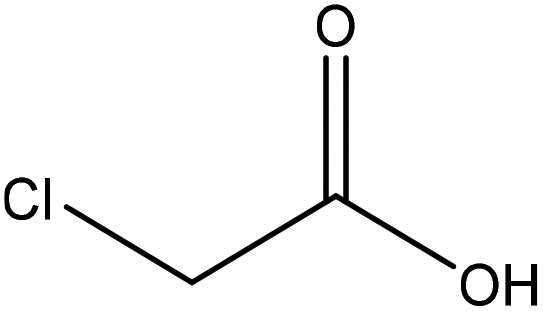
|
Water/ethanol | HCl | Adsorption to fibre-laden | N/A | Stabilizer of fiber-laden foams | 120 |
Carboxymethylated lignin has been proposed as an effective dispersant for oil–water emulsions,109 crude bitumen emulsions,121 and clay,40 cement,116 and graphite suspensions.122 Carboxymethylated lignin was also used as a stabilizer in kerosene-in-water emulsions117 and as a foaming agent.110 The composite of carboxymethylated lignin–tetra ethoxysilane was tested as a packaging and antimicrobial formula as well as in wound dressings. In addition, due to its potential in adsorbing heavy metals, such as nickel and cadmium, this product has been suggested to be used in wastewater treatment and biofilters.119 Carboxymethylated lignosulfonate was also reported to improve the heat capacity of leather when it was used along with tanning chromium in leather production.115
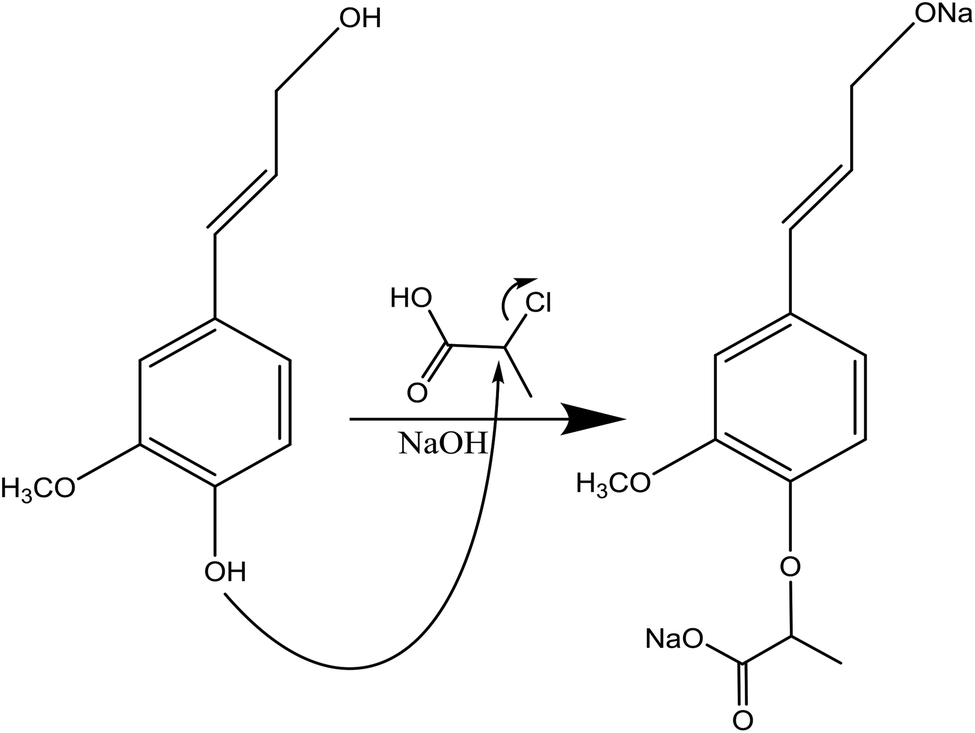 | ||
| Fig. 6 Carboxyethylation of lignin under alkaline conditions by using 2-chloropropionic acid.123 | ||
In this reaction, lignin can be mixed with 2-chloropropionic acid, the donor of the carboxyethyl group, in a mixture of water and isopropyl alcohol in the basic environment at 60–90 °C for 0.5–2 h (Table 8). The production of sodium lactate is the undesired side reaction.123 Due to the insolubility of the produced lignin in solvents, the reaction mixture can be mixed with ethanol for lignin isolation.123 Alternatively, membrane dialysis and filtration can be used for isolating the product from the reaction media.
| Lignin source/type | Reaction conditions | Separation | Advancement in properties | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Lignosulfonate | 0.5–2.0 | 60–90 | Alkaline |
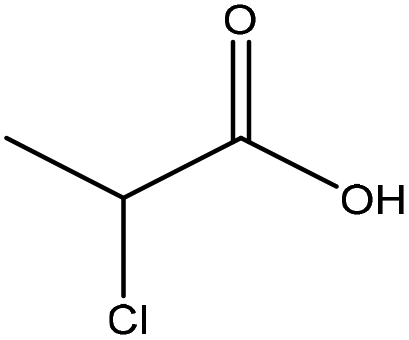
|
Water/isopropyl alcohol | Ethanol/water | Charge density, molecular weight | N/A | Dispersant | 123 |
In opposition to carboxymethylation reactions, carboxyethylation can occur on both aromatic and aliphatic hydroxyl groups of lignin. Therefore, carboxyethylation may be considered as a more influential modification pathway for lignin than carboxymethylation.122,123 However, the main drawback of carboxyethylation is the solvent used in the reaction (e.g., isopropyl alcohol), which may not be an attractive pathway to develop green processes for lignin modification.
Epoxidation
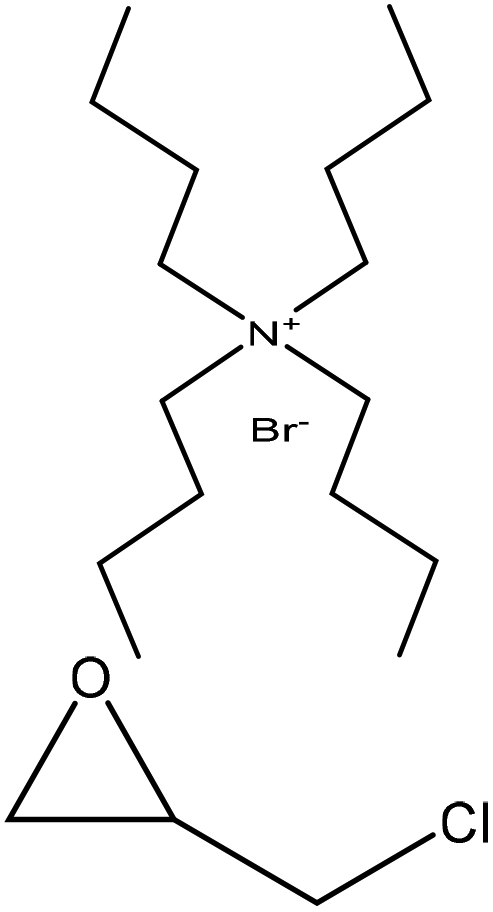
Chemicals possessing amino or hydroxy groups have good reactivity with epoxy groups. Lignin can be rendered lipophilic when it is modified with epoxy containing materials. This reaction facilitates the dissolution of lignin in organic solvents, such as methylene chloride, tetrahydrofuran, acetone, and chloroform for generating value-added products.124 Epoxy resins are used in a broad variety of applications in electrical and electronic laminates, high-performance composites, industrial coatings, adhesives, paving applications, and feedstock for emulsifiers and detergents.122,124–126 Epoxidation of lignin proceeds through the SN2 mechanism as discussed earlier (Fig. 1) under alkaline conditions.112 Epoxidation was reported to occur on the aromatic ring of lignin by the substitution of hydroxy groups with epoxy groups.61,127Table 9 shows the epoxidation reactions of lignin reported in the literature. In this reaction, lignin is mixed with either di-epoxides, such as polyethylene glycol diglycidyl ether (PEGDGE), with different chain lengths, or epichlorohydrin in an alkaline environment (pH > 12) at 30–90 °C for 1–18 h.50,112,126–128 The epoxidized lignin product is then isolated by neutralizing the reaction mixture with sodium dihydrogen phosphate (NaH2PO4) and centrifugation. The solid epoxy lignin can then be recrystallized in chloroform for further use.61–67 Stronger alkalinity could enhance the lignin degradation and produce more phenolic hydroxy groups to react with epichlorohydrin, which increases the reaction yield.112 In this case, lignin macromolecules will be converted to more of lignin monomers, and then monomers would have a higher tendency to epoxidize. However, using epichlorohydrin has some disadvantages, such as toxicity and limited rheological characteristics associated with the gel-like dispersion.129,130
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Alkaline | 5 | 90 | Alkaline |
|
Water | Neutralization and water | Molecular weight | ∼12 | Feedstock for an emulsifier, detergent, and additive | 124 |
| Alkali | 6 | 50–80 | N/A |
|
Diethanolamine, and formaldehyde | N/A | Thermal stability | N/A | Thermal stable resin | 126 |
| Organosolv lignin | 3–5 | 50–90 | N/A |
|
Water | Water and filtration | Molecular weight | 107–126 | Bio-based epoxy resin | 125 |
| Wheat straw | 2 | 60 | Alkaline |
|
Water | N/A | N/A | N/A | Bio-based epoxy resin | 127 |
| Sugarcane bagasse | 3–6 | 70 | Alkaline |
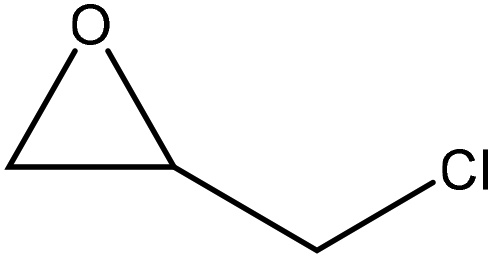
|
Water | Neutralization and centrifugation | Antibacterial activity, lower radical scavenging activity | N/A | Natural antibacterial | 61 |
| Sarkanda, and wheat straw, Protobind 1000 | 3–7 | 50–90 | Alkaline |
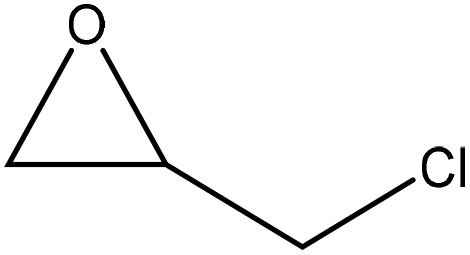
|
Water | Neutralization and centrifugation | Thermal stability decreased | 61–88 | Composite formation | 67 |
| Alkaline | 8 | 50 | N/A |
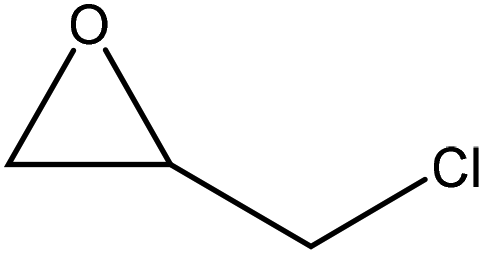
|
Water | Filtration and ethane/water | Decomposition temperature decreased | 37–91 | Epoxy resin additive | 112 |
| Alkali | 3 | 30 | Alkaline |

|
Water | Centrifugation | Viscosity | N/A | Thickener in bio-lubricant | 129 |
Epoxy resins produced by lignin usually have low thermal stability and Tg value over the mercantile ones (i.e., a synthetic bisphenol A). In this regard, the limited number of epoxy rings in lignin prevents the generation of dense crosslinks in cured epoxy systems. Hence, it would be more favorable to produce lignin-based curing agents to be used in generating epoxy systems with efficient performance.131 Similarly, the epoxy lignin was reported to have antibacterial activity and was mostly resistant to Bacillus sp. and Klebsiella sp. strains.61 Nonetheless, the disadvantages associated with lignin-based epoxy resins include low water solubility, slow curing rate, high cost and brittleness.24,132–135
Oxyalkylation/oxypropylation
Oxyalkylation is a process by which hydroxy groups of lignin are converted to oxyalkylated groups. The oxyalkylation of lignin proceeds through the SN2 mechanism (Fig. 7). In the reaction of cyclic organic carbonates with hydroxyl groups of lignin (Fig. 7), cyclic carbonate (e.g., propylene carbonate) can react with aliphatic and aromatic hydroxyl groups136 according to two reaction pathways; aliphatic hydroxy tends to attack the carbonyl carbon atom leading to carbonate linkages, while aromatic hydroxy can attack the alkylene carbon atoms and ether linkage with a subsequent loss of CO2, which allows the production of polyether polyols with primary and secondary hydroxy groups.136,137 At temperature lower than 170 °C, the rate of oxyalkylation trend on the aromatic hydroxy groups is higher than that on the aliphatic ones due to the lower nucleophilicity of aliphatic hydroxy groups. At a higher temperature (>170 °C) and in the presence of the basic catalyst (e.g., K2CO3), only 0.3% of carbonate linkages could be developed, while etherification of the aliphatic hydroxy groups was found to be favored at high temperatures.136,138 Oxyalkylation liberates the lignin's hydroxy groups, especially that of the aromatic OH, and induces moieties of ether that presents uniformity, solubility, and reactivity to lignin. In this case, lignin can be converted to a liquid polyol product139 that could be a potential alternative to replace typical ones in polyurethanes. Among all, oxypropylation is the most popular oxyalkylation reaction, which could be feasible to conduct under both alkaline and acidic conditions, while it was suggested to be more efficient and economically favorable if performed in an alkaline environment.140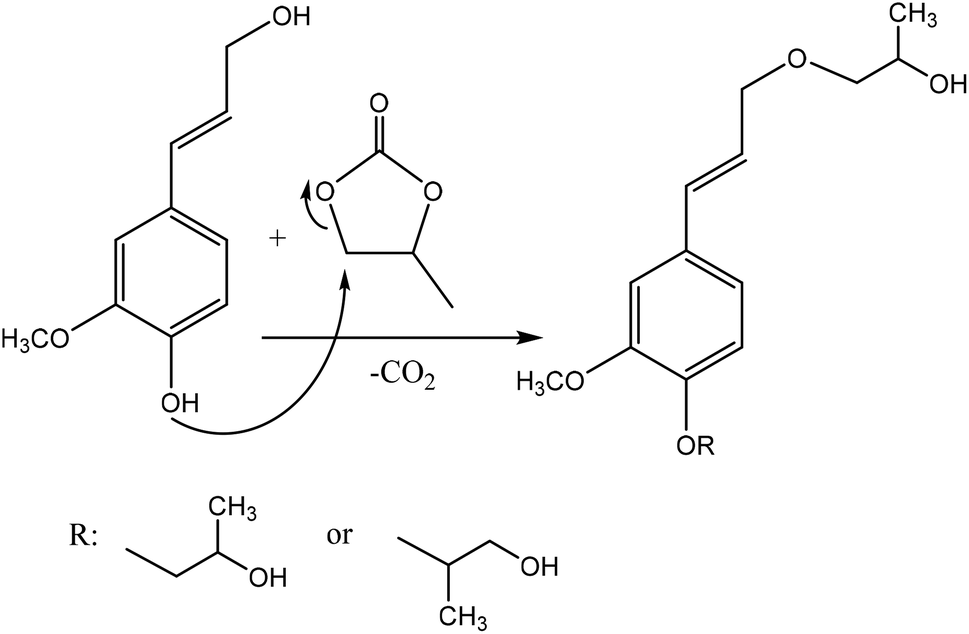 | ||
| Fig. 7 Oxypropylation of lignin by using propylene carbonate.136 | ||
Table 10 shows reports on the oxypropylation of lignin. To produce oxypropylated lignin, lignin is mixed with reagents, such as propylene oxide or propylene carbonate, and NaOH or KOH, and is reacted in the temperature range of 40–285 °C for 4 min to 24 h under atmospheric and pressurized (up to about 40 bar) conditions.136,138,141,144,150 To extract the product from the reaction media, the reaction mixture is acidified to pH 2.5, which facilitates the precipitation of oxypropylated lignin.136,138,141
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Catalyst/solvent | ||||||
| N/A: not available. | ||||||||||
| Kraft | 12 | 40 | Alkaline |
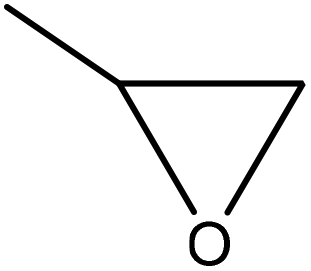
|
Potassium hydroxide | HCl | Molecular weight decreased | N/A | Use in carbon fiber | 147 |
| Soda | 0.08–0.16 | 120–140 | Alkaline |
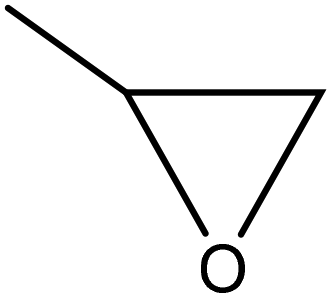
|
Potassium hydroxide | N/A | Molecular weight decreased | N/A | Use in polyurethane foam | 142 |
| Organosolv, kraft, and oxidized organosolv | 0.3–15 | 140–190 | Alkaline |
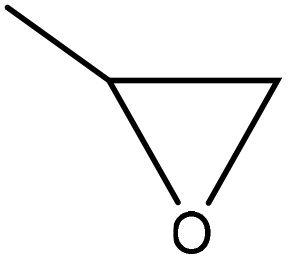
|
Potassium hydroxide | Vacuum removal | Dimensional stability of the foam | N/A | Use in polyurethane foam | 143 |
| Kraft | 2 | 140 | Alkaline |
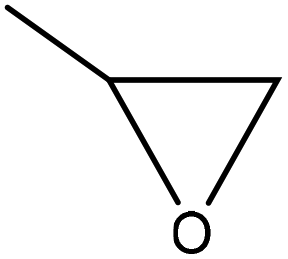
|
Potassium hydroxide | Tartaric acid | Viscosity of the polyester–polyether polyol | N/A | Use in the polyurethane production | 148 |
| Sodium lignosulfonate | 4 | 175 | Alkaline |
|
Sodium carbonate | Distillation | Darkness, viscosity | N/A | As dispersants for carbon black, as emulsifiers | 149 |
| Wheat straw soda | 24 | 70 | Alkaline and acidic |
|
Water | Ethyl ether anhydrous | Molecular weight, smother morphology | N/A | As a substitute for polyols in view of polyurethane | 140 |
| Kraft, soda, organosolv | ∼0.58–1.8 | 169–271 | Alkaline |
|
Potassium hydroxide | N/A | Viscosity | N/A | As a substitute for polyols in view of polyurethane | 140 |
| Kraft | 18 | 40 | Alkaline |
|
Water | HCl | Thermal stability decreased | N/A | Thermoplastic materials | 150 |
| Kraft | 0.15 | 150–285 | Alkaline |
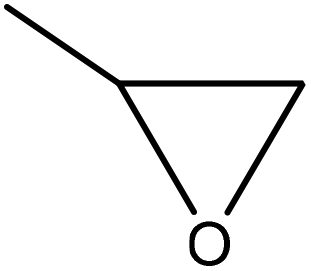
|
Potassium hydroxide | N/A | Molecular weight decreased | N/A | As a substitute for polyols in view of polyurethane | 144 |
| Softwood kraft | 18 | 20–25 | Alkaline |
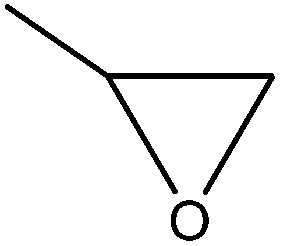
|
Water | N/A | Molecular weight decreased | N/A | N/A | 141 |
| Organosolv | 3 | 170 | N/A |
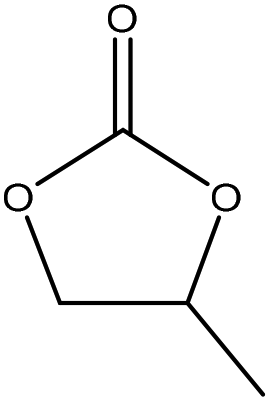
|
Methylimidazole, 1,4-dioxane | Acidification and filtration | Molecular weight | N/A | As a substitute for polyols in view of polyurethane and polyesters | 136 |
| 21 | 50 |
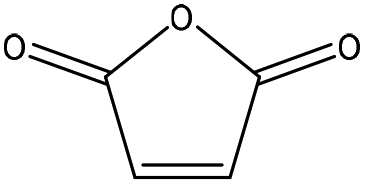
|
||||||||
| Beech wood organosolv | 0.5–24 | 100–170 | N/A |

|
1,8-Diazabicyclo[5.4.0]undec-7-ene/potassium carbonate | Acidification and filtration | Molecular weight | N/A | Substitution of conventional petroleum-based polyols | 137 |
| Beechwood, wheat straw Organosolv | 3 | 170 | N/A |
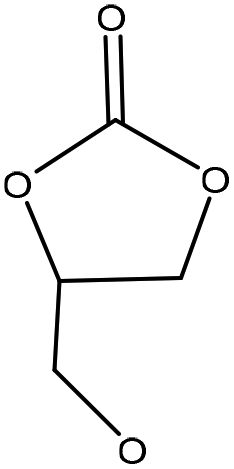
|
1,8-Diazabicyclo[5.4.0]undec-7-ene | Water and filtration | Molecular weight | N/A | Biobased polyols | 138 |
| Organosolv | 3 | 170 | Alkaline |
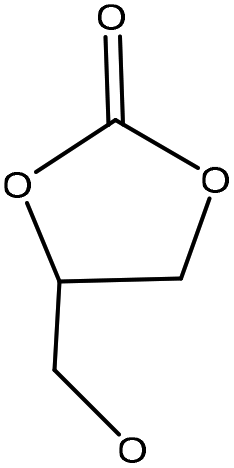
|
1,8-Diazabicyclo[5.4.0]undec-7-ene/dimethyl sulfoxide | Acidification | Molecular weight | 97–99 | Prepolymer for nonisocyanate polyurethanes | 151 |
Oxypropylated lignin has found application in polyurethane foams.142,143 It was reported that oxypropylated lignin has remarkably enhanced the mechanical properties of foams compared to commercial polyols.145 In addition, the produced lignin polyols were suggested to be a valuable substitution for oil-based polyols, which are extensively used in polyester and polyurethane productions.136–138 However, the use of propylene oxide, which is obtained from oil-based chemicals may be unattractive, as the final product may contain less than 50% lignin.145 In addition, this reagent is very expensive which makes the reaction unfavorable for commercial purposes. This reaction also suffers from safety concerns due to the high vapor pressure as well as high toxicity, carcinogenicity, and flammability of propylene oxide in the reaction media. Instead, cyclic organic carbonates, such as propylene carbonate, could be used in this reaction to reduce the precautions since it is non-toxic and eco-friendly.137 Furthermore, high boiling and flash points, as well as low vapor pressure and high solubility make this reagent more attractive than propylene oxide to be used in the oxypropylation of lignin.136,137,146
Esterification
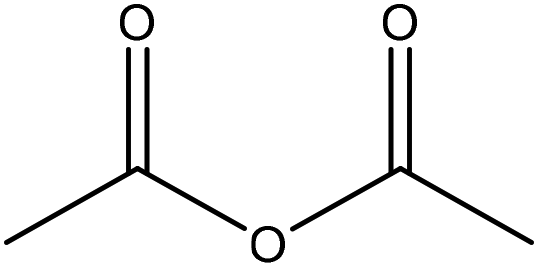
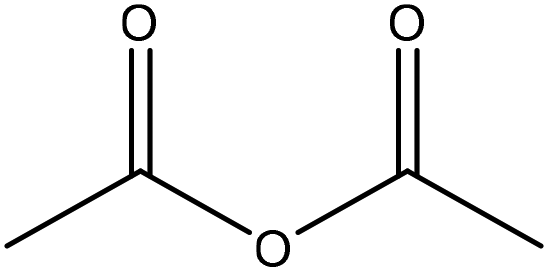
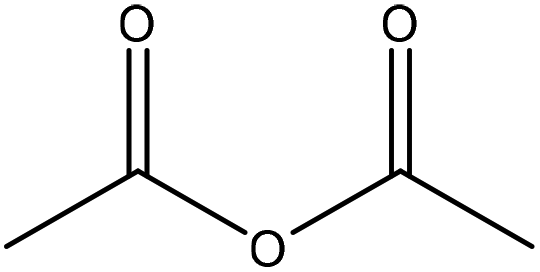
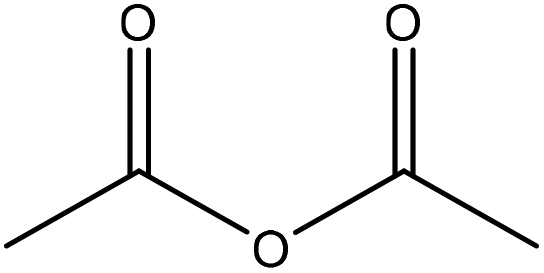
Generally, esterification is the conversion of alcohols to esters.152 The esterification of lignin is performed by nucleophilic substitution (Fig. 8). In this reaction, the lone pair electron of the aromatic hydroxy group will attack the carbon of the ester group on the reagent (Fig. 8). As a result, the carboxylic acid group will leave, and hydroxy groups of the aromatic ring are replaced with carbonyl groups.153 This reaction is feasible using mono- or dicarboxylic acids, their anhydrides, acid chlorides, or via transesterification with carboxylic acid esters. For instance, maleic acid, acetic acid, phthalic acid, fumaric acid, or fatty acids such as oleic acid, lauric acid or their anhydrides, acid chlorides or simple esters can be used in the esterification of lignin.153–155 Esterification occurs on both aromatic and aliphatic hydroxy groups of lignin but is more favored to occur on the aromatic hydroxy group.156 The reason for this tendency could be the lower pKa of the aromatic hydroxy groups than the aliphatic counterparts of lignin since the acetylation reagents, e.g., pyridine or imidazole, act as both catalysts and bases accelerating the production of nucleophiles.157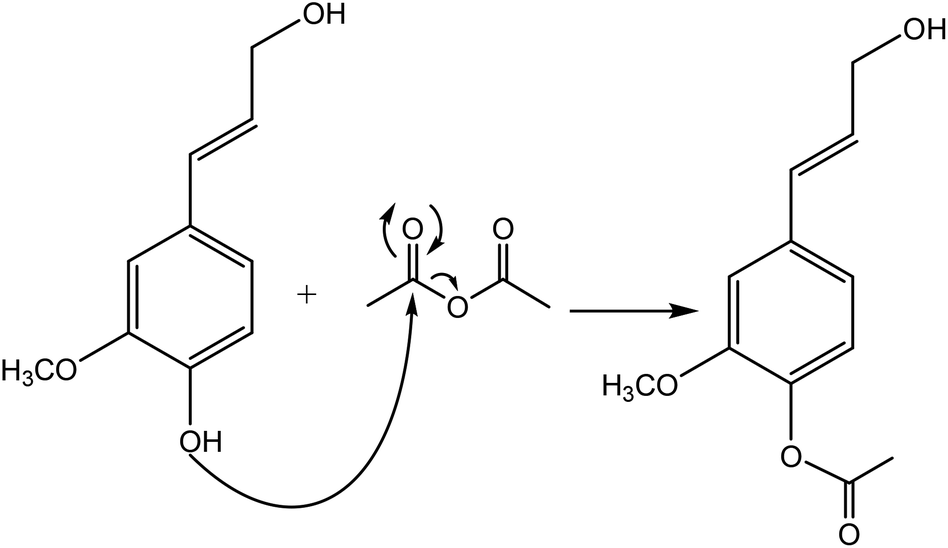 | ||
| Fig. 8 The esterification reaction of lignin with acetic anhydride.61 | ||
Table 11 shows the esterification reaction conducted on lignin. Generally, lignin is mixed with different acid anhydrides (succinic, phthalic, acetic and maleic anhydrides) in solvents, such as pyridine, tetrahydrofuran, dimethylformamide, acetone, dioxin, and 1-methylimidazole, in the temperature range of 25–120 °C for the period of 5 min and 48 h.143,148–151 After the reaction, acidification or solvent (e.g., acetone, ethanol) addition and membrane dialysis were used as means of the product purification strategy.153,158,160,161
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent/Catalyst | ||||||
| N/A: not available. | ||||||||||
| Organosolv | 2 | 20 | N/A |
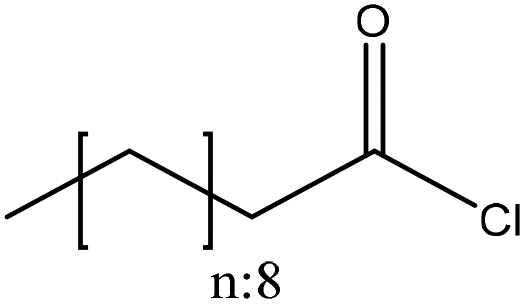
|
N,N,1-Dimethylformamide/pyridine | Filtration and ethanol | Hydrophobicity | N/A | As a dispersant and a protective agent | 173 |
| Pulp and paper industries | 0.6 | 80 | N/A |
|
Pyridine | HCl | Molecular weight | 122 | Mold lubricant | 164 |
| Organosolv | 3 | 100 | N/A |
|
Pyridine containing 4-dimethylaminopyridine | Water or diethyl ether | Strength | N/A | Blend with poly(ε-caprolactone) | 163 |
| Organosolv | 6 | 70 | N/A |
|
Tetrahydrofuran | Methanol and filtration | Surface area, pore volume | ∼36–46 | Lignin-based carbons | 158 |
| Kraft | 5 | 100 | N/A |
|
Triphenyl antimony | Acetone | Thermal stability | N/A | N/A | 153 |
| Softwood and hardwood kraft | 20 | 20–25 | N/A |
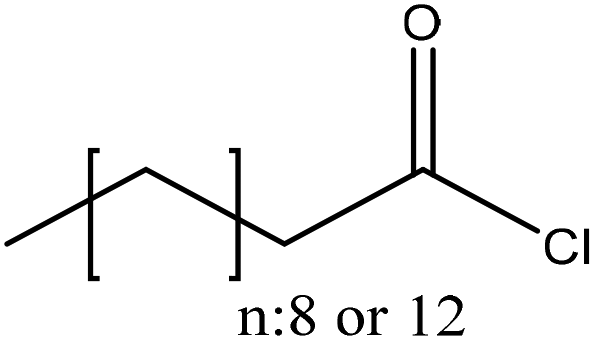
|
Dimethylformamide | Ethanol, Soxhlet extraction in chloroform | Molecular weight | N/A | Oxygen and water vapor barrier | 152 |
| Hardwood and softwood kraft | 3 | 50 | N/A |

|
Acetic, and propionic | Filtration | Molecular weight, reduce water adsorption | 88–99 | Polyethylene blends | 160 |
| Softwood kraft | 24 | 65 | N/A |
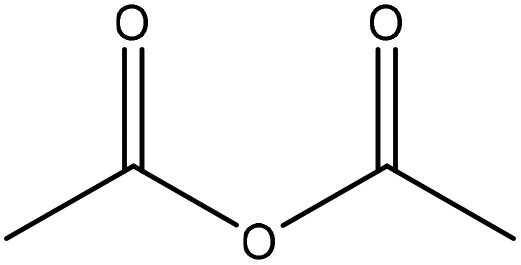
|
1-Methylimidazole | Ethanol and centrifugation | Tensile strength, reduced water adsorption | 24 | Fillers for thermoplastics | 161 |
| 95 |
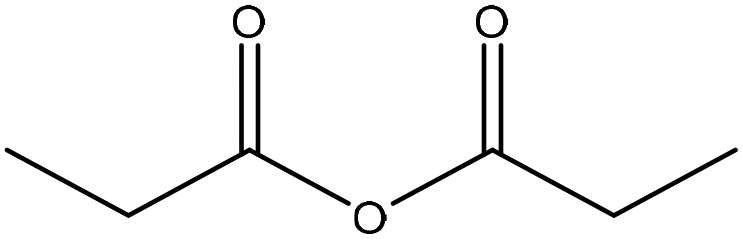
|
29 | ||||||||
| 120 |

|
46 | ||||||||
| Organosolv | 1 | 100 | N/A |
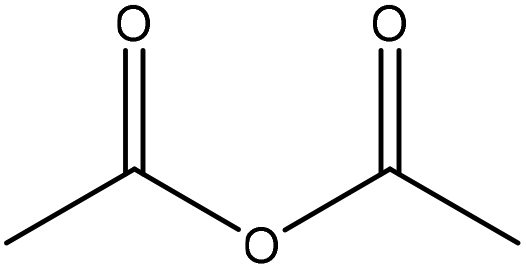
|
Pyridine | N/A | Solubility | ∼96 | N/A | 174 |
| Softwood kraft | 48 | 65 | N/A |
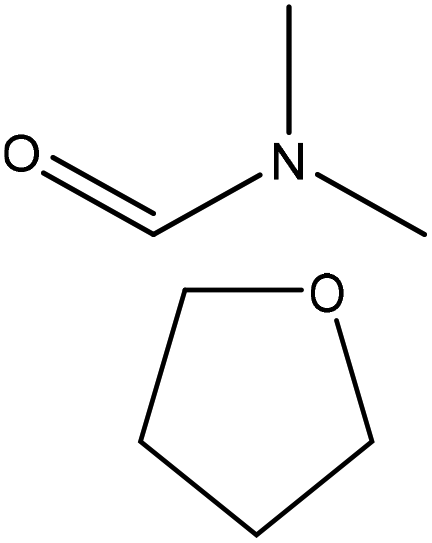
|
Pyridine | Soxhlet extraction using tetrahydrofuran | T g and melt torque reduced | 60–80 | Thermoplastic | 159 |
| Hardwood kraft | Overnight | 20–25 | Alkaline |
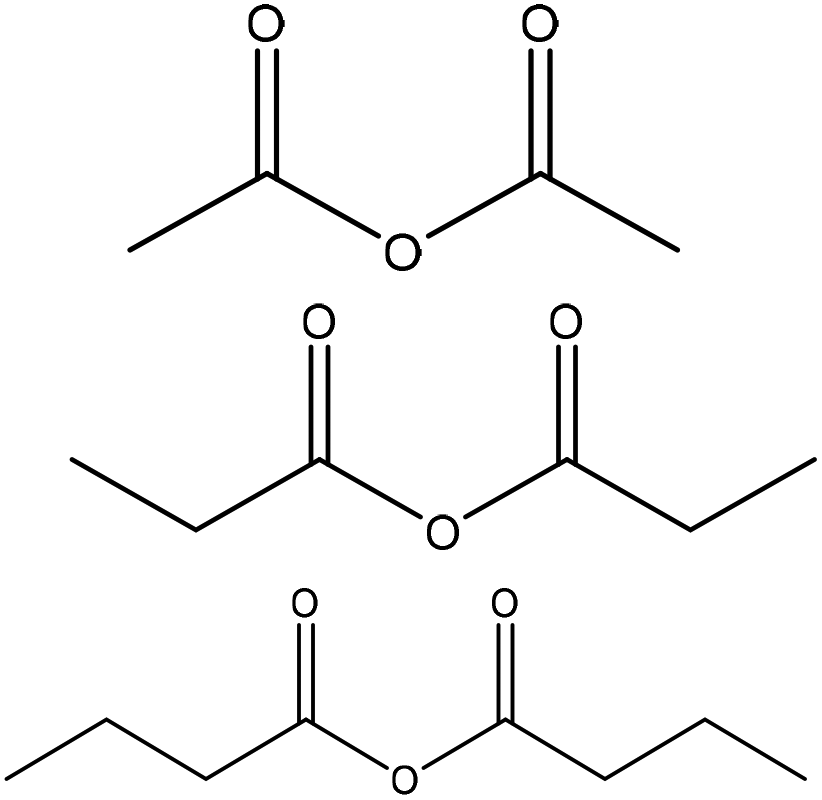
|
Water | Acetone and filtration | Compatibility of lignin | 86 | Polyethylene blends | 168 |
| Hardwood and softwood kraft | Overnight | 50 | N/A |
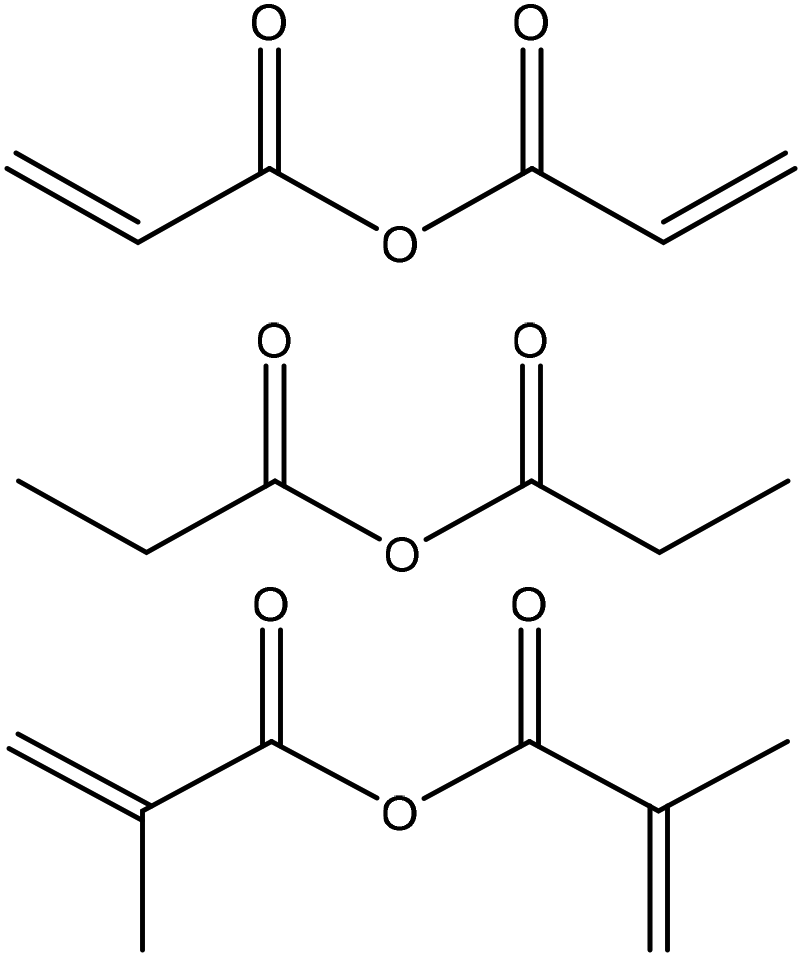
|
1-Methylimidazole/1,4-dioxane | Ethyl ether | Solubility in non-polar solv, molecular weight | N/A | Use in unsaturated thermosets | 169 |
| Enzymatic hydrolysis | 1.5 | 80 | N/A |
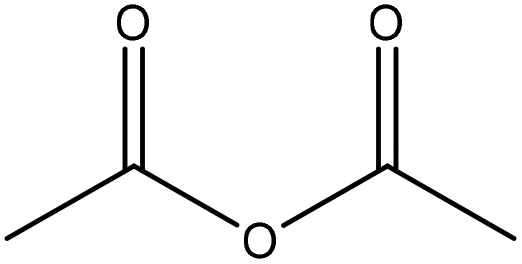
|
4-Dimethylamino pyridine, sodium acetate, and sulfuric acid | Ethanol | N/A | N/A | N/A | 155 |
| Kraft | 7 | 60 | N/A |
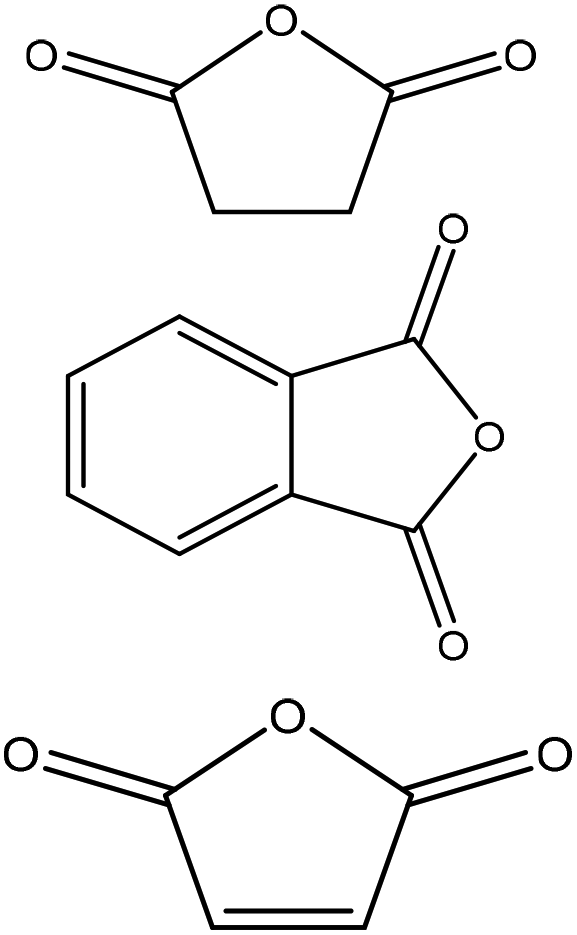
|
Acetone | Filtration, water or toluene | Hydrophobicity, molecular weight, thermal stability | N/A | Reinforced wood plastic composites | 175 |
| Bagasse | 4 | 90 | N/A |
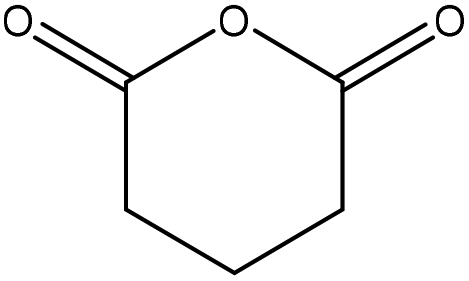
|
1-Allyl-3-methylimidazolium chloride | Ethanol | Surface adhesion compatibility | N/A | N/A | 132 |
| Sugarcane bagasse | 72 | 20–25 | N/A |
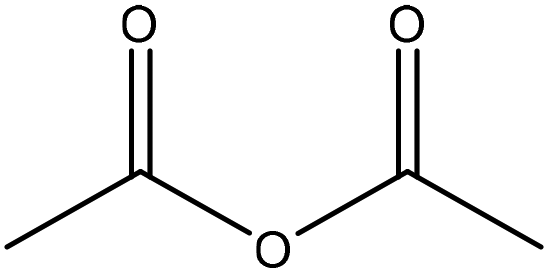
|
Pyridine | Filtration, water | Radical scavenging index decreased | N/A | Antioxidant | 61 |
| Synthetic | 48 | 30 | N/A |
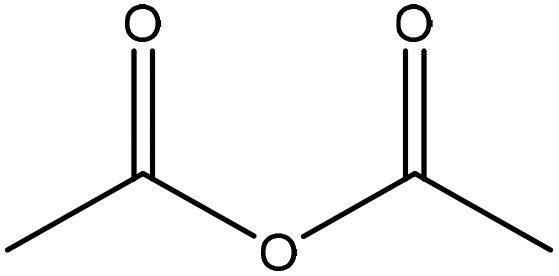
|
Pyridine | HCl, centrifugation | Secondary cinnamaldehydes prohibited | N/A | N/A | 176 |
| Kraft | 48 | N/A | N/A |
|
Pyridine | Methanol | Molecular weight decreased | N/A | Chromatographic eluent | 177 |
| Alkaline soda | 18 | 20–25 | N/A |
|
Pyridine | Ethanol, or HCl | Thermal stability decreased | N/A | In polyolefins | 157 |
| Organosolv | 48 | 25 | N/A |
|
Pyridine | Filtration | Solubility in tetrahydrofuran | N/A | Photosensitizers | 171 |
| Hardwood | N/A | N/A | N/A |
|
Dioxin | N/A | Photodiscoloration behavior under decreased | 77 | Studying photoyellowing properties | 162 |
| Organosolv | 3 | 60 | N/A |
|
1-Methylimidazole | n-Hexane | Viscosity decreased | N/A | Additive for plasticizing | 165 |
| Hardwood kraft | N/A | 50 | N/A |
|
1-Methylimidazole | Ethyl ether | Flexural strength, viscosity | N/A | Resin and flax fibers | 156 |
Esterification has been commonly used to reduce the hydrophilicity and solvophilicity of lignin, which makes it favorable to be used for composite productions.162–164 Lignin esterification by butylation, for instance, transforms the alcohol groups to butyl esters and improves lignin miscibility in low-polar solvents, which would facilitate its use in the construction industry.165–167 Esterifying lignin also enhances its compatibility with plastic blends such as polystyrene, polyethylene, and poly(3-hydroxybutyrate-co-3-hydroxy valerate) blends.160,161,168 In addition, esterified kraft lignin was reported to remarkably improve the interfacial tension between the resin and reinforcing flax fibers.169 Furthermore, lignin esterification increases the thermal mobility of lignin molecules by diminishing the intermolecular interaction, which further leads to a reduction in the glass transition temperature of lignin.170
The esterification was reported to improve the morphology of lignin-based materials. For example, carbon fibers made from phthalic anhydride-modified lignin were revealed to have reasonably high micro-scale porosity in comparison with carbon fibers made from unmodified lignin.158 It is also worth mentioning that the structural properties of carbon fibers produced from lignin depend on the reagent used in the esterification reaction. For instance, lignin with a cyclic anhydride such as succinic, maleic or phthalic may form di-esters, whereas lignin could only form a monoester with acetic anhydride. In addition, the esterification of lignin using phthalic anhydride would render lignin more hydrophobic. Using maleic anhydride, a reagent with a double bond in its structure may increase cross conjugation between lignin's structural units.
These reports also suggested that the esterified lignin could be a green alternative to replace petroleum-based fillers in thermoplastics159,161,169 as well as being a potential photosensitizer.171 Esterification has also promoted lignin's application as an oxygen and water-vapor barrier in the packaging.172 However, esterification was observed to reduce the antioxidant activity of lignin, which may be due to lowering its phenolic hydroxyl groups.61
Propargylation
Propargylation takes place by adding a propargyl group to a molecule. Lignin propargylation occurs via an SN2 mechanism, as shown in Fig. 1. This reaction occurs only on the phenolic hydroxy groups of lignin since these groups have a higher ionization efficiency compared to aliphatic ones.41,147Table 12 shows the propargylation reaction on lignin carried out in the previous studies. In propargylation, lignin is mixed with propargyl containing bromide and NaOH or KOH at 70–90 °C for 1–4 h in an alkaline environment.41,178 Then, the generated product is separated by acidifying the reaction mixture.41,178 Lignin propargylation increases the reactivity of lignin in a uniform and modulated way, thus increasing the potential use of the propargylated lignin in carbon fibers.147,178 According to the US Department of Energy, using propargylated lignin in carbon fibers would reduce the final price of carbon fibers by half.179 Similarly, propargylated lignin has been reported to be used in transportation applications such as tire production and composite production as the curable thermosetting resin.178 However, using propargyl bromide can be considered as the main problem of this modification because it is toxic and may cause environmental issues.
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Kraft | 1 | 90 | 13.7 |
|
Potassium hydroxide | HCl | Molecular weight | N/A | Carbon fiber, tires | 178 |
| Paper factory | 4 | 70 | Alkaline |
|
Water/ethanol | Filtration | Solubility, thermal properties | 98 | Resin for composite matrix | 180 |
| Softwood kraft | 2 | 75 | Alkaline |
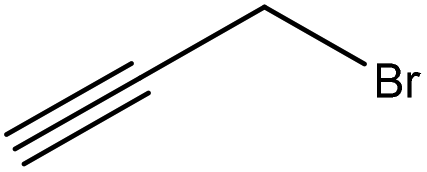
|
Water | Acidification | Thermal stability, molecular weight | 91–96 | To increase thermal stability | 41 |
| Methylated softwood kraft | 2 | 75 | Alkaline |
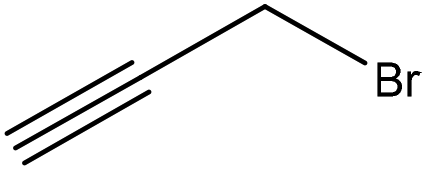
|
Water | Acidification | Molecular weight | N/A | Carbon fiber | 178 |
Methylation
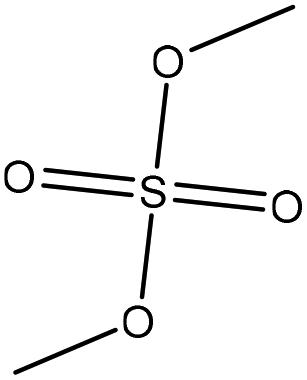
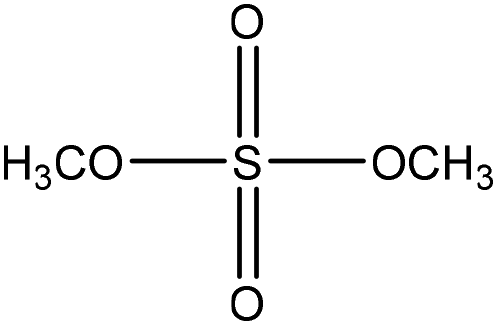
Methylation reaction is the addition of a methyl group (–CH3) to lignin. The methylation of lignin occurs by nucleophilic aromatic substitution (Fig. 9). Under alkaline conditions, the lone pair electron of aromatic hydroxide of lignin attacks the carbon of the methyl group. As a result, hydroxy groups are replaced with a methyl group in lignin. Methylation is considered as an alkylation in which a methyl group is exchanged with a hydrogen atom. This reaction had been used to cover the phenolic hydroxy groups to render lignin hydrophobic, or to analyze whether the desired reaction tends to occur on the phenolic or aliphatic hydroxy groups of lignin. The selective methylation of lignin's phenolic hydroxy groups converts these groups to phenyl methyl ether, which are remarkably less reactive compared to hydroxy groups. | ||
| Fig. 9 Methylation of lignin by dimethyl carbonate.181 | ||
In addition, a carefully controlled and monitored methylation of lignin may reduce lignin's reactivity, which could provide possibilities for its self-polymerization at high temperatures (above 130 °C).147 However, this radically initiated self-polymerization of lignin could be inhibited entirely by methylating the phenolic hydroxyl groups.181 In methylation, methyl groups replace only phenolic hydroxyl groups due to their remarkably higher (about 80 times) ionization efficiency.123,141
Table 13 shows the methylation reaction implemented on lignin. In the past, lignin was mixed with dimethyl sulfate, methyl iodide, diazomethane or tetramethylammonium hydroxide in an alkaline medium or it was dissolved in anhydrous N,N-dimethylformamide (DMF) for methylation. The reaction generally occurs at room temperature for 72 h or at 75–150 °C for 2–24 h.123,141,150,182,183 To collect lignin derivatives, the reaction mixture is acidified, if conducted under the alkaline conditions and purified via filtration, for instance.123,141 The methylation reduces the glass transition temperature of lignin since most of the intra-molecular hydrogen bonding becomes eliminated in this reaction.150
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Milled wood | 72 | 20–25 | Acidic |
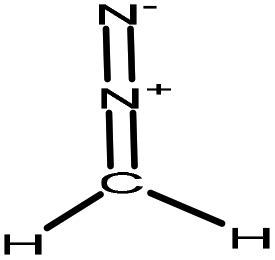
|
Methanol and hydrochloric acid | N/A | N/A | N/A | Mask phenolic hydroxide groups | 182 |
| Lignosulfonate | 2 | 80 | 11–11.5 |
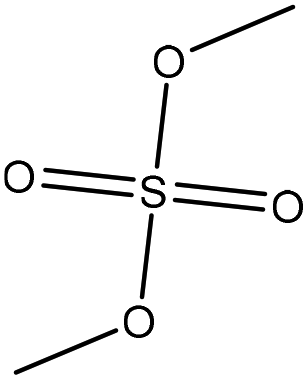
|
Water | HCl | Molecular weight | N/A | Mask phenolic hydroxide groups | 123 |
| Softwood and hardwood kraft | 2, 10 | 25, 80 | Alkaline |
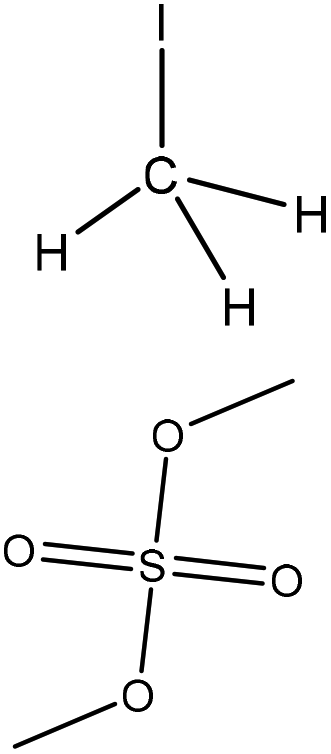
|
N,N-Dimethylformamide | HCl | Molecular weight | N/A | Mask phenolic hydroxide groups | 141 |
| Softwood kraft | 2–24 | 120 and 150 | Alkaline |
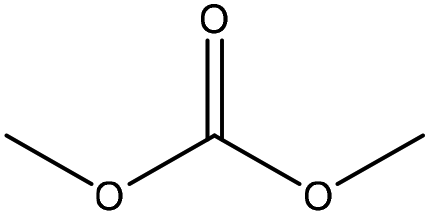
|
Dimethyl sulfoxide | HCl | T g decreased | N/A | N/A | 181 |
| Softwood kraft | 2 | 75 | Alkaline |
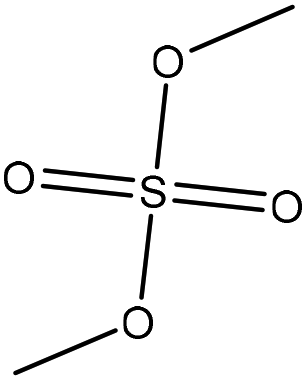
|
Water | HCl | Molecular weight, Tg | N/A | N/A | 150 |
| Softwood kraft | 2 | 75 | Alkaline |
|
Water | HCl | N/A | N/A | Carbon fiber | 178 |
Methylation was reported to enable the use of lignin in thermoplastics and carbon fibers.141,150 However, methyl iodide and dimethyl sulfate, the most common reagents used for methylation, are very toxic and hazardous, which is the major drawback of this process. One advantage of lignin methylation is that the by-products, methanol and carbon dioxide, could be recycled and reused in the production of dimethyl carbonate.181,184 Among other reagents, dimethyl carbonate has the supremacy of safe and straightforward handling since it is not mutagenic or hazardous. However, the chemical reactivity of dimethyl carbonate depends on the temperature in a way that at a temperature higher than 120 °C, it participates in methylation reaction via a base mediated alkyl cleavage nucleophilic substitution mechanism, and at a lower temperature (e.g., 90 °C), it can act as a carboxymethylating agent via a base mediated acyl cleavage nucleophilic substitution mechanism.181 Therefore, to use dimethyl carbonate as a methylating agent, the reaction temperature needs to be higher than 120 °C, and since this temperature is higher than the dimethyl carbonate's boiling point (90 °C), the reaction has to be carried out in a closed reactor and pressurized system.181
Alkylation
Alkylation introduces an alkyl group (–CnH2n+1) to a lignin macromolecule via the SN2 mechanism (Fig. 1). Lignin can be readily alkylated through nucleophilic substitution on its active aromatic hydroxy groups with an alkyl chain having different chain lengths.42 It has been reported that the alkylation reaction increases the thermal resistance of lignin while reducing its hydrophilicity.42,181,185 Alkylated lignin could be used as a plasticizer in polymer blends.186 It was also used in polypropylene composites along with synthetic polymers and it was reported to improve the composite's stiffness and storage moduli.181,185,187Table 14 shows the alkylation reaction of lignin. In general, bromododecane or lead acetate, as the alkylating agent, can be mixed with lignin at pH 8–12 at 25 or 80 °C for 24–72 h in isopropanol, water, and dioxane as a solvent.42,185 As a drawback, alkylated lignin could be quite brittle, which might not be favorable in polymer blends.188
| Lignin source/type | Reaction conditions | Separation | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||
| N/A: not available. | ||||||||
| Unbleached hardwood kraft | 24 | 80 | 8–10 |
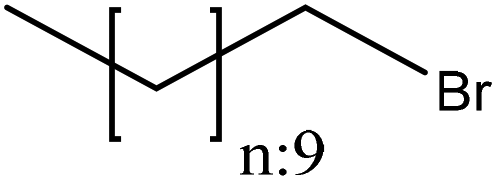
|
Isopropanol | Water | Retardant and toughening agent for polypropylene | 185 |
| Pulping industry | N/A | 50 | N/A |
|
Distilled water | Filtration, water | Surfactants | 42 |
| Kraft | 72 | 20–25 | 11–12 |
|
Dioxane | Water, centrifuged | Plasticizer | 186 |
Halogenation
Halogenation is a method used for introducing a halogen group into the lignin molecule. The halogenation of lignin occurs by electrophilic aromatic substitution. As an example of halogenation, Fig. 10 shows the bromination reaction of lignin with bromine in a hydrophilic (polar) protic solvent of acetic acid. Halogenation has been reported to occur on the aromatic ring of lignin since the aromatic hydroxy group could activate the adjacent positions for the electrophoretic attack.189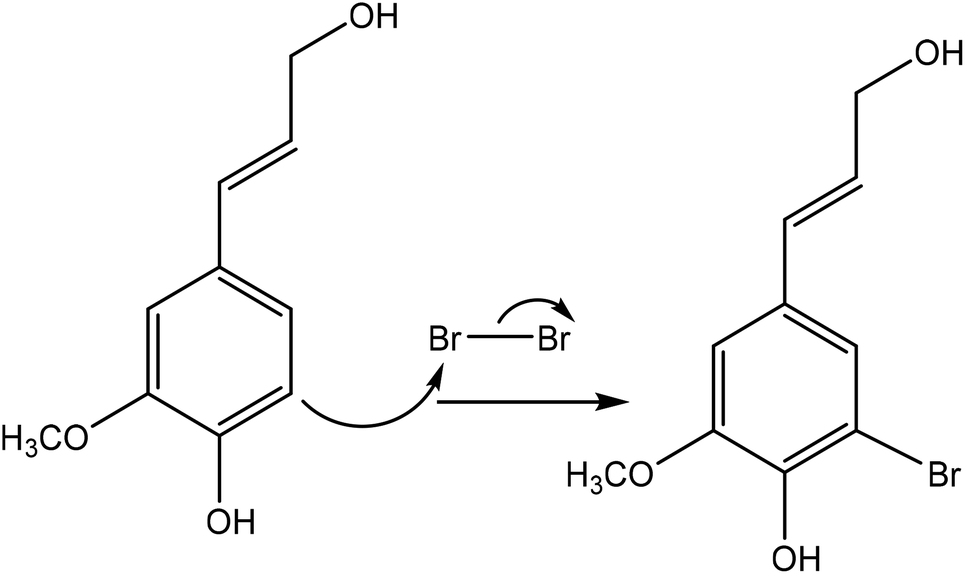 | ||
| Fig. 10 Bromination reaction of lignin by using bromine.191 | ||
Table 15 shows the halogenation reactions of lignin. Typically, lignin is mixed with a halogen (hydrogen bromide, N-bromosuccinimide, and an ionic liquid, liquid chlorine) in the temperature range of 20–164 °C for 1–2 h.189–191 Halogenated lignin can be precipitated using a mixture of diethyl ether and ethyl acetate.191
| Lignin source/type | Reaction conditions | Separation | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||
| N/A: not available. | ||||||||
| Hardwood and softwood | 2 | 163–164 | N/A | Br–Br | Glacial acetic acid | N/A | N/A | 191 |
| Hydrolysis | 1 | 20 | N/A | Br–Br | Carbon tetrachloride and water | Water | N/A | 190 |
| depolymerized | N/A | 20–25 | N/A |
|
1-Butyl-3-methylimidazolium bromide | Diethyl ether and ethyl acetate | Surfactant | 189 |
Bromination of lignin was reported to restrict lignin's agglomeration.192 Meanwhile, halogenated compounds are intensively used as fungicides, herbicides, insecticides, and precursors in the synthesis of pesticides. They could also be used as intermediates in the synthesis of dyes, agricultural chemicals and pharmaceuticals.193 As is well-known, the reagents used in halogenation have different levels of reactivity. For example, chlorine and halogen fluorine are the most aggressive reagents compared to bromine194 due to their high electrophilicity while bromine is a weaker reagent, and iodine is classified as the least reactive reagent of the halogens. It is advised that the chlorination reaction should be carried out with caution due to toxicity issues, which may require additional control. Furthermore, fluorination is mostly used in the production of fluorocarbons.194 Since halogenated organics are highly toxic, the halogenation process has serious disadvantages, which requires health caution.195
Amination
Amination is a simple method for introducing amine groups onto lignin. This reaction occurs through the SN2 route (Fig. 1). Generally, the amination reaction has been applied to lignin with the aim of generating cationic surfactants, slow-release fertilizers, flocculants, heavy metal adsorbents, coagulants, cationic asphalt emulsifiers, curing agents of epoxy resin, anion-exchange resins and retention aids.108,112,196–201 Lignin amination could enhance the surface activity, water solubility, charge density and molecular weight of lignin.69,202Logically, introducing nitrogen-containing groups into lignin can render lignin an efficient adsorbent for heavy metals. This phenomenon arises since the nitrogen-containing bases tend to chelate with acidic metallic ions.203,204
Table 16 shows the amination reactions of lignin. Generally, lignin is mixed with amination agents, such as diethylenetriamine, dimethylamine, methylamine, propane diamine, and triethylamine in water or a solvent, such as formaldehyde or dioxane, in the temperature range of 20–90 °C under both acidic and alkaline conditions for 1–17 h. Produced aminated lignin samples can be precipitated via mixing the reaction mixtures with hydrochloric acid, acetone, ethyl acetate or isopropanol.189 Although inducing amine groups on lignin through amination reaction is selective and straightforward, using the toxic, carcinogenic and mutagenic chemical of formaldehyde in most of the reactions can be unsafe and cause environmental problems.
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Epoxidated | 3 | 80 | Alkaline |
|
Formaldehyde | Filtration and water | N/A | N/A | Heavy metal adsorbent | 197 |
| Oxidized | 5 | 90 | 10 |
|
Formaldehyde | Filtration | N/A | N/A | Heavy metal adsorbent | 60 |
| Alkaline | 2–6 | 40–90 | 8–13 |
|
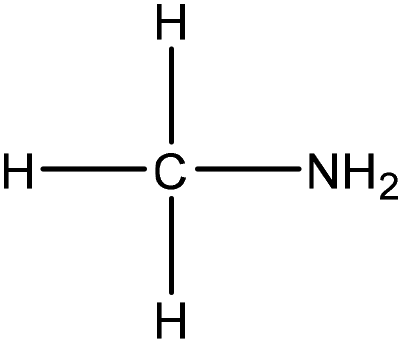
Water | HCl | Molecular weight, nitrogen content | N/A | Lead removal | 108 |
| LignoBoost | 4 | 60 | Acetic |
|
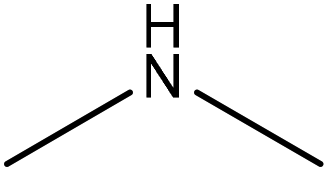
Dioxane, and formaldehyde | Membrane dialysis | Molecular weight, nitrogen content | N/A | Surfactant and slow-release fertilizers | 196 |
| Epoxidated | 4–6 | 80 | Alkaline | H2N–R–NH2 | Water | Acetone | Decomposition temperature decreased | N/A | Curing agents of epoxy resin | 112 |
| Carboxylated | 3 | 80 | Alkaline |
|
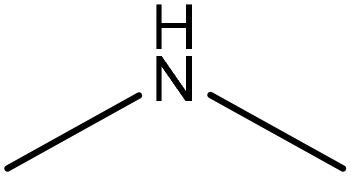
Water | Membrane dialysis | Removing both anionic and cationic dyes | N/A | Flocculant | 205 |
| Sulfuric acid treated | 4–48 | 50 | Acetic |
|
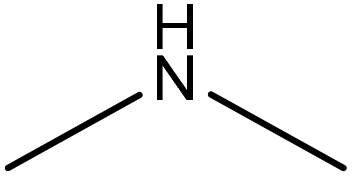
Formaldehyde, and dioxane | Ethyl acetate | Solubility | ∼0.6–63 | Retention aid | 199 |
| Kraft | 1 | 20–25 | N/A |
|
Formaldehyde | Filtration and isopropanol | N/A | 97 | Coagulant in wastewater | 198 |
| Enzymatic hydrolysis | 2 | 20 | 3 |
|
Formaldehyde, and acetone | Membrane dialysis | Efficiency in higher pH | N/A | Flocculant for anionic azo dyes | 201 |
| Sulfonated | 4 | 85 | 12 |
|
Water | Membrane dialysis | Hydrophilicity | N/A | Enhanced the enzymatic hydrolysis of lignocellulose | 206 |
| Lignosulfonate | 4 | 85 | 12 |
|
Water | Membrane dialysis | Fluidity | N/A | Clay-tolerance sacrificial agent | 207 |
| Softwood kraft | 1 | 70 | 12.5 |
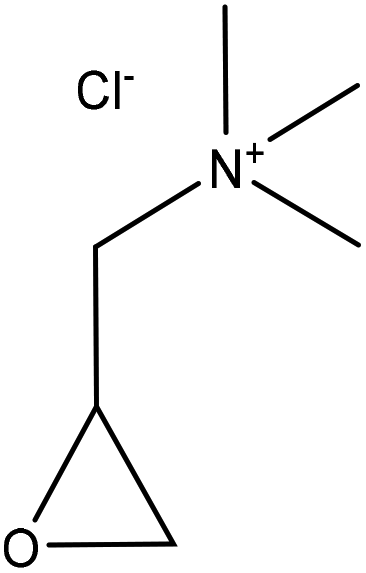
|
Water | Membrane dialysis | Solubility, charge density | N/A | Flocculant for dye removal | 208 |
| Hardwood organosolv, and enzymatic hydrolysis | 20 | 60 | Alkaline |
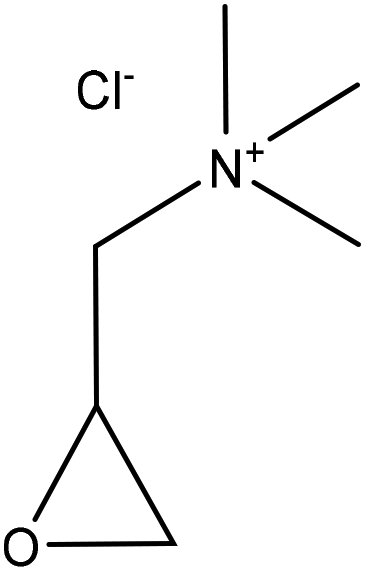
|
Water | Dialyzed | Solubility | N/A | Removal of sulfate, kaolin, and humic acid | 209 |
| Sulfuric acid treated | 4–72 | 60 | N/A |
|
Formaldehyde | Membrane dialysis | Brightness of the paper | 78–90 | Retention aid in papermaking | 200 |
Oxidation
Recently, the oxidation of lignin has become popular because of the extensive need for replacing fossil fuel feedstock with sustainable materials to produce fuels and chemicals. Oxidation occurs through the electron loss of a molecule. In lignin oxidation, the cleavage of C–O bonds occurs leading to the generation of carboxylic acids and aromatic aldehydes.210 The type of catalyst used in the oxidation reaction is responsible for its low molecular weight reduction or phenolic product generation.210–212It is worth mentioning that the reaction pH plays a critical role in lignin oxidation. In one study,212 hydrogen peroxide was used to oxidize lignin under both acidic and alkaline conditions. As a result of the reaction under acidic conditions, formic acid and acetic acid were produced as the main components, while no aromatic acids, aldehydes, chromophoric groups, and phenolic components was detected.212 However, in an alkaline environment, lignin with a high amount of carboxylic acid was produced along with oxalic, formic, acetic, malonic, and succinic, as well as vanillin, syringaldehyde, and chromophoric groups. These chromophoric groups also undergo a ring cleavage reaction and further degradation to form low molecular weight acids. Interestingly, reactions conducted under strong alkaline conditions proceed at low temperatures of 80–90 °C, while those under acidic conditions need higher temperatures of 130–160 °C.212 That being said, most of the oxidation reactions have been performed in an alkaline environment since it helps solubilize lignin and accelerates the deprotonation of hydroxy groups.211,212
| Lignin source/type | Reaction conditions | Separation | Remark | Yield (%) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Catalyst | Oxygen pressure atm | Solvent | |||||
| N/A: not available. | ||||||||||
| Organosolv beech wood | 1.5–3 | N/A | N/A |
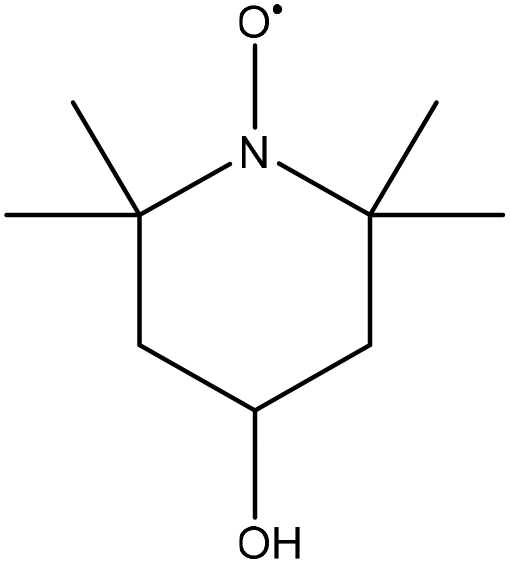
|
1 | Water | Ethyl acetate | Formation of quinones and phenol derivatives | N/A | 231 |
| Precipitated hardwood | 0.08–0.016 | 95–160 | Acidic | HO–OH | N/A | H2SO4 | H2SO4 | Formic acid and acetic acid | 41 | 212 |
| Alkali | 0.15–2 | 20 | N/A |
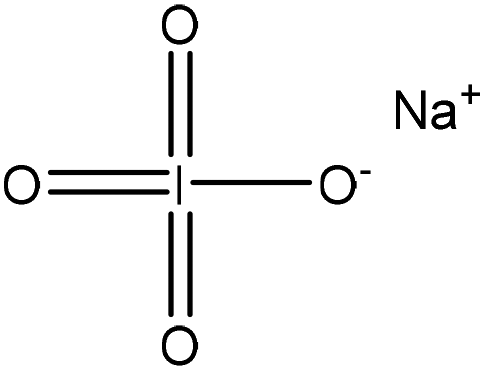
|
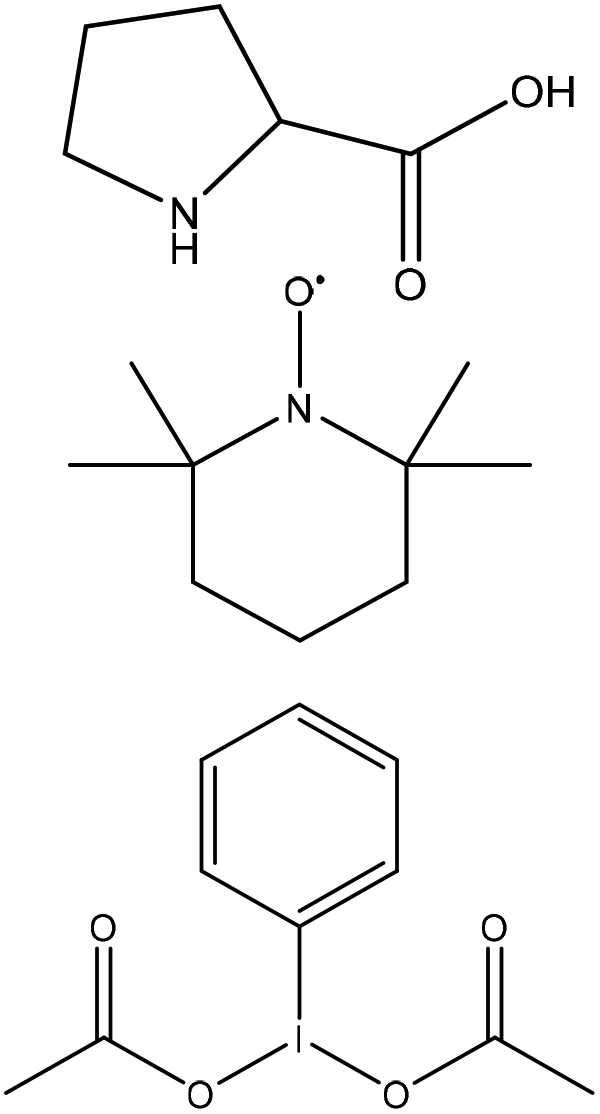
N/A | Acetic acid | N/A | N/A | N/A | 229 |
| Organosolv beech wood | 5 | 20–25 | N/A |
|
N/A | Acetonitrile-d3 | Filtration | One-pot two-step reaction | N/A | 231 |
| Eucalyptus black liquor | 2 | 150–190 | Alkaline | Cu2+, Co2+ | 10–15 | Water | N/A | Phenolic compound production | 3.5 | 211 |
| Aspen | 12–48 | 110 | Acidic |
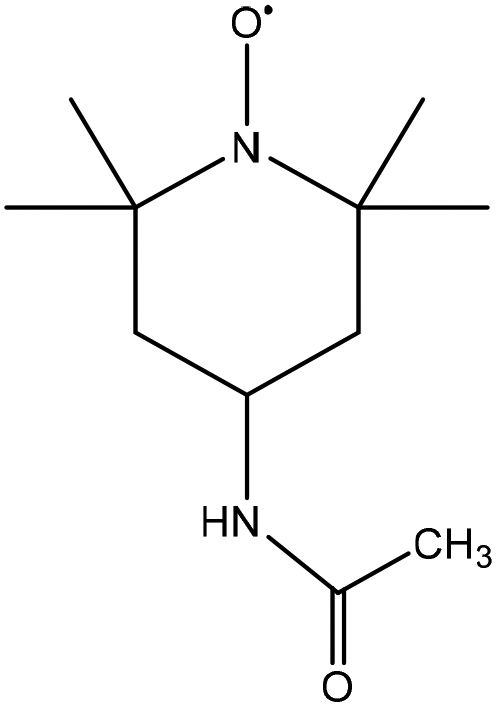
|
2 | HNO3, CH3CN, HCl, water | Evaporation | Two-step reaction | N/A | 232 |
| Curaua fibers and sugar cane bagasse | 0.5 | 55 | N/A | O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ClO ClO |
N/A | CH3COOH | Centrifuging and water | Lower molecular weight products. Elimination of quinones | N/A | 233 |
| Kraft | 1 | 450 | N/A | TiO2, Ta2O5–IrO2 | N/A | Ethanol–isopropanol | N/A | Lower molecular weight products | N/A | 223 |
| Softwood and hardwood | 0.5–12 | 100 | N/A | Polyoxometalate, and HO–OH | N/A | Water | N/A | Vanillin and syringaldehyde | 5 | 7 |
| Softwood and hardwood kraft | 0.08–4 | 90–110 | Alkaline | O2 | ∼5.9–8.8 | Water | HCl | Lower molecular weight products | N/A | 220 |
| Softwood kraft | 0.5 | 95 | Alkaline | Fe3+ | ∼9.8 | Water containing Fe3+ | H2SO4 | Lower molecular weight products and vanillin | 20 | 219 |
| Organosolv | 40 | 135 | N/A | (HTc) and V(acac)3/Cu(NO3)2·3H2O | 9.8 | Pyridine | N/A | Veratraldehyde and veratric acid | 22–30 | 234 |
| Softwood kraft | N/A | 150 | 14 | O2 | ∼9.8 | Water | N/A | Vanillin | ∼3.5–8 | 235 |
| Softwood | N/A | 160 | 13 | CuSO4 | ∼1.9 | Water | N/A | Vanillin | N/A | 217 |
| Poplar, maple, and maize | 28 | 65 | Acetic |
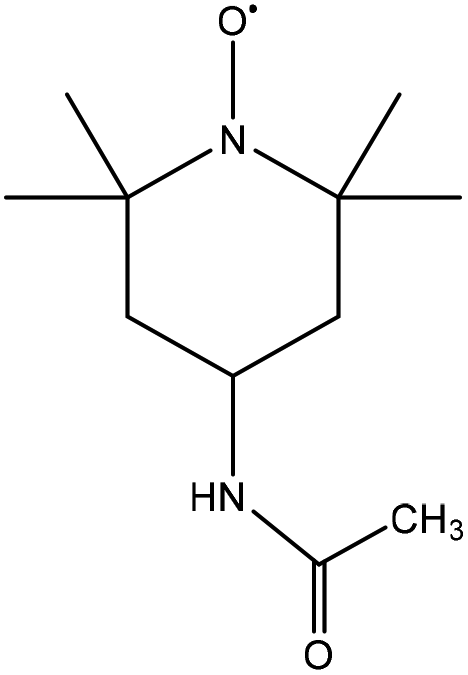
|
2 | HNO3, CH3CN, HCl | Evaporation | Low-molecular-weight aromatics | N/A | 236 |
| Hydrolyzed | N/A | 80 | Acidic | HO–OH | N/A | Sulfuric acid | Filtration and acidification | Forming soluble fraction of oxidized hydrolyzed lignin | 22–78 | 237 |
| Sugar-cane | N/A | ∼99.8–139.8 | N/A | PdCl2, and γ-alumina | ∼1.9–9.8 | Water | N/A | Aromatic aldehyde | 12 | 221 |
| Soda | 10–120 | N/A | Acidic | HO–OH | N/A | Water | Filtration | More carboxylic groups rather than ketones or aldehydes | N/A | 238 |
Lignin oxidation by the aliphatic and aromatic hydroxy groups of lignin leads to the generation of ketones, quinones, aldehydes, vanillin (shown in Table 17).7,222,223 Also, some of the most advanced oxidative routes are used in pulp and paper industries to depolymerize or remove lignin from cellulosic materials.224,225
Based on the literature reports,226,227 a correlation could be found between the resulting product and the oxidative breaking of specific linkage. Fig. 11 depicts this correlation under aerobic oxidation conditions. As depicted, the cleavage of the Cα–Cβ bond forms phenolic aldehydes, while Cph–Cα bond cleavage leads to the generation of para-quinones and oxirane structures. The cleavage of the lignin aromatic ring also yields the production of muconic acid derivatives.226,227
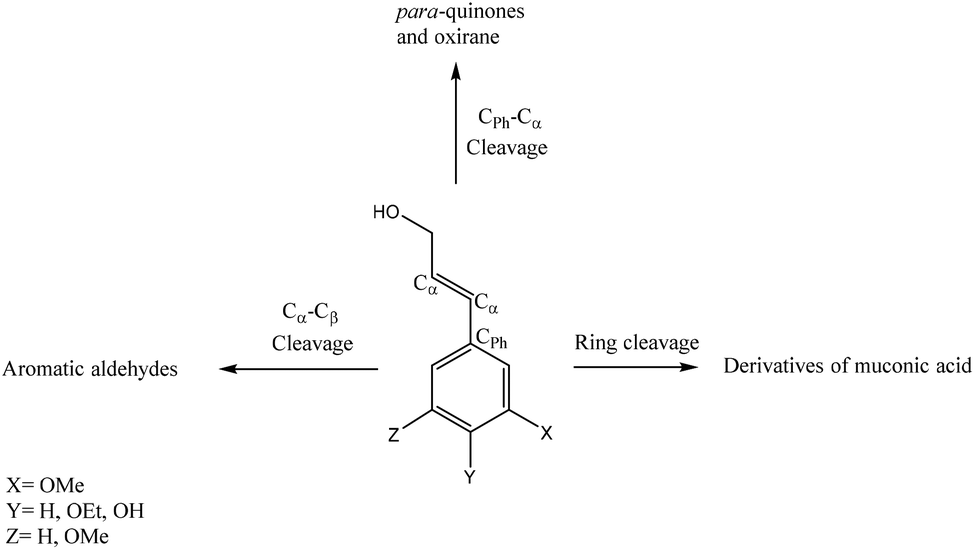 | ||
| Fig. 11 Potential bond cleavage pathways in lignin aerobic oxidation.226,227 | ||
Meanwhile, aromatic aldehydes, such as vanillin, could be the main product of lignin oxidation.228 Vanillin is the only mercantile product achieved from lignin through oxidation with a market volume of around 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes per year. Nonetheless, a majority (90%) of the synthetic vanillin used today is oil-based implying a need to improve and develop the lignin-based vanillin production, which is closer in flavor/taste to natural vanilla than vanillin produced from petrochemical guaiacol.8 However, these oxidation pathways are accompanied by some disadvantages, such as long reaction times and use of toxic catalysts, such as sodium periodate or palladium chloride.229,230
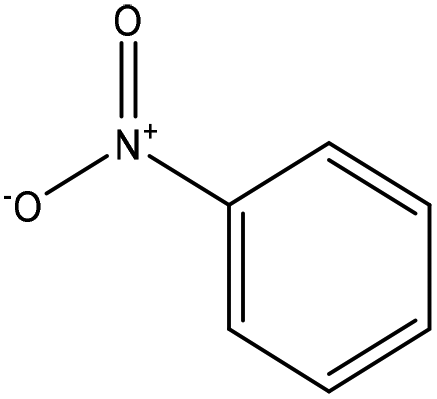
000 tonnes per year. Nonetheless, a majority (90%) of the synthetic vanillin used today is oil-based implying a need to improve and develop the lignin-based vanillin production, which is closer in flavor/taste to natural vanilla than vanillin produced from petrochemical guaiacol.8 However, these oxidation pathways are accompanied by some disadvantages, such as long reaction times and use of toxic catalysts, such as sodium periodate or palladium chloride.229,230
Hydrogen peroxide is widely available in pulp mills and is extensively used for bleaching pulp worldwide. As an oxidant, it can also be used for oxidizing lignin to introduce carboxylate groups. It is reported that hydrogen peroxide generally decomposes the phenolate group of lignin, whereas it induces the carboxylate group into lignin.42,240,241 It was reported that the majority of hydrogen peroxide molecules was utilized for the partial decomposition of the lignin structure by the bond cleavage of lignin's ether bonds.240
Fig. 12 shows the mild oxidation by using different sources of lignin and oxidizing reagents. Lignin undergoes two different reaction sets in lignin oxidation with hydrogen peroxide; perhydroxyl anions attack nucleophilically while removing lignin chromophores. Meanwhile, free radical species generated by the decomposition of hydrogen peroxide yield oxidative degradation of the phenolic structures of lignin and converts them to carboxylic acid groups.242 The perhydroxyl anion cleaves the side chains of lignin, opens the benzene ring, and produces new compounds, which have carboxylate or chromophore groups.212,242,243 These groups may undergo the ring cleavage reaction under severe reaction conditions and further degrade to form different low molecular weight compounds, such as oxalic acid, formic acid, and malonic acid.212
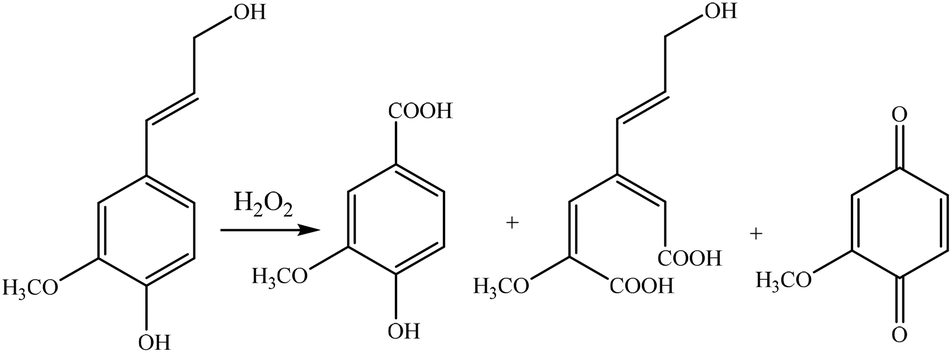 | ||
| Fig. 12 Mild oxidation of lignin by hydrogen peroxide under alkaline conditions.240 | ||
The oxidation of lignin via hydrogen peroxide would promote the solubility and increase the charge density of lignin, and therefore it could be used as an anionic dispersant for kaolin and other suspensions.240 Employing nitrobenzene to oxidize lignin generates aromatic aldehydes as main products; however, using nitrobenzene have some disadvantages, such as difficulties in recovering the oxidant as well as the complexity of the reaction raised from the formation of phenylhydroxylamine, aniline, and nitrobenzene products, leading to a condensation among them. Also, the respective carboxylic acids produced in mild oxidation have lower yields than in harsh oxidation.218
Oxygen, Cu(II), Co(II), and CuO have also been used to oxidize lignin (Table 18). Protolignin oxidation with CuO has claimed to have a lower yield compared with nitrobenzene as the oxidant.218,244,245 Co(II) is a better oxidant than nitrobenzene, as the oxidant recovery is easier and harmful byproducts are not produced in the reaction.246 Although oxygen (or air) as a catalyst will not contribute significantly to the oxidation cost, it has lower selectivity and produces low molecular weight acids.
| Lignin source/type | Reaction conditions | Separation | Remark | Yield (%) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Catalyst | Oxygen pressure atm | Solvent | |||||
| N/A: not available. | ||||||||||
| Proto | 2.5 | 170–180 | Alkaline |
|
N/A | Water | N/A | Product ratio depends on the material source | Syringaldehyde 36%, vanillin 15% | 244 |
| Alkali | Syringaldehyde 25%, vanillin 10% | |||||||||
| Hardwood | N/A | 80–160 | Alkaline | HO–OH | N/A | Water | N/A | Lower molecular weight products | 30–50 | 212 |
| Calcium lignosulfonate | 1.5 | 80 | 9 | HO–OH | N/A | Water | N/A | Foam height and half-life decreased | N/A | 57 |
| Eucalyptus black liquor | 2–2.30 | 150 | Alkaline | Cu(II), and Co(II) | 10–15 | Water | N/A | Lower phenolic aldehyde compared to other lignin oxidants CuO and nitrobenzene | 4 | 211 |
| Eucalyptus black liquor | 2–4 | 170–190 | Alkaline |
|
N/A | Water | N/A | Nitrobenzene is more effective than copper(II) oxide | 15–18% by using nitrobenzene, 7–8% by using CuO | 218 |
| Alkali | 2 | 180 | Alkaline |
|
N/A | Water | Filtration | Product ratio depends on the material source | Vanillin 17% | 248 |
| Thio | Vanillin 6.6% | |||||||||
| Softwood kraft | 1 | 78–110 | Alkaline | MgSO4 | 2.72 | Water | Water | Increase in tensile strength | 43–50 | 249 |
| Proto | 4 | 160 | Alkaline |
|
N/A | Water | N/A | N/A | Vanillin 4% | 245 |
| Alkali | Vanillin 3% | |||||||||
| Sugar cane, red spruce kraft, and hardwood organosolvent | N/A | 20–25 | N/A |
|
N/A | Acetic acid | Filtration | Increase in carboxylic acid content | 98 | 243 |
| Softwood kraft | 1–3 | 60–100 | Alkaline | HO–OH | N/A | Water | Membrane | Decrease in molecular weight, increase in charge density and degree of the carboxylate group. Dispersant for kaolin suspensions | N/A | 240 |
| Proto | 3 | 160 | Alkaline |
|
N/A | Water | Filtration | Half of lignin combined through C–C linkages | Syringaldehyde 13%, vanillin 8.5% | 250 |
| Native | 2 | 180 | Alkaline |
|
8.9 | Water | Filtration | N/A | Vanillin 25% | 251 |
| Alkali | Vanillin 14% | |||||||||
Generally, although the mild oxidation pathways could be affordable, they are not effective in significantly altering the hydrophobicity of lignin.239 Therefore, the lignin-based materials produced in some pulping and biorefining processes may need a stronger oxidizing agent.42,247
Silylation
Silica can be introduced on both phenolic and aliphatic hydroxy groups of lignin252 under an SN2 reaction mechanism (Fig. 1). Silica and derivatives of silane have been used as flame retardants. Dissimilar to carbon-based polymers, silicas generate inorganic silica under combustion. Silica residues could hamper fire deployment by restraining mass and heat transfer. Basically, no toxic emissions are produced from the combustion of silicas.253,254 Hence, silica foams are highly favorable to be used in applications, such as construction building (e.g., acoustic designs). Lignin–silica products have been used as gaskets, sealing materials, metal and organic adsorbents, as well as adsorbents for heavy metal ions and dyes.43,252,255 They have also been reported as effective electrochemical sensors, polymer fillers,252 and biosorbents for removing toxic substances from aqueous solutions.42,252,255Table 19 shows the grafting of various silica on lignin. This reaction is generally conducted by introducing reagents including tetraethyl orthosilicate, silicon dioxide, sodium metasilicate, and tetraethoxysilane into lignin in a solvent (pyridine, ethanol, water, and dioxane) environment at 25–35 °C for 1.5 to 24 h. Although the lignin–silica reaction is popular in producing composites, these reactions are associated with drawbacks. For example, the two-step method used to produce lignin/silica composites intensely agglomerates composite particles. Also, the consumption of surfactants and various costly coupling agents in the preparation process of lignin/silica composites makes this process complicated and costly. Thus, there is a need for the expansion of a one-pot method for the lignin/silica composites’ preparation, mainly in aqueous media.256
| Lignin source/type | Reaction conditions | Separation | Property improvement | Yield (%) | Application | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time (h) | Temperature (°C) | pH | Reagent | Solvent | ||||||
| N/A: not available. | ||||||||||
| Alkali | 24 | 20–25 | N/A |
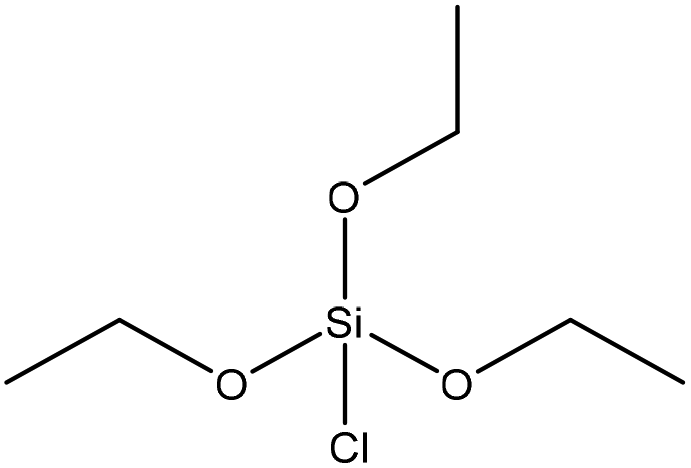
|
Pyridine | Pyridine | Thermal stability, surface area | N/A | Wastewater treatment | 43 |
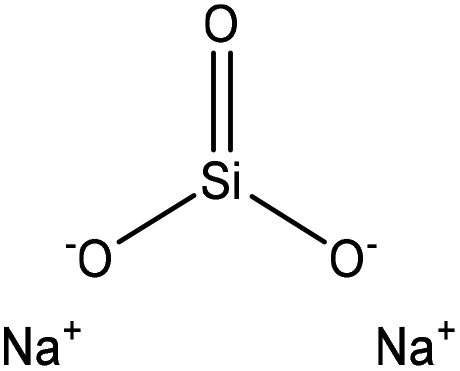
| Magnesium lignosulfonate | 1.5 | N/A | N/A | OSiO |
Water | Vacuum evaporator | Thermal stability | N/A | Polymer filler, and metal adsorbent | 252 |
| Quaternized alkali | 4 | 35 | Alkaline |
|
Ethanol and water | Water | UV-absorption ability, mechanical properties | N/A | Blend with polyurethane films | 256 |
| Ultrafiltered LignoBoost kraft | 24 | N/A | Acetic |
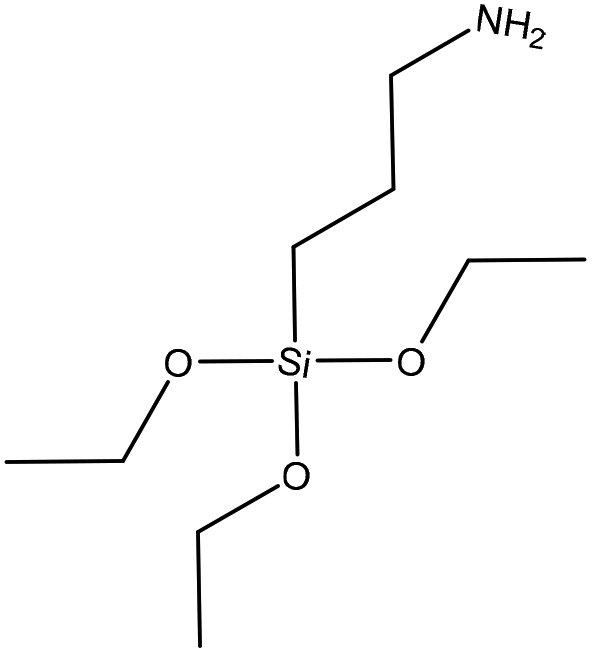
|
Dioxane and water | Ethanol and water | Thermal stability surface area | N/A | Sorbents for organic molecules | 255 |
Modified lignin's characteristics
Hydrophilic lignin
Lignin is known for its complexity due to having hydrophilic groups attached to its hydrophobic rings. Chemical modifications (e.g., carboxyalkylation and sulfoalkylation) would render it more hydrophilic.40,100,175 The applications for hydrophilic lignin products can expand extensively to an adsorbent, stabilizer, and dispersant for emulsions, clay suspensions, coal/water slurry, dye suspensions, and cement admixtures. However, it is mostly favored to be used as a dispersant or flocculant for emulsion and suspension systems.Various reactions render lignin anionic by introducing a negative charge into its backbone, of which carboxyalkylation, sulfoalkylation, and oxidation are the most common ones. The reaction temperature preferred for performing carboxymethylation, carboxyethylation, and sulfobutylation is mostly below 90 °C, but sulfomethylation is generally carried out at higher temperature (Tables 5–8). In addition, sulfobutylation has mostly been performed in a solvent-free environment, which is highly favorable.
Comparing the reaction routes stated above, while both carboxyalkylation and sulfomethylation are conducted in alkaline media, sulfomethylation seems to be less favorable due to several reasons: (1) an oxidation reaction is suggested to perform prior to sulfomethylation to increase the reaction yield, which is unfavorable since it has a dramatic impact on the performance of the sulfomethylation reaction; (2) the use of formaldehyde in sulfomethylation is a major downside due to its toxicity; and (3) the sulfomethylation reaction is a slow reaction that occurs at high temperature.
Carboxyethylation and sulfobutylation have more advantages than carboxymethylation and sulfomethylation, since they grant lignin with a higher anionic charge density stemming from the occurrence of the reaction on both aromatic and aliphatic hydroxy groups of lignin.110,122,123
Cationic lignin is produced by the addition of a positively charged group to its backbone. While lignin cationization has not been carried out as extensive as the anionization in the past, producing cationic lignin through amination is a well-known method of cationization, which further fosters its potential use in various applications, e.g., flocculant, adsorbent, surfactant.108,196–198
Hydrophobic lignin
Hydrophobic lignin and its derivatives have a strong interaction with other materials in organic solvents,12 and are almost insoluble in water. Reactions improving the hydrophobicity of lignin include esterification, alkylation, methylation, epoxidation, propylation, and oxypropylation, as they introduce hydrophobic groups into lignin's structure.In comparing these reaction routes, while all are mostly conducted under alkaline conditions, alkylation and epoxidation benefit from a relatively lower reaction temperature (below 90 °C). On the other hand, a broad and high reaction temperature range in oxypropylation (40–285 °C) and prolonged reaction time (72 h) in alkylation, as well as in methylation (if conducted at room temperature) might be the drawbacks of some of these modification pathways. In addition, oxypropylation could render lignin more hydrophobic than esterification,12 which is favorable for some applications, such as foams and composites.
Although the abovementioned reactions improve the hydrophobicity of lignin, each endows lignin with different features; the thermal resistance of lignin was reported to be improved via alkylation, and thus it promotes the application of alkylated lignin as a plasticizer in polymer blends.42,181 On the other hand, epoxy lignin was reported to have antibacterial activity,61 while esterified and methylated lignin were both reported to have a lower glass transition temperature than untreated lignin. Therefore, these reactions make products suitable for thermoplastics, plastic blends and carbon fibers.150,158 However, none of the mentioned reactions are environmentally friendly, as reagents in these reactions are mostly toxic and carcinogenic.
Applications for modified lignin
Lignin is a polymer with tremendous potential for its use in various industries. Table 20 shows the reactions conducted on lignin for specific applications, as well as drawbacks of lignin properties for the desired applications, which were improved through modification reactions. As seen, various reactions have been carried out on lignin to improve its charge density and solubility/hydrophilicity to be used as a dispersant, such as sulfomethylation for coal–water slurry and concrete admixture, sulfobutylation for coal–water slurry and carbendazim, carboxymethylation for oil–water emulsions, crude bitumen emulsion, clay, cement, and graphite suspensions, halogenation for surfactant, animation of cationic surfactants, and cationic asphalt emulsifier productions.| Application | Shortcomings of lignin | Reaction | Ref. |
|---|---|---|---|
| Dispersant | Charge density | Sulfomethylation | 20, 21, 57, 100, 101, 104 and 107 |
| Solubility/wettability | Sulfonation | 95 | |
| Sulfobutylation | 109, 110 and 111 | ||
| Carboxymethylation | 40, 116 and 120–122 | ||
| Carboxyethylation | 123 | ||
| Oxidation | 240 | ||
| Esterification | 173 | ||
| Oxyalkylation | 149 | ||
| Surfactant | Hydrophilicity/phobicity | Halogenation | 189 |
| Molecular weight | Sulfonation | 92 | |
| Alkylation | 42 | ||
| Amination | 196 | ||
| Plasticizer | Charge density | Esterification | 159 and 175 |
| Hydrophilicity | Sulfonation | 90 and 91 | |
| Oxyalkylation | 150 | ||
| Esterification | 159 | ||
| Alkylation | 186 | ||
| Flame-retardant | Thermal and oxidative stability | Phosphorylation | 38, 39, 45, 47, 48, 53 and 139 |
| Lignin–silica | 256 | ||
| Additive in polyurethane | Tensile strength | Phosphorylation | 47 |
| Glass-transition temperature | Phenolation | 77 | |
| Molecular weight | Oxypropylation | 136–140, 142–144 and 148 | |
| Viscosity | |||
| Adsorbent of metal ions in wastewater treatment | Selectivity | Phosphorylation | 49 and 51 |
| Thermal stability | Lignin–silica | 252 | |
| Sulfomethylation | 60 | ||
| Amination | 60 and 197 | ||
| Flocculant/coagulant | Molecular weight | Sulfomethylation | 105 |
| Charge density | Amination | 198, 201, 205, 208 and 209 | |
| Epoxy | Thermal stability | Phosphorylation | 46 |
| Molecular weight | Epoxidation | 112 and 125 | |
| Amination | 112 | ||
| Adhesive | Viscosity | Hydroxymethylation | 54, 66 and 68–72 |
| Molecular weight | Sulfonation | 88 and 93 | |
| Anti-oxidant and anti-bacterial | High radical-scavenging activity | Epoxidation | 61 |
| Sulfonation | 96 | ||
| Resin | Thermal stability | Phenolation | 78, 80 and 83 |
| Cross-linking |
Phosphorylation, hydroxymethylation, and oxypropylation reactions make modified lignin a good alternative for oil-based polyols used in polyester and polyurethane productions through improving lignin properties, such as tensile strength, molecular weight, viscosity, and glass-transition temperature. In addition, phosphorylation, carboxyethylation, amination, silylation, and sulfomethylation make lignin a polyelectrolyte with applications in aqueous systems by enhancing its selectivity and thermal stability.
Phosphorylation, epoxidation, and amination (curing agents of epoxy resin) increase lignin's molecular weight and thermal stability, which further promote lignin's application in the epoxy resin industry. Epoxy resins possess a wide range of applications, such as flooring, electronic laminates, industrial coatings and adhesives and high-performance composites. However, a slow curing rate, limited water solubility, and brittleness are the negative aspects of lignin-based epoxy resins. Lignin-based adhesives could be produced by hydroxymethylation and epoxidation. They could also be applied to mimic lignin antioxidant and anti-bacterial properties.
Miscibility of the polymers is a critical factor in applications such as polymer blends. Although two polymers’ miscibility is not favorable entropically, it could be improved if the polymers involved in blending have intermolecular interactions since the negative enthalpy of mixing overcomes the opposite entropy.257 Using lignin in polymer blends develops a convenient and powerful pathway to produce novel and functional green materials. It should be stated that polar synthetic polymers generate an intermolecular hydrogen bonding with lignin, while non-polar ones generally generate immiscible blends, showing distinct Tg points for two immiscible phases. In the past, lignin had been used in blending with synthetic polymers.181 Lignin's phenolic hydroxy groups tend to contribute more in forming hydrogen bonds with other polymers in a blend than its aliphatic ones due to the higher acidity of lignin's phenolic hydroxy group.257 Previously, alkylation, acetylation, methylation, and esterification of lignin have been carried out to increase the compatibility of lignin with various synthetic polymers in polymer blend applications.
Lignin alkylation and acetylation have reported modulating its chemical and thermal reactivities, which further leads to a thermal improvement in a polymer blend. Comparing alkylated and acetylated lignin with the unmodified lignin, it is found that the thermal stability of the polymer blends was improved when modified lignins were used in blends’ composition.185
Lignin methylation was also observed to impact the thermal stability of the lignin/polyethylene blend, diminishing the degradation temperature of the polyethylene remarkably. Nevertheless, since the lignin's phenolic hydroxyl groups become entirely masked through methylation, the tendency to form intermolecular hydrogen bonding in polymer blends can be reduced significantly.257
Lignin esterification was also performed to increase lignin compatibility in polymer blends.163 On the other hand, the esterified lignin's miscibility strongly depends on the carbon numbers presented in the side chain of the ester in that the miscibility improves with an increase in the ester groups’ chain length. However, the esterified lignin's interaction in a polymer blend might be similar to the methylated lignin's interaction due to the masking of the phenolic hydroxy groups via esterification, hindering the hydrogen bonding development.
Current and future trends
Lignin has been used as fuel. The price of lignin as fuel varies between 70–150 USD per ton, which is dependent on its moisture content and contaminants.258 Two technologies have been developed to generate lignin in dried powder forms. Lignoboost™ that has been implemented at Stora Enso, Sweden, with the production capacity of 50000 ton per year, and Domtar, NC, USA with the production capacity of 25000 ton per year. Lignoforce™ has also been commercialized at West Fraser Inc, AB, Canada, with the production capacity of 30 t per day. The production of lignin in such quantities will undeniably pave the way to generate lignin derivatives at commercial scales. Table 21 shows the addressable markets of lignin-based products. This table also comprises the estimated market values and capacities as well as the price of the lignin or fossil-based products. As seen, there are products, such as vanillin and phenol, which have been commercially available since 1933 and 2015, respectively, with lower or equal prices to their fossil-based counterparts. Lignin-based carbon fiber is also estimated to be produced commercially in 2020–2025.
| Industrial products from lignin | Est. total market value per year (billion dollar) | Est. scale produced per year (lignin-based) (MTon) | Est. lignin-based price ($ per kg) | Est. fossil-based price ($ per kg) | Ref. |
|---|---|---|---|---|---|
| N/A: not available. | |||||
| Carbon fiber | 4.5 | N/A | 6.5–12 | 17–26 | 261 and 262 |
| Phenol | 12–15 | 8 | 1–1.5 | N/A | 261–263 |
| Vanillin | 0.2 | 0.016 | 12 | 12 | 261 and 262 |
While the utilization of lignin might seem limited with current technologies, it is anticipated that lignin would be even more available in future due to the production of lignin in commercial processes, such as LignoForce and LignoBoost,259,260 which can pave the way for its further valorization. Although various modifications have been performed on lignin, there are still some unexplored reactions that could further improve the properties of lignin for different applications. For example, in carboxyalkylation, there is room for carboxypropylation and carboxybutylation of lignin to tune its charge density and hydrophilicity/hydrophobicity. Although sulfomethylation and sulfobutylation reactions have been conducted on lignin, no specific reports have been found in the literature on sulfoethylation and sulfopropylation of lignin, which could be a case of study since different reactions of carboxyalkylation or sulfoalkylation lead to the production of lignin with different properties as they introduce different carbon chain lengths and mimic the hydrophilicity/phobicity of lignin alterably. In the case of oxyalkylation, by far, oxypropylation has been the only reaction carried out on lignin, leaving room for investigating other routes in oxyalkylation. Generally, the solvent use and recovery impact the operation costs and ultimately the price of lignin derivatives and the environmental footprints of the developed technologies. If solvent use is necessary for lignin modification, the solvent recovery process is an important aspect of the process from finance and environment perspectives. For developing more industrially attractive and environmentally friendly pathways for lignin valorization, non-toxic reagents and chemistry should be discovered. Furthermore, the use of aqueous systems for lignin alteration would help reduce the environmental impacts of any lignin valorization processes.
As mentioned earlier, lignin has been studied in many applications, such as a dispersant, flocculant, adsorbent, and flame retardant (Table 20), while biological applications for lignin have been barely touched in the literature. For example, lignin's interaction with organic molecules, such as proteins, antibiotics, bacteria, and viruses could be studied for expanding either lignin-based biomedical applications as well as water and wastewater treatment systems. Using lignin in drug delivery systems, wound dressing, tissue engineering, and pharmaceutical applications could also be investigated. However, proceeding with such applications for lignin requires broad and detailed studies on the toxicity analysis of lignin, which has not been covered extensively as of yet.
Conclusions
Lignin is the most abundant aromatic polymer in nature, while its utility has been untapped in industry. Thanks to the numerous studies conducted in exploring lignin chemistry and structure over the last few years, the applications of lignin have been growing tremendously. This trend shows the increasing level of enthusiasm and interest of researchers in developing lignin-based applications and more importantly, the replacement of aromatic substances with petrochemical origins. Thus, various modifications have been performed on lignin to alleviate or remove the restrictions in the nature of lignin that limits its applications in industry. Some of these reactions are very promising to improve lignin's reactivity with other materials and compatibility in different environments. This work comprehensively reviewed and compared various modification routes while highlighting the strength and weak points. Despite the reactions mentioned, there is still room for expanding the other possible modification methods. In addition, biological applications of lignin could be studied more extensively to explore further and expand the functionality of this biobased and abundant material. In conclusion, distinguishing the modifications occurring on different parts of lignin, knowing the precise manner of reactions and reaction sites of the modifications, as well as pros and cons of each reaction, can pave the way for understanding and expanding the use of lignin in various processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Leah Nadin for her guidance with language proficiency while drafting this review.References
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Heroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- A. Scalbert, C. Morand, C. Manach and C. Rémésy, Biomed. Pharmacother., 2002, 56, 276–282 CrossRef CAS.
- G. Janusz, A. Pawlik, J. Sulej, U. Świderska-Burek, A. Jarosz-Wilkołazka and A. Paszczyński, FEMS Microbiol. Rev., 2017, 41, 941–962 CrossRef CAS PubMed.
- J. C. M. S. Moura, C. A. V. Bonine, J. de Oliveira Fernandes Viana, M. C. Dornelas and P. Mazzafera, J. Integr. Plant Biol., 2010, 52, 360–376 CrossRef CAS.
- J. Pérez, J. Muñoz-Dorado, T. de la Rubia and J. Martínez, Int. Microbiol., 2002, 5, 53–63 CrossRef.
- J. Chen, A. Eraghi Kazzaz, N. AlipoorMazandarani, Z. Hosseinpour Feizi and P. Fatehi, Molecules, 2018, 23, 868 CrossRef PubMed.
- G. F. D. Gregorio, R. Prado, C. Vriamont, X. Erdocia, J. Labidi, J. P. Hallett and T. Welton, ACS Sustainable Chem. Eng., 2016, 4, 6031–6036 CrossRef.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- M. Alekhina, O. Ershova, A. Ebert, S. Heikkinen and H. Sixta, Ind. Crops Prod., 2015, 66, 220–228 CrossRef CAS.
- Y. Liu and K. Li, J. Adhes., 2006, 82, 593–605 CrossRef CAS.
- M. Zhou, X. Qiu, D. Yang, H. Lou and X. Ouyang, Fuel Process. Technol., 2007, 88, 375–382 CrossRef CAS.
- S. Laurichesse and L. Averous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- V. P. Saraf and W. G. Glasser, J. Appl. Polym. Sci., 1984, 29, 1831–1841 CrossRef CAS.
- N. Alwadani and P. Fatehi, Carbon Resour. Convers., 2018, 1, 126–138 CrossRef.
- A. Lourenco and H. Pereira, Lignin: Trends and Applications, InTech, Croatia, 2018 Search PubMed.
- L. Christopher, J. H. Clark and G. A. Kraus, Integrated forest biorefineries, Royal Society of Chemistry, Cambridge, 2012 Search PubMed.
- A. G. Vishtal and A. Kraslawski, BioResources, 2011, 6, 3547–3568 Search PubMed.
- Z. H. Feizi, A. E. Kazzaz, F. Kong and P. Fatehi, Sep. Purif. Technol., 2019, 222, 254–263 CrossRef CAS.
- D. Tarasov, M. Leitch and P. Fatehi, Biofuels, 2018, 11, 269 CrossRef CAS PubMed.
- M. K. Konduri and P. Fatehi, ACS Sustainable Chem. Eng., 2015, 3, 1172–1182 CrossRef CAS.
- Y. Qin, D. Yang, W. Guo and X. Qiu, J. Ind. Eng. Chem., 2015, 27, 192–200 CrossRef CAS.
- M. A. Hubbe, R. Alén, M. Paleologou, M. Kannangara and J. Kihlman, BioResources, 2018, 14, 2300–2351 Search PubMed.
- A. N. Evdokimov, A. V. Kurzin, O. V. Fedorova, P. V. Lukanin, V. G. Kazakov and A. D. Trifonova, Wood Sci. Technol., 2018, 52, 1165–1174 CrossRef CAS.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2015, 116, 2275–2306 CrossRef PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, A. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem. – Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- J. Zhang, ChemSusChem, 2018, 11, 3071–3080 CrossRef CAS PubMed.
- S. Gharehkhani, Y. Zhang and P. Fatehi, Prog. Energy Combust. Sci., 2019, 72, 59–89 CrossRef.
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Lukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- X. Chen, L. Song and Y. Hu, J. Appl. Polym. Sci., 2010, 115, 3332–3338 CrossRef CAS.
- H. Vothi, C. Nguyen, K. Lee and J. Kim, Polym. Degrad. Stab., 2010, 95, 1092–1098 CrossRef CAS.
- P. A. Song, L. Xu, Z. Guo, Y. Zhang and Z. Fang, J. Mater. Chem., 2008, 18, 5083–5091 RSC.
- G. L. Bykov and B. G. Ershov, Russ. J. Appl. Chem., 2010, 83, 316–319 CrossRef CAS.
- B. Prieur, M. Meub, M. Wittemann, R. Klein, S. Bellayer, G. Fontaine and S. Bourbigot, Polym. Degrad. Stab., 2016, 127, 32–43 CrossRef CAS.
- G. P. Mendis, S. G. Weiss, M. Korey, C. R. Boardman, M. Dietenberger, J. P. Youngblood and J. A. Howarter, Green Mater., 2016, 4, 1–11 CrossRef.
- M. K. Konduri, F. Kong and P. Fatehi, Eur. Polym. J., 2015, 70, 371–383 CrossRef CAS.
- S. Sen, H. Sadeghifar and D. S. Argyropoulos, Biomacromolecules, 2013, 14, 3399–3408 CrossRef CAS PubMed.
- L. R. Morrow, M. G. DaGue and L. E. Whittington, US Pat, 4790382, 1988 Search PubMed.
- R. Saad and J. Hawari, J. Porous Mater., 2013, 20, 227–233 CrossRef CAS.
- L. Liu, G. Huang, P. Song, Y. Yu and S. Fu, ACS Sustainable Chem. Eng., 2016, 4, 4732–4742 CrossRef CAS.
- L. Liu, M. Qian, P. A. Song, G. Huang, Y. Yu and S. Fu, ACS Sustainable Chem. Eng., 2016, 4, 2422–2431 CrossRef CAS.
- A. A. Alalykin, R. L. Vesnin and D. A. Kozulin, Russ. J. Appl. Chem., 2011, 84, 1616 CrossRef CAS.
- L. Ferry, G. Dorez, A. Taguet, B. Otazaghine and J. M. Lopez-Cuesta, Polym. Degrad. Stab., 2015, 113, 135–143 CrossRef CAS.
- B. Prieur, M. Meub, M. Wittemann, R. Klein, S. Bellayer, G. Fontaine and S. Bourbigot, RSC Adv., 2017, 7, 16866–16877 RSC.
- A. M. Nada, N. F. Kassem and S. H. Mohamed, BioResources, 2008, 3, 538–548 Search PubMed.
- J. Qin, M. Woloctt and J. Zhang, ACS Sustainable Chem. Eng., 2014, 2, 188–193 CrossRef CAS.
- A. A. M. Nada and M. L. Hassan, J. Appl. Polym. Sci., 2003, 89, 2950–2956 CrossRef CAS.
- G. B. Quistad, N. Zhang, S. E. Sparks and J. E. Casida, Chem. Res. Toxicol., 2000, 13, 652–657 Search PubMed.
- Y. Yu, S. Fu, P. A. Song, X. Luo, Y. Jin, F. Lu and J. Ye, Polym. Degrad. Stab., 2012, 97, 541–546 CrossRef CAS.
- M. R. Clarke and A. J. Dolenko, U.S. Patent, 4113675, 1978 Search PubMed.
- A. M. Capraru, V. I. Popa, T. Malutan and G. Lisa, Cellul. Chem. Technol., 2009, 43, 409–418 CAS.
- A. R. Goncalves and P. Benar, Bioresour. Technol., 2001, 79, 103–111 CrossRef CAS PubMed.
- Y. X. Pang, X. Q. Qiu, D. J. Yang and H. M. Lou, Colloids Surf., A, 2008, 312, 154–159 CrossRef CAS.
- X. L. Liu, Y. H. Liao, Z. J. Wu, L. F. Cun, X. M. Zhang and W. C. Yuan, J. Org. Chem., 2010, 75, 4872–4875 CrossRef CAS PubMed.
- T. Malutan, R. Nicu and V. I. Popa, BioResources, 2007, 3, 13–20 Search PubMed.
- Y. Ge, Z. Li, Y. Kong, Q. Song and K. Wang, J. Ind. Eng. Chem., 2014, 20, 4429–4436 CrossRef CAS.
- R. Kaur, S. K. Uppal and P. Sharma, Sugar Tech, 2017, 19, 675–680 CrossRef CAS.
- R. V. Gadhave, P. A. Mahanwar and P. T. Gadekar, Open J. Polym. Chem., 2018, 8, 1 CrossRef CAS.
- S. Pietarinen, O. Ringena, K. Eskelinen and S. Valkonen, US Pat, 9464219, 2016 Search PubMed.
- P. Benar, A. R. Gonçalves, D. Mandelli and U. Schuchardt, Bioresour. Technol., 1999, 68, 11–16 CrossRef CAS.
- A. M. Capraru, E. Ungureanu, L. Trinca, T. Malutan and V. I. Popa, Cellul. Chem. Technol., 2012, 46, 589–597 CAS.
- M. E. Taverna, F. Felissia, M. C. Area, D. A. Estenoz and V. V. Nicolau, J. Appl. Polym. Sci., 2019, 47712 CrossRef.
- T. Malutan, R. Nicu and V. I. Popa, BioResources, 2008, 3, 1371–13767 Search PubMed.
- E. Ungureanu, O. Ungureanu, A. M. Capraru and V. I. Popa, Cellul. Chem. Technol., 2009, 43, 263 CAS.
- X. Wang, Y. Zhang, C. Hao, F. Feng, H. Yin and N. Si, Ind. Eng. Chem. Res., 2014, 53, 6585–6592 CrossRef CAS.
- A. Ang, Z. Ashaari, E. S. Bakar and N. A. Ibrahim, BioResources, 2015, 10, 4795–4810 CAS.
- N. E. E. Mansouri, A. Pizzi and J. Salvado, J. Appl. Polym. Sci., 2007, 103, 1690–1699 CrossRef.
- H. Younesi-Kordkheili and A. Pizzi, J. Adhes., 2017, 14, 1120–1130 CrossRef.
- F. Zhang, X. Jiang, J. Lin, G. Zhao, H. M. Chang and H. Jameel, New J. Chem., 2019, 43, 2238–2246 RSC.
- A. Effendi, H. Gerhauser and A. V. Bridgwater, Renewable Sustainable Energy Rev., 2008, 12, 2092–2116 CrossRef CAS.
- X. Jiang, J. Liu, X. Du, Z. Hu, H. M. Chang and H. Jameel, ACS Sustainable Chem. Eng., 2018, 6, 5504–5512 CrossRef CAS.
- L. Hu, H. Pan, Y. Zhou and M. Zhang, BioResources, 2011, 6, 3515–3525 CAS.
- T. T. M. Tan, Polym. Int., 1996, 41, 13–16 CrossRef.
- N. S. Cetin and N. Özmen, Int. J. Adhes. Adhes., 2002, 22, 477–480 CrossRef CAS.
- N. S. Cetin and N. Özmen, Int. J. Adhes. Adhes., 2002, 22, 481–486 CrossRef CAS.
- S. Kamel, Int. J. Polym. Mater., 2005, 55, 283–291 CrossRef.
- Y. Matsushita and S. Yasuda, Bioresour. Technol., 2005, 96, 465–470 CrossRef CAS PubMed.
- J. Podschun, B. Saake and R. Lehnen, Eur. Polym. J., 2015, 67, 1–11 CrossRef CAS.
- G. Vazquez, J. Gonzalez, S. Freire and G. Antorrena, Bioresour. Technol., 1997, 60, 191–198 CrossRef CAS.
- M. Funaoka, M. Matsubara, N. Seki and S. Fukatsu, Biotechnol. Bioeng., 1995, 46, 545–552 CrossRef CAS PubMed.
- J. Podschun, A. Stücker, R. I. Buchholz, M. Heitmann, A. Schreiber, B. Saake and R. Lehnen, Ind. Eng. Chem. Res., 2016, 55, 5231–5237 CrossRef CAS.
- E. Rojo, M. S. Peresin, W. W. Sampson, I. C. Hoeger, J. Vartiainen, J. Laine and O. J. Rojas, Green Chem., 2015, 17, 1853–1866 RSC.
- D. Fengel and G. Wegener, Wood: chemistry. ultrastructure. Reaction, Walter de Gruyter, New York, 2011 Search PubMed.
- C. I. Chiwetelu, V. Hornof, G. H. Neale and A. E. George, Can. J. Chem. Eng., 1994, 72, 534–540 CrossRef CAS.
- G. Telysheva, T. Dizhbite, E. Paegle, A. Shapatin and I. Demidov, J. Appl. Polym. Sci., 2001, 82, 1013–1020 CrossRef CAS.
- A. Macias and S. Goni, Mater. J., 1999, 96, 40–46 CAS.
- A. Kamoun, A. Jelidi and M. Chaabouni, Cem. Concr. Res., 2003, 33, 995–1003 CrossRef CAS.
- W. L. Ng, D. Rana, G. H. Neale and V. Hornof, J. Appl. Polym. Sci., 2003, 88, 860–865 CrossRef CAS.
- R. Chen and Q. Wu, J. Appl. Polym. Sci., 1994, 52, 437–443 CrossRef CAS.
- T. Aro and P. Fatehi, ChemSusChem, 2017, 10, 1861–1877 CrossRef CAS.
- P. Dilling, US Pat, 4546173, 1985 Search PubMed.
- S. E. Lebo Jr., J. D. Gargulak and T. J. McNally, Encycl. Polym. Sci. Technol., 2002 DOI:10.1002/0471440264.pst179.
- H. Zhang, Y. Bai, W. Zhou and F. Chen, Int. J. Biol. Macromol., 2017, 97, 201–208 CrossRef CAS PubMed.
- A. J. Latibari, M. Roohnia, A. Tajdini, F. Darvishqadima and M. Moradbak, J. Appl. Chem. Res., 2008, 2, 34–44 Search PubMed.
- P. Dilling, US Pat, 4732572, 1988 Search PubMed.
- H. Wu, F. Chen, Q. Feng and X. Yue, BioResources, 2012, 7, 2742–2751 CAS.
- W. He and P. Fatehi, RSC Adv., 2015, 5, 47031–47039 RSC.
- P. Dilling, US Pat, 4670482, 1987 Search PubMed.
- D. Yang, Y. Chang, X. Wu, X. Qiu and H. Lou, RSC Adv., 2014, 4, 53855–53863 RSC.
- H. Zhou, Y. Chang, X. Wu, D. Yang and X. Qiu, ACS Sustainable Chem. Eng., 2015, 3, 518–523 CrossRef CAS.
- A. E. Kazzaz, Z. H. Feizi and P. Fatehi, Colloid Polym. Sci., 2018, 296, 1867–1878 CrossRef CAS.
- B. Zhang, D. Yang, H. Wang, Y. Qian, J. Huang, L. Yu and X. Qiu, ACS Sustainable Chem. Eng., 2018, 7, 1120–1128 CrossRef.
- C. Huang, J. Ma, W. Zhang, G. Huang and Q. Yong, Polymer, 2018, 10, 841 Search PubMed.
- Y. Ge, Q. Song and Z. Li, J. Ind. Eng. Chem., 2015, 23, 228–234 CrossRef CAS.
- X. Qiu, W. Zeng, W. Liang, Y. Xue, N. Hong and Y. Li, J. Dispersion Sci. Technol., 2016, 37, 472–478 CrossRef CAS.
- X. Qiu, W. Zeng, W. Yu, Y. Xue, Y. Pang, X. Li and Y. Li, ACS Sustainable Chem. Eng., 2015, 3, 1551–1557 CrossRef CAS.
- Y. Li, Y. Wu, W. Zeng, Y. Li, L. Xu, X. Qiu and W. Huang, ACS Sustainable Chem. Eng., 2016, 4, 2004–2011 CrossRef CAS.
- H. Pan, G. Sun and T. Zhao, Int. J. Biol. Macromol., 2013, 59, 221–226 CrossRef CAS.
- H. Zhang, B. Yu, W. Zhou, X. Liu and F. Chen, Int. J. Biol. Macromol., 2018, 109, 1232–1238 CrossRef CAS PubMed.
- Y. Xue, X. Qiu, Y. Wu, Y. Qian, M. Zhou, Y. Deng and Y. Li, Polym. Chem., 2016, 7, 3502–3508 RSC.
- V. I. Chursin, Russ. J. Appl. Chem., 2010, 83, 312–315 CrossRef CAS.
- B. M. Cerrutti, C. S. de Souza, A. Castellan, R. Ruggiero and E. Frollini, Ind. Crops Prod., 2012, 36, 108–115 CrossRef CAS.
- S. Li, J. A. Willoughby and O. J. Rojas, ChemSusChem, 2016, 9, 2460–2469 CrossRef CAS.
- W. Lange and W. Schweers, Wood Sci. Technol., 1980, 14, 1–7 CrossRef CAS.
- K. Shweta and H. Jha, Bioresour. Bioprocess., 2016, 3, 31 CrossRef.
- S. Li, W. Xiang, M. Jarvinen, T. Lappalainen, K. Salminen and O. J. Rojas, ACS Appl. Mater. Interfaces, 2016, 8, 19827–19835 CrossRef CAS.
- S. Li, D. Ogunkoya, T. Fang, J. Willoughby and O. J. Rojas, J. Colloid Interface Sci., 2016, 482, 27–38 CrossRef CAS PubMed.
- L. H. Gan, M. S. Zhou and X. Q. Qiu, Adv. Mater. Res., 2012, 550, 1293–1298 Search PubMed.
- K. Bahrpaima and P. Fatehi, ChemSusChem, 2018, 11, 2967–2980 CrossRef CAS.
- C. Chen, M. Zhu, M. Li, Y. Fan and R. C. Sun, Biotechnol. Biofuels, 2016, 9, 87 CrossRef.
- F. Ferdosian, Z. Yuan, M. Anderson and C. C. Xu, J. Sci. Technol. For. Prod. Processes, 2012, 2, 11–15 Search PubMed.
- N. Ding, X. Wang, Y. Tian, L. Yang, H. Chen and Z. Wang, Polym. Eng. Sci., 2014, 54, 2777–2784 CrossRef CAS.
- G. H. Delmas, B. Benjelloun-Mlayah, Y. L. Bigot and M. Delmas, J. Appl. Polym. Sci., 2013, 127, 1863–1872 CrossRef CAS.
- Y. Nonaka, B. Tomita and Y. Hatano, Holzforschung, 1997, 51, 183–187 CrossRef CAS.
- E. Cortes-Trivino, C. Valencia, M. Delgado and J. Franco, Polymer, 2018, 10, 670 Search PubMed.
- A. L. Brocas, G. Cendejas, S. Caillol, A. Deffieux and S. Carlotti, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2677–2684 CrossRef CAS.
- S. Nikafshar, O. Zabihi, Y. Moradi, M. Ahmadi, S. Amiri and M. Naebe, Polymer, 2017, 9, 266 Search PubMed.
- S. Wang, S. Ma, C. Xu, Y. Liu, J. Dai, Z. Wang and J. Zhu, Macromolecules, 2017, 50, 1892–1901 CrossRef CAS.
- K. Hofmann and W. G. Glasser, J. Adhes., 1993, 40, 229–241 CrossRef CAS.
- T. A. Khan, J. H. Lee and H. J. Kim, Lignocellulose for Future Bioeconomy, Elsevier, Amsterdam, 2019 Search PubMed.
- Q. Yin, W. Yang, C. Sun and M. Di, BioResources, 2012, 7, 5737–5748 CrossRef.
- I. Kuhnel, J. Podschun, B. Saake and R. Lehnen, Holzforschung, 2015, 69, 531–538 Search PubMed.
- I. Kuhnel, B. Saake and R. Lehnen, React. Funct. Polym., 2017, 120, 83–91 CrossRef.
- I. Kuhnel, B. Saake and R. Lehnen, Ind. Crops Prod., 2017, 101, 75–83 CrossRef.
- C. A. Cateto, M. F. Barreiro, A. E. Rodrigues and M. N. Belgacem, Ind. Eng. Chem. Res., 2009, 48, 2583–2589 CrossRef CAS.
- B. Ahvazi, O. Wojciechowicz, T. M. Ton-That and J. Hawari, J. Agric. Food Chem., 2011, 59, 10505–10516 CrossRef CAS PubMed.
- H. Sadeghifar, C. Cui and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2012, 51, 16713–16720 CrossRef CAS.
- B. Berrima, G. Mortha, S. Boufi, E. El Aloui and M. N. Belgacem, Cellul. Chem. Technol., 2016, 50, 941–950 CAS.
- H. Nadji, C. Bruzzèse, M. N. Belgacem, A. Benaboura and A. Gandini, Macromol. Mater. Eng., 2005, 290, 1009–1016 CrossRef CAS.
- Y. Li and A. J. Ragauskas, J. Wood Chem. Technol., 2012, 32, 210–224 CrossRef CAS.
- A. Arbenz and L. Averous, RSC Adv., 2014, 4, 61564–61572 RSC.
- J. Busch, J. Am. Coll. Toxicol., 1987, 6, 23–51 CrossRef.
- D. S. Argyropoulos, US Pat, 61601181, 2012 Search PubMed.
- W. G. Glasser and O. H. H. Hsu, US Pat, 4017474, 1977 Search PubMed.
- L. T. Monson and W. J. Dickson, US Pat, 2854444, 1958 Search PubMed.
- C. Cui, H. Sadeghifar, S. Sen and D. S. Argyropoulos, BioResources, 2013, 8, 864–886 Search PubMed.
- I. Kühnel, B. Saake and R. Lehnen, Macromol. Chem. Phys., 2018, 219, 613 CrossRef.
- M. Wanga, Z. Wanga, Z. Sun and H. Jiang, React. Kinet. Catal. Lett., 2005, 84, 223–228 CrossRef.
- J. Lisperguer, C. Nunez and P. Perez-Guerrero, J. Chil. Chem. Soc., 2013, 58, 1937–1940 CrossRef CAS.
- N. Blank, I. Schober, P. R. Von Rohr and T. Voitl, US Pat, 13257733, 2010 Search PubMed.
- X. Zhao, Y. Zhang, L. Wie, H. Hu, Z. Huang, M. Yang, A. Huang, J. Wua and Z. Fenga, RSC Adv., 2017, 7, 52382–52390 RSC.
- L. Li, Y. Hu and F. Cheng, BioResources, 2015, 10, 3181–3196 CAS.
- P. Buono, A. Duval, P. Verge, L. Averous and Y. Habibi, ACS Sustainable Chem. Eng., 2016, 4, 5212–5222 CrossRef CAS.
- S. Chatterjee, A. Clingenpeel, A. McKenna, O. Rios and A. Johs, RSC Adv., 2014, 4, 4743–4753 RSC.
- K. A. Koivu, H. Sadeghifar, P. A. Nousiainen, D. S. Argyropoulos and J. Sipilä, ACS Sustainable Chem. Eng., 2016, 4, 5238–5247 CrossRef CAS.
- L. Dehne, C. V. Babarro, B. Saake and K. U. Schwarz, Ind. Crops Prod., 2016, 86, 320–328 CrossRef CAS.
- S. Luo, J. Cao and A. G. McDonald, Ind. Crops Prod., 2017, 97, 281–291 CrossRef CAS.
- Y. Pu and A. J. Ragauskas, Can. J. Chem., 2005, 83, 2132–2139 CrossRef CAS.
- Y. Teramoto, S. H. Lee and T. Endo, Polym. J., 2009, 41, 219 CrossRef CAS.
- H. F. Lewis and F. E. Brauns, US Pat, 2429102, 1947 Search PubMed.
- R. Ding, H. Wu, M. Thunga, N. Bowler and M. R. Kessler, Carbon, 2016, 100, 126–136 CrossRef CAS.
- W. G. Glasser and R. K. Jain, Holzforschung, 1993, 47, 225–233 CrossRef CAS.
- M. Thunga, K. Chen, D. Grewell and M. R. Kessler, Carbon, 2014, 68, 159–166 CrossRef CAS.
- L. Dehne, C. Vila, B. Saake and K. U. Schwarz, J. Appl. Polym. Sci., 2017, 134, 44582 CrossRef.
- W. Thielemans and R. P. Wool, Composites, Part A, 2004, 35, 327–338 CrossRef.
- J. H. Lora and W. G. Glasser, J. Polym. Environ., 2002, 10, 39–48 CrossRef CAS.
- G. Marchand, C. A. Calliste, R. M. Williams, C. McLure, S. Leroy-Lhez and N. Villandier, ChemistrySelect, 2018, 3, 5512–5516 CrossRef CAS.
- E. L. Hult, K. Koivu, J. Asikkala, J. Ropponen, P. Wrigstedt, J. Sipilä and K. Poppius-Levlin, Holzforschung, 2013, 67, 899–905 CAS.
- O. Gordobil, R. Herrera, R. Llano-Ponte and J. Labidi, Prog. Org. Coat., 2017, 103, 143–151 CrossRef CAS.
- N. Cachet, S. Camy, B. Benjelloun-Mlayah, J. S. Condoret and M. Delmas, Ind. Crops Prod., 2014, 58, 287–297 CrossRef CAS.
- Y. Chen, N. M. Stark, Z. Cai, C. R. Frihart, L. F. Lorenz and R. E. Ibach, BioResources, 2014, 9, 5488–5500 Search PubMed.
- T. Sonoda, T. Ona, H. Yokoi, Y. Ishida, H. Ohtani and S. Tsuge, Anal. Chem., 2001, 73, 5429–5435 CrossRef CAS PubMed.
- N. E. Mansouri and J. Salvadó, Ind. Crops Prod., 2006, 24, 8–16 CrossRef.
- H. Sadeghifar, S. Sen and S. V. Patil, ACS Sustainable Chem. Eng., 2016, 4, 5230–5237 CrossRef CAS.
- D. A. Baker and T. G. Rials, J. Appl. Polym. Sci., 2013, 130, 713–728 CrossRef CAS.
- M. Wang and L. Yang, J. Polym. Environ., 2012, 20, 783–787 CrossRef CAS.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 1077–1087 RSC.
- K. Iiyama and R. Pant, Wood Sci. Technol., 1988, 22, 167–175 CrossRef CAS.
- D. E. McKinney, D. M. Carson, D. J. Clifford, R. D. Minard and P. G. Hatcher, J. Anal. Appl. Pyrolysis, 1995, 34, 41–46 CrossRef CAS.
- D. Delledonne, F. Rivetti and U. Romano, J. Organomet. Chem., 1995, 488, 15–19 CrossRef.
- F. Chen, H. Dai, X. Dong, J. Yang and M. Zhong, Polym. Compos., 2011, 32, 1019–1025 CrossRef CAS.
- S. Sarkanen and Y. Li, US Pat, 6172204, 2001 Search PubMed.
- M. Tajvidi, R. H. Falk and J. C. Hermanson, J. Appl. Polym. Sci., 2006, 101, 4341–4349 CrossRef CAS.
- Y. Li and S. Sarkanen, Macromolecules, 2002, 35, 9707–9715 CrossRef CAS.
- A. Sequeiros, L. Serrano and J. Labidi, J. Chem. Technol. Biotechnol., 2016, 91, 1809–1815 CrossRef CAS.
- N. N. Shorygina and L. I. Kolotova, Russ. Chem. Bull., 1953, 2, 505–508 CrossRef.
- S. C. Puri, S. M. Anand and C. K. Atal, Indian J. Chem., 1985, 16, 294–295 Search PubMed.
- W. Zhao, L. P. Xiao, G. Song, R. C. Sun, L. He, S. Singh and G. Cheng, Green Chem., 2017, 19, 3272–3281 RSC.
- A. Longoria, R. Tinoco and R. Vázquez-Duhalt, Chemosphere, 2008, 72, 485–490 CrossRef CAS PubMed.
- J. G. Speight, Environmental organic chemistry for engineers, Butterworth-Heinemann, Oxford, 2016 Search PubMed.
- D. N. S. Hon, Chemical modification of lignocellulosic materials, CRC Press, New York, 1995 Search PubMed.
- X. Du, J. Li and M. E. Lindstrom, Ind. Crops Prod., 2014, 52, 729–735 CrossRef CAS.
- X. Liu, H. Zhu, C. Qin, J. Zhou, J. R. Zhao and S. Wang, BioResources, 2013, 8, 2257–2269 CAS.
- H. Dong, W. Peng and L. N. Lewis, US Pat, 9181405, 2013 Search PubMed.
- Y. Matsushita and S. Yasuda, J. Wood Sci., 2003, 49, 166–171 CrossRef CAS.
- Y. Matsushita, A. Iwatsuki and S. Yasuda, J. Wood Sci., 2004, 50, 540–544 CrossRef CAS.
- R. Fang, X. Cheng and X. Xu, Bioresour. Technol., 2010, 101, 7323–7329 CrossRef CAS PubMed.
- H. Ruihua, Y. Bingchao, D. Zheng and B. Wang, J. Mater. Sci., 2012, 47, 845–851 CrossRef.
- H. Harmita, K. G. Karthikeyan and X. Pan, Bioresour. Technol., 2009, 100, 6183–6191 CrossRef CAS PubMed.
- J. Aguado, J. M. Arsuaga, A. Arencibia, M. Lindo and V. Gascón, J. Hazard. Mater., 2009, 163, 213–221 CrossRef CAS PubMed.
- J. Tian, S. Ren, G. Fang, Y. Ma and Q. Ai, BioResources, 2014, 9, 6290–6303 CrossRef.
- C. Cai, X. Zhan, M. Zeng, H. Lou, Y. Pang, J. Yang and X. Qiu, Green Chem., 2017, 19, 5479–5487 RSC.
- T. Zheng, D. Zheng, X. Li, C. Cai, H. Lou, W. Liu and X. Qiu, ACS Sustainable Chem. Eng., 2017, 5, 7743–7750 CrossRef CAS.
- F. Kong, K. Parhiala, S. Wang and P. Fatehi, Eur. Polym. J., 2015, 67, 335–345 CrossRef CAS.
- R. Wahlstrom, A. Kalliola, J. Heikkinen, H. Kyllonen and T. Tamminen, Ind. Crops Prod., 2017, 104, 188–194 CrossRef CAS.
- C. Cheng, J. Wang, D. Shen, J. Xue, S. Guan, S. Gu and K. Luo, Polymer, 2017, 9, 240 Search PubMed.
- J. C. Villar, A. Caperos and F. Garcia-Ochoa, Wood Sci. Technol., 2001, 35, 245–255 CrossRef CAS.
- Q. Xiang and Y. Y. Lee, Appl. Biochem. Biotechnol., 2000, 84, 153–162 CrossRef PubMed.
- J. M. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369–1377 CrossRef CAS.
- S. K. Hanson, R. Wu and L. A. P. Silks, Angew. Chem., 2012, 124, 3466–3469 CrossRef.
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS PubMed.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef CAS.
- V. E. Tarabanko, N. A. Fomova, B. N. Kuznetsov, N. M. Ivanchenko and A. V. Kudryashev, React. Kinet. Catal. Lett., 1995, 55, 161–170 CrossRef CAS.
- J. C. Villar, A. Caperos and F. García-Ochoa, React. Kinet. Catal. Lett., 1997, 17, 259–285 CAS.
- G. Tong, Y. Matsumoto and G. Meshitsuka, J. Wood Sci., 2000, 46, 371–375 CrossRef CAS.
- S. Rovio, S. Kuitunen, T. Ohra-aho, S. Alakurtti, A. Kalliola and T. Tamminen, Holzforschung, 2011, 65, 575–585 CAS.
- F. G. Sales, C. A. M. Abreu and J. A. F. R. Pereira, Braz. J. Chem. Eng., 2004, 21, 211–218 CrossRef CAS.
- T. A. D. Nguyen, A. M. Wright, J. S. Page, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 11377–11387 CrossRef CAS PubMed.
- M. Tian, J. Wen, D. MacDonald, R. M. Asmussen and A. Chen, Electrochem. Commun., 2010, 12, 527–530 CrossRef CAS.
- C. Zhu, W. Ding, T. Shen, C. Tang, C. Sun, S. Xu and H. Ying, ChemSusChem, 2015, 8, 1768–1778 CrossRef CAS PubMed.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- J. Gierer, Wood Sci. Technol., 1986, 20, 1–33 CrossRef CAS.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- R. Prado, A. Brandt, X. Erdocia, J. Hallet, T. Welton and J. Labidi, Green Chem., 2016, 18, 834–841 RSC.
- J. Marton, S. C. Charleston and E. Adler, US Pat, 3071570, 1963 Search PubMed.
- T. Okamoto, H. Takeda, T. Funabiki, M. Takatani and R. Hamada, React. Kinet. Catal. Lett., 1996, 58, 237–242 CrossRef CAS.
- S. Dabral, H. Wotruba, J. G. Hernández and C. Bolm, ACS Sustainable Chem. Eng., 2018, 6, 3242–3254 CrossRef CAS.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249 CrossRef CAS PubMed.
- W. Hoareau, W. G. Trindade, B. Siegmund, A. Castellan and E. Frollini, Polym. Degrad. Stab., 2004, 86, 567–576 CrossRef CAS.
- J. Mottweiler, M. Puche, C. Räuber, T. Schmidt, P. Concepción, A. Corma and C. Bolm, ChemSusChem, 2016, 8, 2106–2113 CrossRef.
- J. D. Araújo, C. A. Grande and A. E. Rodrigues, Chem. Eng. Res. Des., 2010, 88, 1024–1032 CrossRef.
- A. Das, A. Rahimi, A. Ulbrich, M. Alherech, A. H. Motagamwala, A. Bhalla and B. E. Dale, ACS Sustainable Chem. Eng., 2018, 6, 3367–3374 CrossRef CAS.
- E. I. Evstigneyev, O. S. Yuzikhin, A. A. Gurinov, A. Y. Ivanov, T. O. Artamonova, M. A. Khodorkovskiy and A. V. Vasilyev, J. Wood Chem. Technol., 2016, 36, 259–269 CrossRef CAS.
- A. Mancera, V. Fierro, A. Pizzi, S. Dumarçay, P. Gérardin, J. Velásquez and A. Celzard, Polym. Degrad. Stab., 2010, 95, 470–476 CrossRef CAS.
- R. L. Couch, J. T. Price and P. Fatehi, ACS Sustainable Chem. Eng., 2016, 4, 1954–1962 CrossRef CAS.
- W. He, W. Gao and P. Fatehi, ACS Sustainable Chem. Eng., 2017, 5, 10597–10605 CrossRef CAS.
- J. J. Meister, J. Macromol. Sci., Polym. Rev., 2002, 42, 235–289 CrossRef.
- Y. Sun, M. Fenster, A. Yu, R. M. Berry and D. S. Argyropoulos, Can. J. Chem., 1999, 77, 667–675 CrossRef CAS.
- C. Crestini, P. Pro, V. Neri and R. Saladino, Bioorg. Med. Chem., 2005, 13, 2569–2578 CrossRef CAS.
- K. R. Kavanagh and J. M. Pepper, Can. J. Chem., 1955, 33, 24–30 CrossRef CAS.
- S. Kagawa, Jpn. TAPPI J., 1970, 24, 424–428 CrossRef CAS.
- Y. Sang, B. Wang, Q. Wang, G. Zhao and P. Guo, Sci. Rep., 2014, 4, 6321 CrossRef CAS PubMed.
- M. N. Alam and T. G. Van De Ven, J. Sci. Technol. For. Prod. Processes, 2014, 4, 22–26 CAS.
- D. E. Bland, G. Ho and W. E. Cohen, Aust. J. Chem., 1950, 3, 642–648 CrossRef.
- R. Yang, L. Lucia, A. J. Ragauskas and H. Jameel, J. Wood Chem. Technol., 2003, 23, 13–29 CrossRef CAS.
- T. Higuchi, J. Biochem., 1985, 45, 675–685 CrossRef.
- J. C. Pew, J. Am. Chem. Soc., 1955, 77, 2831–2833 CrossRef CAS.
- L. Klapiszewski, J. Zdarta, T. Szatkowski, M. Wysokowski, M. Nowacka, K. Szwarc-Rzepka and T. Jesionowski, Open Chem., 2014, 12, 719–735 CAS.
- S. Hamdani, C. Longuet, D. Perrin, J. M. Lopez-cuesta and F. Ganachaud, Polym. Degrad. Stab., 2009, 94, 465–495 CrossRef CAS.
- J. Zhang, E. Fleury, Y. Chen and M. A. Brook, RSC Adv., 2015, 5, 103907–103914 RSC.
- T. Budnyak, S. Aminzadeh, I. Pylypchuk, A. Riazanova, V. Tertykh, M. Lindström and O. Sevastyanova, Nanomaterials, 2018, 8, 950 CrossRef PubMed.
- W. Xiong, X. Qiu, D. Yang, R. Zhong, Y. Qian, Y. Li and H. Wang, Chem. Eng. J., 2017, 326, 803–810 CrossRef CAS.
- J. F. Kadla and S. Kubo, Composites, Part A, 2004, 35, 395–400 CrossRef.
- P. Bruijnincx, B. Weckhuysen, G. J. Gruter and E. Engelen-Smeets, Lignin valorisation: The importance of a full value chain approach, Utrecht University, Google Books, 2016 Search PubMed.
- L. Kouisni, A. Gagné, K. Maki, P. Holt-Hindle and M. Paleologou, ACS Sustainable Chem. Eng., 2016, 4, 5152–5159 CrossRef CAS.
- J. Kihlman, Nord. Pulp Pap. Res. J., 2016, 31, 573–582 CAS.
- M. Abraham, Encyclopedia of sustainable technologies. Elsevier, Cambridge, 2017 Search PubMed.
- N. Smolarski, Frost & Sullivan, https://www.greenmaterials.fr/wp-content/uploads/2013/01/High-value-Opportunities-for-Lignin-Unlocking-its-Potential-Market-Insights.pdf, (accessed August 2019).
- LigniMATCH online, https://www.gmv.gu.se/digitalAssets/1448/1448662_roadmap.pdf, (accessed August 2019).
| This journal is © The Royal Society of Chemistry 2019 |