
“Water soluble” palladium nanoparticle engineering for C–C coupling, reduction and cyclization catalysis†

Abstract
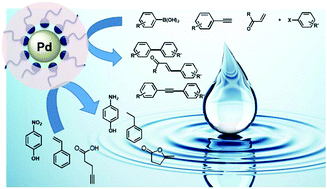
The use of Pd nanoparticles (Pd-NPs) to realize several important organic reactions allows efficient catalysis with low metal loading (<1000 ppm), hence providing a greener catalytic system. However, to be truly green Pd-NPs need to be synthesized in a sustainable manner and be able to react in aqueous media in order to avoid the use of organic solvents. Here we describe an original and eco-friendly synthesis of Pd-NPs (using benign reactants and simple conditions) perfectly stable in water. Remarkably, this synthesis allows for control over their size and morphology by simply tuning the pH of the stabilizer. We then evaluate the catalytic efficiency of these Pd-NPs on six different model reactions (Suzuki Miyaura, Sonogashira, Heck, nitrophenol reduction and pentynoic cycloisomerization) in aqueous media. We show that the stabilizer structure influences the activity owing to its ability to promote the mass transfer of the organic substrates towards the NP surface in the aqueous environment. Finally, catalytic evaluations show that our nano-catalysts prepared in an eco-friendly manner are among the best catalysts described so far in the literature in each case, with high turnover frequencies reached with a low loading of palladium.
- This article is part of the themed collection: International Symposium on Green Chemistry 2019
 Please wait while we load your content...
Please wait while we load your content...

