
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering of a fungal laccase to develop a robust, versatile and highly-expressed biocatalyst for sustainable chemistry†
Felipe
de Salas
 a,
Pablo
Aza
a,
Joan F.
Gilabert
b,
Gerard
Santiago
bc,
Sibel
Kilic
d,
Mehmet E.
Sener
d,
Jesper
Vind
e,
Víctor
Guallar
bf,
Angel T.
Martínez
a and
Susana
Camarero
*a
a,
Pablo
Aza
a,
Joan F.
Gilabert
b,
Gerard
Santiago
bc,
Sibel
Kilic
d,
Mehmet E.
Sener
d,
Jesper
Vind
e,
Víctor
Guallar
bf,
Angel T.
Martínez
a and
Susana
Camarero
*a
aCentro de Investigaciones Biológicas, CSIC. Ramiro de Maeztu 9, 28040 Madrid, Spain. E-mail: susanacam@cib.csic.es
bBarcelona Supercomputing Center, Jordi Girona 29, E-08034 Barcelona, Spain
cNostrum Biodiscovery, Jordi Girona 29, Nexus II D128, 08034, Barcelona, Spain
dSetas Kimya San AS., Karanfil Sok 34, 34330 Istanbul, Turkey
eNovozymes A/S, Krogshoejvej 36, 2880, Bagsvaerd, Denmark
fICREA, Passeig Lluís Companys 23, E-08010 Barcelona, Spain
First published on 3rd September 2019
Abstract
Fungal laccases can play an important role as biocatalysts in organic chemistry to replace chemical synthesis. In a previous work we synthesized conductive polyaniline using a high-redox potential laccase from our collection of recombinant fungal variants. Still, the oxidation of aniline is hindered by the reaction conditions (low pH and presence of anionic surfactants). Thus, we tackle here the directed evolution of the enzyme assisted by computational simulation aiming at improving aniline oxidation at the required polymerization conditions while maintaining the enzyme's substrate promiscuity. Simultaneously, its secretion by the host used for the engineering (Saccharomyces cerevisiae) was enhanced. Then, the improved laccase variant was overproduced in the industrial host Aspergillus oryzae and assayed for one-pot synthesis of polyaniline and naphtol-derived dyes whose textile dyeing properties were verified in an industrial environment. Finally, modification of its C-terminal tail further enhanced laccase stability by flexibilization of the region. The resulting biocatalyst displays noticeable stability at high temperature and extreme pH while shows improved kcat values on the different substrates tested. Moreover, it is remarkably produced in S. cerevisiae at rates not formerly reported in the literature. These facts, together with the overexpression in A. oryzae opens new scenarios for its further development and application.
Introduction
Laccases (EC 1.10.3.2, benzenediol: O2 oxidoreductase) are multicopper oxidases that contain four catalytic copper ions involved in the one-electron oxidation of substrates coupled to the four-electron reduction of molecular oxygen to water. The T1 copper, responsible for the characteristic blue colour of laccases (absorbance 600 nm), catalyzes the oxidation of the reducing substrate. The electrons are sequentially transferred through cysteine and histidine ligands to the other three copper ions (one T2/two T3) arranged in a trinuclear cluster (TNC) where the reduction of O2 takes place.1,2 Laccases are widely distributed in fungi, plants, bacteria and some insects, playing diverse physiological roles. In fungi they are involved in lignin degradation, morphogenesis, pathogenesis (fungal–plant interaction) and stress defense.3 The redox potential of laccases at the T1 site ranges from near 0.4 V in plant and bacterial laccases to up to 0.8 V in some fungal laccases. Saprobic basidiomycetes degrading lignin during decay of wood and leaf litter produce high redox potential laccases (HRPLs), with (E° = 0.720–0.790 V).4,5 Laccases are promiscuous oxidizing a broad spectrum of phenols, aryl amines, substituted N-heterocycles, thiols and some metal ions.4,6,7 The use of atmospheric oxygen for their activation, the production of water as sole by-product, and the possibility to catalyze either degradation reactions (such as lignin depolymerization) or synthesis reactions depending on the conditions used, make these enzymes ideal biocatalysts for a number of applications.8–10 Their use in single or multistep biocatalytic processes for organic synthesis has drawn increasing interest due to the considerable advantages obtained from the milder reaction conditions and lower environmental impact than equivalent chemical methods.11Polyaniline (PANI) is a conducting polymer discovered over 150 years ago. Because of its rich chemistry, high electrical conductivity and attractive processing properties, it is one of the most studied conducting polymers of the past 50 years. Polyaniline shows high stability, simple production, low cost of the monomer and the ability to change its optic, structural and conductivity capabilities depending on the synthesis conditions and protonation state.12 Broad range high-value applications of PANI include supercapacitors, solar panels, biosensors, static insulators etc. Electroconductive PANI is formed after oxidative p-coupling (without branching) of aniline, an aromatic amine with applications in dye, rubber or urethane production. Besides linearity, a half oxidized (imine) – half reduced (amine) protonated state of the polymer is required to obtain electroconductive PANI (Emeraldine salt). To obtain this green PANI, aniline polymerization has to be performed at acidic conditions (below the pKa 4.6 of aniline), which increase the redox potential of the monomer (from E° = 0.63 V of non-protonated aniline to E° = 1.05 V of anilinium cation).13 Nowadays, the industrial synthesis of PANI follows chemical processes with ammonium peroxydisulfate as oxidizer and extremely acidic conditions. By contrast, the enzymatic synthesis of PANI catalyzed by laccase allows the use of milder conditions and reduces pollution.14 The addition of anionic surfactants as doping templates in PANI synthesis prevents polymer branches while, acting as amphiphilic systems, they solubilize the polymer in water by forming micelles or vesicles.15 Besides, the use of different templates results in diverse nano-structured polymers.16,17
In a previous work, we set the optimal conditions for the enzymatic synthesis of electroconductive polyaniline, obtaining a nano-fibered water-soluble polymer with excellent electrochemistry and conductivity.17 The laccase used as biocatalyst had been developed by DNA shuffling of two fungal laccases expressed in Saccharomyces cerevisiae,5 and it was selected for this target due to acidic pH profile and better activity on aromatic amines than commercial laccases.17 Nevertheless, the reaction is still demanding for laccases due to their poor stability to acid pH and anionic surfactants, and the difficult oxidation of the protonated aniline. Protein engineering can help us to improve the catalytic activity or stability of the enzyme at target conditions.18–21
The easy manipulation, high recombination frequency and feasible secretion of heterologous proteins, make S. cerevisiae the preferred host for the directed evolution of fungal oxidorreductases.20,22–25 However, the low protein yields provided by S. cerevisiae as expression system is a major bottleneck to evaluate the biotechnological potential of the enzymes engineered in the lab. Consequently, their up-scale production in other fungal hosts such as Pichia pastoris or Aspergillus is commonly pursued.26,27
In this work, laccase directed evolution and computational design have been combined to improve the acidic synthesis of conductive polyaniline, while maintaining the generalist catalytic activity of the enzyme, and to increase laccase expression in S. cerevisiae. Then, the improved laccase variant was overexpressed in the industrial host Aspergillus oryzae (Novozymes A/S, Denmark), and used as biocatalyst for the synthesis of polyaniline and dyes to be tested on textiles at an industrial environment (SETAŞ AS, Turkey). Finally, engineering of the C-terminal tail significantly raised laccase stability and activity.
Results and discussion
Laccase directed evolution and semi-rational design
The laccase used here as the starting point for enzyme engineering was selected among a set of fungal laccase variants previously evolved in S. cerevisiae in our group. This enzyme, laccase 7D5, possesses high redox potential (E° = 0.76 V referred to NHE standard electrode), activity towards anilines, acidic pH profile, and better stability to the reaction conditions than other counterparts developed in the same directed evolution campaign,5 being, therefore, selected for PANI synthesis.17To direct laccase evolution towards better activity on anilines, we used a high throughput screening (HTS) colorimetric assay with N,N-dimethyl-p-phenylenediamine (DMPD) as a surrogate substrate less-toxic than aniline to explore laccase activity in the mutant libraries expressed in yeast. Laccase oxidizes DMPD to the stable Würstern dye at pH 3, with ε550 = 4134 M−1 cm−1 (Fig. S1A†). The low coefficient of variation (CV = 12%) of the response given by a certain clone cultured in the 96 wells of the same microplate proved the reproducibility of the colorimetric assay (Fig. S1B†). Also, the direct correlation of the response with increasing volumes of the same supernatant confirmed the linearity of the HTS assay (Fig. S1C†). Finally, its sensitivity was verified on a laccase mutant library of 1800 clones generated by epPCR and expressed in the yeast (Fig. S1D†).
Besides, a stability assay at pH 3 was performed during the HTS of laccase libraries to avoid a significant loss of enzyme stability during the evolution pathway. The oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) was also used as a reference assay for laccase activity to maintain the generalist activity of the enzyme while increasing its activity on aromatic amines. In each evolution round, improvements in total activity (TAI) for the oxidation of DMPD and ABTS and in stability to pH 3 were calculated for each clone compared with the parent laccase. Finally, the oxidation of aniline in the presence of the anionic surfactant SDBS was used to evaluate the activities and stabilities of the new variants to the reaction conditions for PANI synthesis.
Laccase engineering started with two rounds of random mutagenesis over the laccase CDS fused to the mutated S. cerevisiae alpha mating factor pre-proleader, as signal sequence.5 After screening over 4000 clones, the highest activity improvement (8.5-fold TAI) was obtained with variant 9H2 which held two mutations in the α pre-proleader (Aα20T and Qα32H) plus F454S mutation in the mature protein (Fig. 1). The new variant was produced in flask cultures and though laccase activity detected in the yeast culture broth was significantly higher than that of parent 7D5, the stability at pH 3 was remarkably low (Fig. 2). Mutation F454S had appeared likewise during the evolution pathway of PM1 basidiomycete laccase (one of the 7D5 parents) for expression in S. cerevisiae, although it was reverted due to its destabilizing effect.23,28 Molecular dynamics (MD) simulation of Phe454 and Ser454 in 7D5 laccase showed only 4 hydrogen bonds in the T1 coordination site for the F454S mutant, 2 less than the parental protein. This correlates with the result obtained from the server ENCoM29 that shows a flexibility increase in this protein region, opening the T1 site into solvent and affecting the hydrogen bond network. This reduction in the number of hydrogen bonds correlates with an increase in activity and loss of stability.30
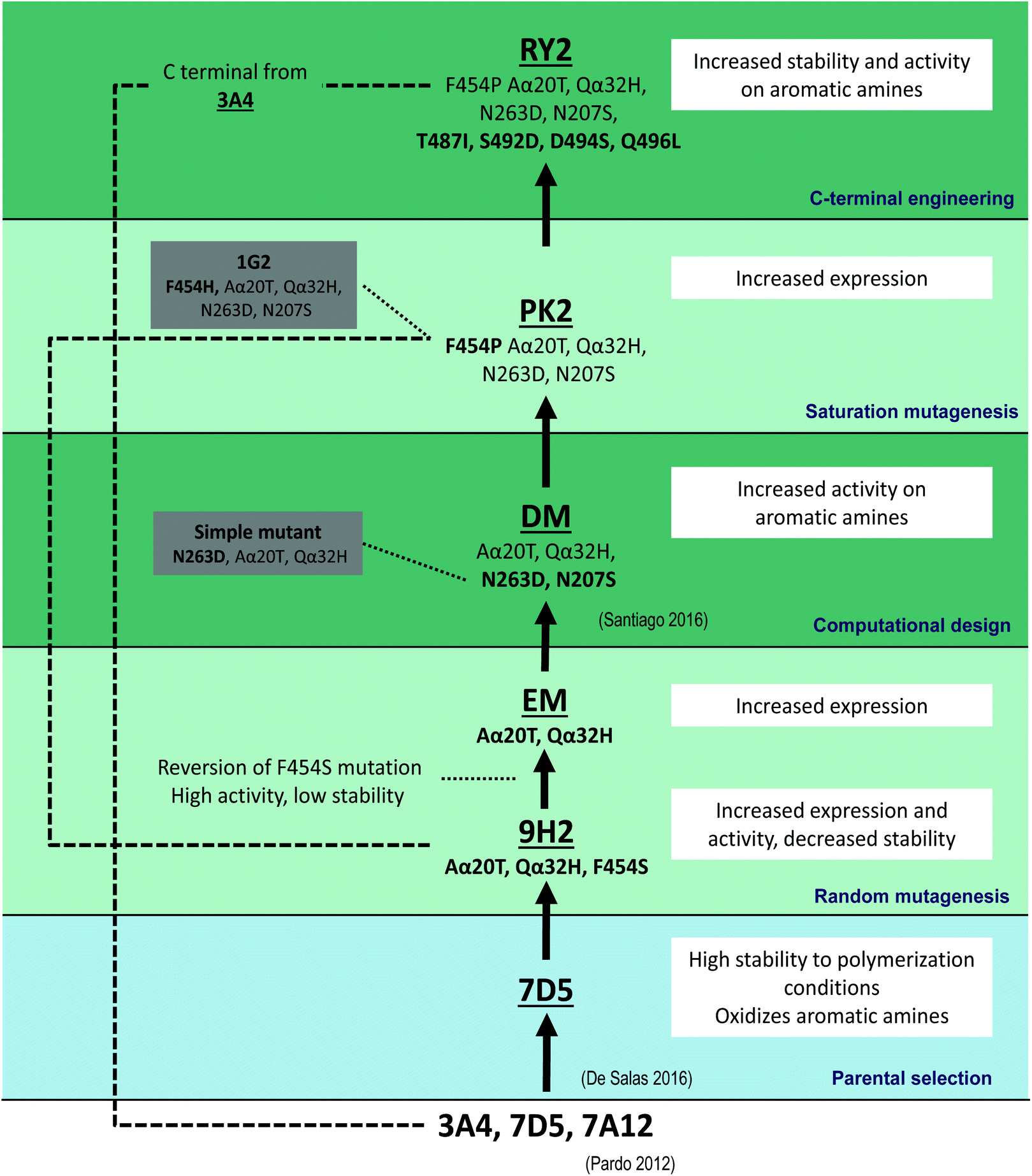 | ||
| Fig. 1 Laccase engineering pathway consisting of: (i) selection of parent laccase 7D5; (ii) random mutagenesis; (iii) computational design; (iv) saturation mutagenesis; and (v) C-terminal engineering. The mutations accumulated in the improved variants and the main results attained in each consecutive mutational step are shown. | ||
 | ||
| Fig. 2 Laccase production in flask cultures of recombinant S. cerevisiae (A), and acidic (pH 3) stability (B) of laccase 7D5 (red) compared with 9H2 (purple) and EM (brown) variants from random mutagenesis (crude enzymes). | ||
Our attempts to offset the destabilizing effect in 9H2 variant were unsuccessful. Thus, we reverted F454S mutation maintaining the two mutations of the signal sequence (Aα20T and Qα32H), giving rise to the expression mutant (EM). The EM variant produced in S. cerevisiae flasks cultures (Fig. 2A) recovered the stability of 7D5, although the total activity improvement (TAI) was lowered respecting 9H2. TAI is the result of joint contribution of enhanced secretion or/and activity in crude extracts. By contrast, the 5-fold TAI detected for EM respecting 7D5 is just the result of better secretion due to the two new mutations in the α pre-proleader, which increased laccase production from 3 g l−1 (7D5) to 16 g l−1 (EM).
Next, to enhance the difficult oxidation of aniline at pH 3, we focused the engineering of laccase on the catalytic pocket. With this purpose, we took advantage of computational simulation by using PELE (Protein Energy Landscape Exploration) and QM/MM (quantum mechanics/molecular mechanics) calculations.31 Two mutations, N207S and N263D, were predicted to lower the interaction energy of the protonated aniline in the binding pocket, and increase the spin density and electron transfer from the anilinium cation to the catalytic T1 through His455. Both mutations were introduced in EM to obtain the double mutated variant (DM) which was produced in S. cerevisiae, purified and characterized.32 The latter showed improved catalytic activity towards aromatic amines, without jeopardizing laccase stability. By contrast, N263D mutation alone decreased 4-fold the production of the enzyme, and reduces in 27% the activity with aniline respecting DM (Fig. S2†) confirming the synergism between both mutations.
Thereafter, the repeated occurrence of mutations in residue 454 during this and related evolution campaigns led us to explore this position to enhance the catalytic activity towards anilines at pH 3 (Fig. 3).23,28 Residue 454 is adjacent to His 455 that coordinates T1 copper and it is involved in the binding of the reducing substrate and electron withdrawal and transfer to T1 copper.33 As aforementioned, mutation F454S (appeared in 9H2) induced an important activity improvement in 7D5 laccase, although it entailed a significant decrease of stability (Fig. 2). Saturation mutagenesis of Phe 454 in DM variant led to selection of mutations F454H, F454T, F454P and, again, F454S, with significant TAI values on aromatic amines at pH 3. These 454-mutated variants were produced in flasks and the activities and acidic stabilities of the crude enzymes were compared with aniline as substrate (Fig. 3A). Mutation F454H (variant 1G2) followed by F454S (variant 2H8) enhanced the most the oxidation of aniline at acid pH. However, the 12-fold increment of activity of 1G2 variant respecting DM significantly jeopardized the stability to acid pH and thermostability (T50) (Fig. 3B and D). This destabilizing trend was even more pronounced in variants 2H8 (F454S) and 1E2 (F454T). Conversely, mutation F454P in PK2 variant induced a 6-fold increment of activity, while maintained the stability to pH 3 and kept the T50 value closer to that of DM.
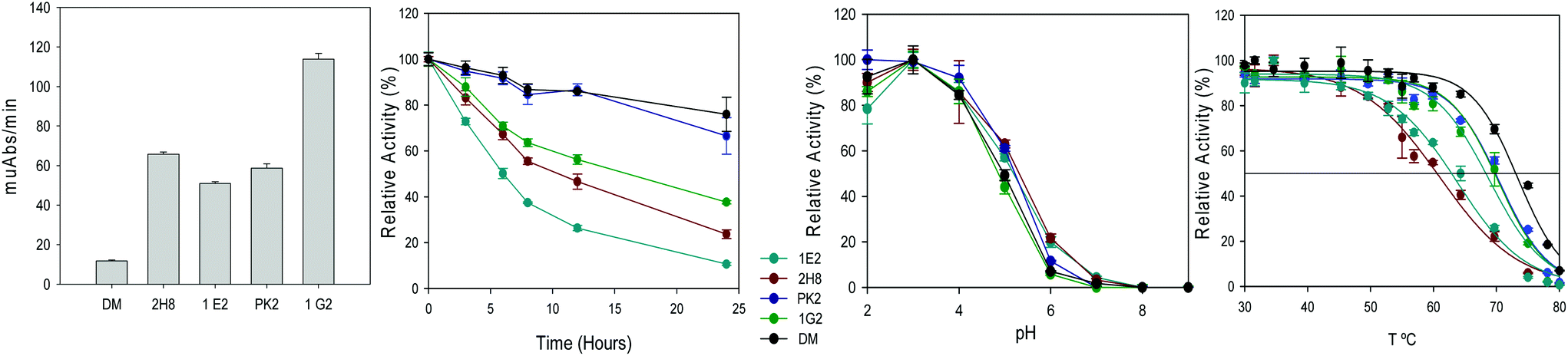 | ||
| Fig. 3 Oxidation of 300 mM aniline at pH 3 (A), residual activity after incubation at pH 3 (B), optimum pH with ABTS (C), and T50 (10 min) curves (D) for DM (F454) and its 454-mutated variants 2H8 (F454S), 1E2 (F454 T), PK2 (F454P) and 1G2 (F454H) (crude enzymes). | ||
To sum up, serine and threonine polar residues in position 454 heavily jeopardize the enzyme stability at acid pH and at high temperature, while basic histidine or nonpolar proline, especially the latter, scarcely affect protein stability. Previous studies have demonstrated that thermophilic enzymes usually are mostly made of hydrophobic and charged residues, showing smaller proportion of uncharged polar residues.34 Recently, a highly stable laccase towards acid pH and high temperature has been developed by directed evolution through increasing the hydrophobicity of the T1 copper environment.25
Variant PK2 was selected as parent for the next evolution round. In order to improve its performance as biocatalyst, we put the attention on former results obtained during the development of 7D5 laccase.5 In that study, most of the stable chimeras obtained from the DNA shuffling of PM1 and Pycnoporus cinnabarinus laccases shared a number of residues from the C-terminal tail of P. cinnabarinus parent, although they had higher total sequence identity with PM1 laccase. Since the C-terminal tail of 7D5 laccase differs from those of other counterparts obtained in the same directed evolution campaign, we modified it accordingly. Specifically, the C-terminal tail of the stable 3A4 laccase differs in four mutations from 7D5 (T487I, S492D, D494S, Q496L). We introduced these mutations in PK2 by replacing the last ten amino acids by those from 3A4 using in vivo overlap extension, IVOE (Fig. 4).35 The new variant RY2 showed a remarkable stability at the target reaction conditions and also at high temperature compared with its parent PK2. In line with these finding, a small library of chimeric laccases has been recently generated from three fungal laccases, two of which are the 7D5 parents, using SCHEMA RASPP to guide the recombination of protein blocks. Most of the stable variants selected shared also the C-terminal block from P. cinnabarinus laccase.36
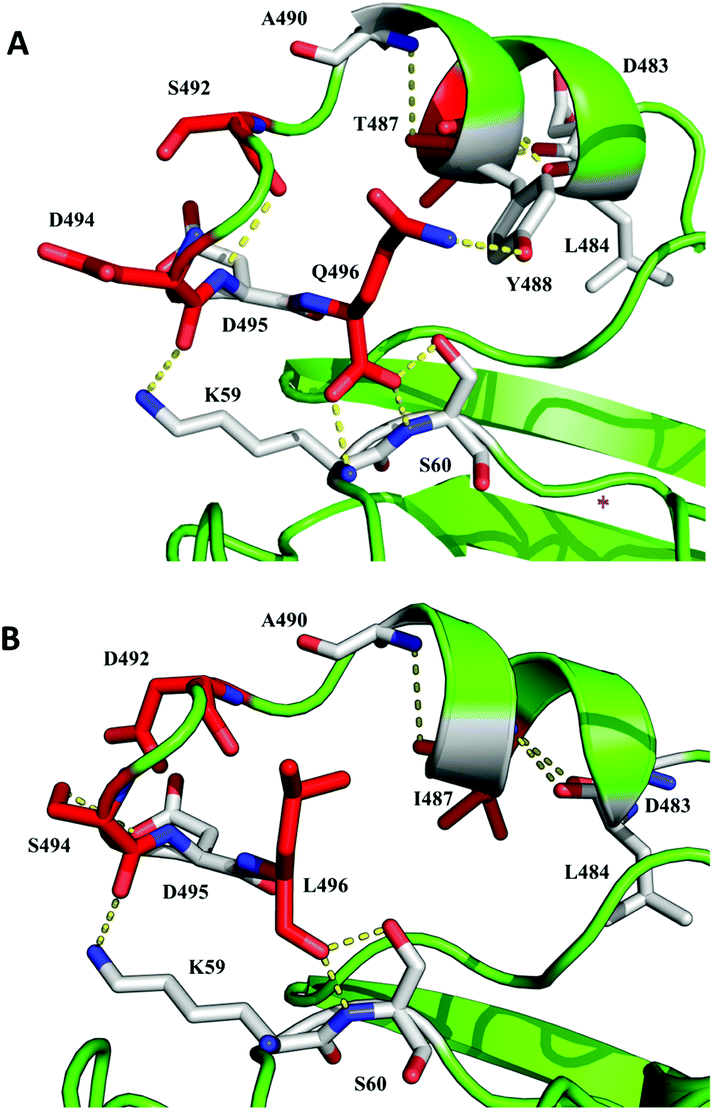 | ||
| Fig. 4 Detail of the C-terminal tail in parent laccase 7D5 (A) and final variant RY2 engineered in this study (B). Mutated residues are shown as red-carbon sticks and residues with which they establish polar contacts are depicted as white-carbon sticks. Based on PDB entry 6H5Y. | ||
Laccase production was significantly raised through 7D5 engineering pathway. To the major contribution of mutations Aα20T and Qα32H (in the α pre-proleader), which upgraded secretion in flask cultures from 3 mg l−1 of 7D5 parent to 16 mg l−1 of EM and DM variants, the added effect of mutation F454P further raised enzyme production up to 25 mg l−1 in PK2 and RY2 variants. This production rate is, as far as we know, the highest ever reported for the heterologous expression of basidiomycete laccases in S. cerevisiae.26,37 The yeast offers remarkably advantages as host for the engineering of eukaryotic enzymes,22 but the expression yields are noticeably poor. Thanks to elevated cell densities under the control of strong promoters, P. pastoris offers superior expression yields and is frequently used as expression system for the production of fungal enzymes,26,27,38 including up-scaling of those engineered in S. cerevisiae.37 By contrast, the protein yields obtained here in S. cerevisiae are similar or even greater than some reported for P. pastoris.37–40 Moreover, 7D5 laccase and evolved variants can be overproduced at relevant scale in an industrial strain of A. oryzae.17
In total 9 new mutations were selected during the 7D5 engineering pathway: N207S, N263D, F454P, T487I, S492D, D494S and Q496L in the mature laccase sequence (Fig. 5), together with Aα20T and Qα32H in the signal peptide. Altogether, these mutations remarkably boost the activity, stability and production of the enzyme by S. cerevisiae and, consequently, its biotechnological value as biocatalyst (see sections below).
 | ||
| Fig. 5 Cartoon representation of the 3D-structures of the parent laccase 7D5 (A) and the final variant RY2 engineered in this study (B) showing the four copper ions as orange spheres and the residues mutated during the evolution pathway as wheat-colored sticks. Based on PDB entry 6H5Y. | ||
Characterization of the engineered variants
The final laccase RY2, together with PK2, DM and EM variants, were produced in S. cerevisiae, purified to homogeneity, and characterized. EM was used as reference of the parent type, because it has the same mature sequence as 7D5 but it is 5-fold better produced (due to the two mutations of the signal peptide). According to the NetNGlyc 1.0 server the parent laccase and the engineered variants hold two conserved N-glycosylation sites, N54 and N433.41,42 SDS-PAGE of purified enzymes before and after Endo-H deglycosylation showed around a 10% N-glycosylation for all variants (Fig. S3†). However, some hyperglycosylation of RY2 was evidenced by a faint smear at 150–100 kDa that disappeared after treatment with Endo-H, resulting in a MW for deglycosylated RY2 (55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 187 Da) similar to that of deglycosylated EM (54658 Da) according to MALDI/TOF-TOF analysis.
187 Da) similar to that of deglycosylated EM (54658 Da) according to MALDI/TOF-TOF analysis.
Catalytic activity
Laccase activity was evaluated with different substrates: aniline, DMPD and ABTS (used during the screening of the mutant libraries), and 2,6-dimethoxyphenol -DMP- (a phenolic substrate not targeted during the engineering of the enzyme).Optimal pH for the oxidation of ABTS, aniline and DMP by DM, PK2 and RY2 variants were compared with those of the parent type (EM) (Fig. S4A–C,† respectively). The shift in maximum absorbance of Würsten dye with pH precluded the use of DMPD in this comparison. In general, the parent type showed a more acidic profile than the rest. Nevertheless, all variants maintained the maximum ABTS activity at the lowest pH, with some increment of activity at pH 3–5 for the engineering variants (Fig. S4A†). The optimal pH with aniline was shifted from 4 to 5 by mutations N263D and N207S first introduced in DM, although the activity at pH 3 was maintained (Fig. S4B†). Changes in pH profile were more pronounced with DMP, with a clear shift of the optimum pH from 4 to 5 through enzyme evolution. Mutations of DM notably raised laccase activity at pH 5, and mutation F454P, first selected in PK2, further shifted and narrowed the pH profile, with a clear maximum at pH 5 (Fig. S4C†). Mutations of C-terminal tail did not modify the pH profile for DMP of RY2 respecting PK2 variant.
The final variant RY2 displayed improved catalytic constants with all the substrates tested as compared with the parent type (and also with the other variants) (Fig. 6, Table S1†). The oxidation rate (kcat values) of aromatic amines was 3–5 fold enhanced, and around 2 fold for ABTS. Taking into account the shift in optimal pH for DMP through the engineering pathway, reactions were carried out at pH 4 (parent type's optimum) and pH 5 (optimum for PK2 and RY2). RY2 maintained the oxidation rate of the parent type with DMP, which widens its applicability as biocatalyst. In general, Km was not improved during the enzyme engineering (the use of saturation concentrations of substrate during the screenings is likely to contribute to this). However, this is not crucial for the industrial application of biocatalysts taking into account substrate is added in excess. Finally, none of the modifications observed in the kinetic constants were related to changes in the laccase's redox potential, given the equal E° of the T1 copper (0.76 ± 0.01 V vs. NHE) of the parent laccase and DM and PK2 variants (RY2's redox potential is supposed to be also unaltered due to the distal location of C-terminal mutations respecting the T1 site).
 | ||
| Fig. 6 Enhancement of catalytic activity (kcat) from the parent laccase (EM) to the last engineered variant (RY2) for the oxidation of different substrates. | ||
The active-site mutations of DM and the C-terminal mutations of RY2 made the most important contributions to expand the oxidation of aromatic amines (pH 3) by laccase. Surprisingly, we found no progress in the kcat values of PK2 variant with respect to DM for DMPD and aniline (or ABTS), although we had observed important improvement on laccase activity during the screening of the saturation mutagenesis library (on Phe454 of DM) and also during the comparison of the selected 454-mutated variants produced in flask cultures (Fig. 3).
To rationalize the molecular mechanism behind the activity of PK2 and the other 454-mutated variants, we used PELE to study the substrate migration and binding. Substrate positioning has been previously shown to correlate with increases in activity of laccases.31 From PELE simulations we extracted a profile of the ligand binding energy with respect to the distance between the N1 atom of aniline and the NE2 atom of His455, which we will refer as N–N distance (Fig. 7A). This shows the profile for DM simulation, but those from the 454 mutations show a remarkable similarity in terms of minima values and topology. The profiles for the five laccase variants feature two main minima (marked as 1 and 2 in Fig. 7A). The first one consists of those poses with a low N–N distance that correspond to catalytic-like conformation, with a peak in interaction energy of −60 kcal mol−1. The second minimum, a non-catalytic one, is located at approximately 12 Å of N–N distance and a peak in interaction energy of −70 kcal mol−1. While the shape and energy values of these regions are very similar for both DM and the 454-mutated variants, the population of each minimum, measured as the number of simulation frames included in each region, is different from DM to the mutated systems. In Fig. 7B, the percentage of the simulation frames located in regions 1 and 2 is shown for all the enzyme variants. For DM, the two percentages are almost exactly equal, with a difference of 1%. This ratio is shifted in the 454-mutated variants, favoring the catalytic minimum with varying values. The notable shift in population for 2H8 (F454S) and 1E2 (F454T) variants indicates an important increase in catalytic conformations. By contrast, these mutations are the most deleterious for enzyme stability (as shown in Fig. 3B and D). The poor increment in population in the catalytic minima for PK2 correlates with the null progress of their kcat values respecting DM, but it does not correlate with the activity improvement observed with crude enzymes (Fig. 3A) These differences are explained by the effect that mutations on Phe454 may exert on laccase production (together with laccase activity). In fact, laccase yields rose from 16 mg l−1 in DM to 25 mg l−1 in PK2 (F454P), thus explaining the selection of PK2 during the screening.
 | ||
| Fig. 7 Analysis of minima during PELE diffusion of aniline on the 454-laccase variants. Interaction energy vs distance between aniline N1 and His455 NE2 atoms in laccase DM. Region 1 includes 0–8.5 Å distances (catalytic and close to catalytic poses) while region 2 includes poses at 8.5–15 Å distances. The two regions are delimited by interaction energies between −10 and −80 kcal mol−1. Similar minima were obtained for the 2H8 (F454S), 1E2 (F454T), PK2 (F454P) and 1G2 (F454H) variants (A). Percentage population of each region in the number of simulation frames for laccase DM and the above variants (B). | ||
Enzyme stability
The stability to pH 2–9 of the final variant RY2 was compared with those of the parent type (EM) and former variant PK2 (Fig. 8, purified enzymes). The loss of stability at basic pH observed in PK2 was recovered in RY2 variant to values even higher than those of the parent type. Besides, the final variant showed superior stability at pH 3. | ||
| Fig. 8 Residual activities of parent type EM (A), and PK2 (B) and RY2 (C) variants during 24 h incubation at pH 2–9. Activities of the purified enzymes at different incubation times (triplicates measured with ABTS, pH 3) are depicted as percentages of the initial activity at pH 3. | ||
The engineering of laccase C-terminal tail also produced a noteworthy increment of T50 (10 min) in RY2 variant with T50 = 73 °C, whereas the rest of purified variants (EM, DM and PK2) displayed T50 values around 65 °C (Fig. 9). Thermal inactivation assays at 50–80 °C for the four variants showed slightly improvement of half-lives from EM to DM that were decreased in PK2 variant (Table 1). Thereafter, the mutations introduced in PK2 C-terminal tail seemed to stabilize the enzyme, increasing the half-lives and lowering the inactivation constants in RY2 variant. The significantly lower activation energy (Ea), calculated from the slope of Arrhenius plot, for the last evolved variant, indicates a lesser sensitivity to temperature changes as compared with the parental and the intermediate variants (Table 1).
 | ||
| Fig. 9 T 50 (10 min) curves of purified parent type EM (circles), and DM (triangles), PK2 (diamonds) and RY2 (squares) variants. | ||
| T (°C) | EM | DM | PK2 | RY2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t 1/2 (h) | k d (h−1) | E a (kJ mol−1) | t 1/2 (h) | k d (h−1) | E a (kJ mol−1) | t 1/2 (h) | k d (h−1) | E a (kJ mol−1) | t 1/2 (h) | k d (h−1) | E a (kJ mol−1) | |
| 50 °C | 13.05 | 0.05 | 201.1 | 19.25 | 0.04 | 209.8 | 13.59 | 0.05 | 204.3 | 22.43 | 0.03 | 162.1 |
| 60 °C | 2.88 | 0.24 | 3.04 | 0.23 | 2.01 | 0.34 | 2.03 | 0.34 | ||||
| 70 °C | 0.29 | 2.43 | 0.31 | 2.23 | 0.18 | 3.76 | 0.36 | 1.93 | ||||
| 80 °C | 0.02 | 29.60 | 0.03 | 27.35 | 0.02 | 30.56 | 0.04 | 19.30 | ||||
We evaluated the possible contribution of protein hiperglycosylation to enhanced thermostability in RY2 laccase by comparing the stability at 65 °C of the glycosylated and deglycosylated forms of this variant with those from the glycosylated and deglycosylated forms of the parent type (EM) (Fig. S5†). The deglycosylated form of RY2 was slightly less stable than the glycosylated form, exactly the same as observed in the parent type (which is not hyperglycosylated), thus evidencing hyperglycosylation is not the main responsible for RY2's thermostability.
The effect of temperature on laccase structure was analyzed by circular dichroism (CD) by monitoring the changes in the ellipticity spectrum of the protein that are produced by perturbations in the secondary structures.43 First, a ramp temperature between 50 and 95 °C was recorded at 220 nm (characteristic band of α-helices) for RY2 and PK2 variants and parent type (EM). No loss of secondary structures could be observed and consequently no apparent Tm could be calculated for any of the laccase variants assayed. Therefore, we incubated the enzymes at 100 °C during 24 h and measured the changes in their far UV CD spectra at different times (Fig. 10). The initial spectra presented two dichroic bands, a strong minimum at 216 nm and a maximum around 196 nm, typical of antiparallel β-sheet proteins.44 The conformation of the parent type (EM) was maintained during the first 2 h of incubation at 100 °C. Thereafter, the gradual loss of ellipticity revealed the loss of secondary structures until total denaturalization of the enzyme after 24 h at 100 °C. The drop in ellipticity observed in PK2 CD spectrum after 2 h at 100 °C and its total denaturalization after 5 h, denotes the significantly diminished structural stability of this variant. On the contrary, an important recovery of stability was observed in the final variant, RY2 whose ellipticity CD spectrum was slightly modified during the first 8 h of incubation at 100 °C (Fig. 10C). These results suggest that the four mutations of RY2 C-terminal tail contribute to the structural stabilization of the enzyme hindering the prompt denaturalization of the protein.
 | ||
| Fig. 10 CD spectra for the thermal denaturation assay of parent type EM (A), and PK2 (B) and RY2 (C) variants after different incubation times at 100 °C. | ||
To understand the reason for this stability improvement, we run MD simulations using OpenMM as the MD engine and Amber ff14SB for proteins. Using these simulations we analyzed the flexibility of the C-terminal residues (defined as the residues between Ala480 and Gln496, or Leu496 in the mutated variant) by calculating the root-mean square fluctuation (RMSF) of those residues in PK2 and RY2 variants, at 27 °C or 67 °C (Fig. 11). To calculate the RMSF values we: (i) discarded the first 100 ns of each trajectory, avoiding possible biases imposed by the initial structures; and (ii) used the average structure as a representative reference. The RMSF plot of RY2 variant showed significantly larger mobility of C-terminal residues at 27 °C, which did not increase too much when moving to 67 °C. PK2, however, shows a large increase in mobility when increasing the temperature. The large increase in RY2's mobility at 27 °C seems to originate the loosening of the secondary structure of the C-terminal tail, most likely as a result of the Thr487 mutation. When monitoring the three hydrogen bonds of this short alpha helix: Ala480-Leu484, Asp483-Thr487 (Ile487 in the RY2 variant) and Pro486-Ala490, the original PK2 variant maintains the three H bonds 24.1% of the time, while for RY2 species it only happens 3.7% of the simulation time. In addition, the mutations of the C-terminal entail the loss of H bonds with neighbor residues (Fig. 4), contributing to increase the mobility of the region. Such an increase in mobility in flexible regions has recently been observed when analyzing other thermostable enzymes.45 Thus, higher flexibility of the C-terminal region in RY2 helps in neutralizing the destabilization caused by the larger thermal fluctuations at higher temperatures, which could allow the rest of the protein to maintain the native structure and remain active. The effect of C-terminal on the enzymatic thermal stability was described in 1985 by Arnold and collaborators.46 In that study, the thermostability of an esterase was enhanced due to six different mutations clustered in the C-terminal region of the enzyme.
 | ||
| Fig. 11 RMSF per residue of the C-terminal region, defined as the last 16 residues, from Ala480 to Gln496 (Leu496 in the mutated variant). The RMSF values are shown for each variant at temperatures of 27 and 67 °C. (A) RMSF for RY2 variant (B) RMSF for PK2 variant. Histogram of the minimum distance between residues 494, 495 or 496 to the TNC for PK2 and RY2 at 27 °C (C). Snapshot showing the closer position of the C-terminal tail for RY2 system, copper ions are shown in VDW representation, the TNC channel is shown in a white mesh surface and C-terminal residues are shown in ball-and-stick representation and labeled (D). | ||
As regards the increment of activity showed by RY2 towards all the substrates assayed, the analysis of the MD trajectories reveal that the new C-terminal region directly interacts with the entrance of the water channel that forms the access route to the type-3 copper sites in the TNC (Fig. 11C).2,47 In particular, we observed how the hydrophobic substitution at position 496 partially occupies the channel (Fig. 11D), which significantly modifies its hydrophobicity and could affect O2/H2O traffic.2 In fact, we observed a better interaction between the mutated C-terminal and Asp 457 (Fig. S6†). Mutation D494S changes the charge of the distal region of the C-terminal from negative to neutral, which might lead to modifications in the electrostatic environment of TNC. A more positive charge of TNC environment has been recently described to increase the redox potential of the catalytic native intermediate (NI), strongly influencing the overall laccase activity.2
The relationship of the C-terminal tail with the enzyme kinetic behavior has been observed in other fungal laccases. Ascomycete laccases are characterized by elongated C-terminal tails, which block the TNC tunnel if they are not correctly processed, leading to dramatic changes in the catalytic behavior of these enzymes.48,49 On the other hand, substitution of the 11 amino acids of the C-terminal region of T. versicolor basidiomycete laccase with a single cysteine residue significantly reduces the redox potential of the copper T1.50 While, the last 18 amino acids in the C-terminal tail of other basidiomycete laccase from P. eryngii seem to play a critical role in the activity, stability and kinetics of the enzyme.51 Although the above laccases hold elongated C-terminal tails that lie closer to the entrance of TNC than in our case, mutations added to PK2 C-terminal tail make it more flexible and may entail a displacement that could improve the access to the TNC channel.52,53 This hypothesis will be verify when the RY2 variant is crystallized, taking advantage of the high production yields obtained in yeast.
Application case studies
Crude (unpurified) variants of laccase 7D5 developed and produced in S. cerevisiae were tested for the synthesis of green polyaniline and compared with the parent laccase using described conditions (Fig. 12A).17 The synthesis of the polymer was monitored by the increase of absorbance at 800 nm, typical for the conductive form (Emeraldine salt).54 The use of EM entailed an important increment of polymerization (respecting the use of 7D5 laccase) due to its superior secretion by the yeast. Variant DM also raised the polymerization rate attained with EM after 8 h reaction due to the better oxidation of aniline. Finally, PK2 and RY2 variants slightly increased the polymerization rates obtained with DM. | ||
| Fig. 12 Polymerization of 15 mM aniline by 0.594 mg ml−1 of 7D5 (red), EM (purple), DM (orange), PK2 (green) and RY2 (blue) crude enzymes in the presence of 5 mM SDBS as template (A). Multifiber test from PANI synthesized with the engineered laccase variant (B), and multifiber test of the acid dye synthesized by the engineered laccase variant and Novozyme 51003 laccase with 1-naphthol as precursor (C). | ||
In a next step, the last variant engineered towards aniline oxidation, that is PK2, was expressed in the industrially relevant host A. oryzae (Novozymes), under the same conditions used for the parent 7D5 laccase.17 The purified engineered variant expressed in A. oryzae augmented the PANI yields from 75% to 87% after 24 h of polymerization reaction.
Aniline is an important precursor for the synthesis of different dyestuff as indo dyes, aniline black (pernigraline), mauveine or aniline blue.55–57 Then, performance as biocatalyst of the enzyme overproduced in A. oryzae for the synthesis of polyaniline and a naphtol-derived compound as dyes was assessed at relevant industrial conditions at SETAŞ A.S (Turkey), a leader company in chemistry and industrial colour processes. The textile dyeing capacities of the resulting colorants were evaluated using standard industrial tests. The type of dye, the reproducibility of the colour and colour depth, exhaustion of the bath at the end of the dyeing process and fastness properties of the new dyes were determined.
According to the multifiber dyeing test, the enzymatically synthesized polyaniline produced diverse colour strengths on different fabrics. The best dyeing efficiency was obtained on acrylic, nylon and acetate fabrics (Fig. 12B). A subsequent fiber dyeing test on acrylic fiber demonstrated the excellent dyeing efficiency of the enzymatic synthesized PANI, with high exhaustion of the dyeing bath and strong colour fastness to light (Fig. S7A†).
As regards the synthesis of naphtol-derived dye, the performance of the engineered laccase variant was compared with that of the commercial laccase Novozyme 51003. The new dye is in the yellow scale of the spectra (maximum absorbance at 470 nm) and it's defined as acid dye by its dyeing properties. The new acid dye obtained with both laccases rendered an intense dye fixation for wool and nylon fabrics in the multifiber test (Fig. 12C). Due to the colour shade similarity with the new dye, Nyloset Brown N2R from SETAŞ dye range was used as reference to compare the dyeing efficiency of the new colorant. The exhaustion of the bath at the end of the second dyeing process of nylon fibres with the dye synthesized with the engineered laccase showed a 55% STR-SUM, which indicates lower dye efficiency compared with PANI (Fig. S7B†). The colour fastness to light was also lower than the observed for polyaniline. However, the dye efficiency of the new acid dye was higher when it was synthesized by the engineered enzyme than by the commercial laccase, and very close to the dye efficiency of Nyloset Brown N2R (Fig. S7C†). Also, the colour strengths of the new dye and the Nyloset Brown N2R are comparable, even when the new dye are not concentrated enough. It is worth mentioning that Nyloset Brown N2R synthesis requires very acid pH (<1) and very toxic reagents as NaNO2.58 By contrast, we demonstrate here the enzymatic synthesis of similar performing dyes using notably milder conditions.
Conclusions
The intrinsic low catalytic requirements, broad substrate promiscuity, high redox potential and stability of certain basidiomycete laccases make them excellent candidates to develop new tailor-made biocatalysts for different oxidation reactions. However, their difficult heterologous expression constitutes a main bottleneck first for engineering and then for application. So far, they are not actively expressed in E. coli and the enzyme yields rendered by S. cerevisiae, our preferred host for laccase engineering, are very low. By contrast, the variants engineered here are produced by the yeast at remarkable levels and can be overexpressed by A. oryzae at relevant industrial scale. This allowed us to prove their excellent properties as biocatalysts for feasible synthesis of high-performing organic dyes without adding any redox mediators. The viable application of the engineered laccases in an industrial environment to replace toxic chemical catalysts and harsh industrial conditions by milder ones is an important advance towards the development of green chemistry industrial processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Patrizia Gentili (Università degli Studi La Sapienza, Roma) for the redox potential measurements and Dr Iván Ayuso (CIB, CSIC, Madrid) for the assistance with CD assays. This work has been funded by the INDOX EU project (KBBE- 2013-7-613549), the Spanish projects BIO2017-86559-R and CTQ2016-79138-R and the BBI JU project WoodZymes (H2020-BBI-JU-792070). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- R. Mehra, J. Muschiol, A. S. Meyer and K. P. Kepp, Sci. Rep., 2018, 8, 1–16 CrossRef CAS PubMed.
- A. Sekretaryova, S. M. Jones and E. I. Solomon, J. Am. Chem. Soc., 2019, 141, 11304–11314 CrossRef CAS PubMed.
- P. Baldrian, FEMS Microbiol. Rev., 2006, 30, 215–242 CrossRef CAS PubMed.
- C. M. Rivera-Hoyos, E. D. Morales-Álvarez, R. A. Poutou-Piñales, A. M. Pedroza-Rodríguez, R. RodrÍguez-Vázquez and J. M. Delgado-Boada, Fungal Biol. Rev., 2013, 27, 67–82 CrossRef.
- I. Pardo, A. I. Vicente, D. M. Mate, M. Alcalde and S. Camarero, Biotechnol. Bioeng., 2012, 109, 2978–2986 CrossRef CAS PubMed.
- S. Riva, Trends Biotechnol., 2006, 24, 219–226 CrossRef CAS PubMed.
- D. Rodríguez-Padrón, A. D. Jodlowski, G. De Miguel, A. R. Puente-Santiago, A. M. Balu and R. Luque, Green Chem., 2018, 20, 225–229 RSC.
- A. Franco, S. Cebrián-García, D. Rodríguez-Padrón, A. R. Puente-Santiago, M. J. Munoz-Batista, A. Caballero, A. M. Balu, A. A. Romero and R. Luque, ACS Sustainable Chem. Eng., 2018, 6, 11058–11062 CrossRef CAS.
- D. M. Mate and M. Alcalde, Microb. Biotechnol., 2017, 10, 1457–1467 CrossRef CAS PubMed.
- A. R. Puente-Santiago, D. Rodríguez-Padrón, X. Quan, M. J. Muñoz Batista, L. O. Martins, S. Verma, R. S. Varma, J. Zhou and R. Luque, ACS Sustainable Chem. Eng., 2019, 7, 1474–1484 CrossRef CAS.
- M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS.
- G. Shumakovich, A. Streltsov, E. Gorshina, T. Rusinova, V. Kurova, I. Vasil'eva, G. Otrokhov, O. Morozova and A. Yaropolov, J. Mol. Catal. B: Enzym., 2011, 69, 83–88 CrossRef CAS.
- J. Zhang, F. Zou, X. Yu, X. Huang and Y. Qu, Colloid Polym. Sci., 2014, 292, 2549–2554 CrossRef CAS.
- I. S. Vasil'eva, O. V. Morozova, G. P. Shumakovich, S. V. Shleev, I. Y. Sakharov and A. I. Yaropolov, Synth. Met., 2007, 157, 684–689 CrossRef.
- T. Hino, T. Namiki and N. Kuramoto, Synth. Met., 2006, 156, 1327–1332 CrossRef CAS.
- Z. Wei, Z. Zhang and M. Wan, Langmuir, 2002, 18, 917–921 CrossRef CAS.
- F. de Salas, I. Pardo, H. J. Salavagione, P. Aza, E. Amougi, J. Vind, A. T. Martínez and S. Camarero, PLoS One, 2016, 11, 1–18 Search PubMed.
- A. M. Wallraf, H. Liu, L. Zhu, G. Khalfallah, C. Simons, H. Alibiglou, M. D. Davari and U. Schwaneberg, Green Chem., 2018, 20, 2801–2812 RSC.
- S. Scheiblbrandner, E. Breslmayr, F. Csarman, R. Paukner, J. Führer, P. L. Herzog, S. V. Shleev, E. M. Osipov, T. V. Tikhonova, V. O. Popov, D. Haltrich, R. Ludwig and R. Kittl, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- M. Zumárraga, T. Bulter, S. Shleev, J. Polaina, A. Martinez, F. J. Plou, A. Ballesteros, A. Martínez-Arias, F. J. Plou, A. Ballesteros and M. Alcalde, Chem. Biol., 2007, 14, 1–18 CrossRef PubMed.
- I. Pardo, D. Rodríguez-Escribano, P. Aza, F. de Salas, A. T. Martínez and S. Camarero, Sci. Rep., 2018, 8, 1–10 CrossRef CAS PubMed.
- D. Gonzalez-Perez, E. Garcia-Ruiz and M. Alcalde, Bioeng. Bugs, 2012, 3, 172–177 Search PubMed.
- D. Maté, C. García-Burgos, E. García-Ruiz, A. O. Ballesteros, S. Camarero and M. Alcalde, Chem. Biol., 2010, 17, 1030–1041 CrossRef PubMed.
- S. Camarero, I. Pardo, A. I. Cañas, P. Molina, E. Record, A. T. Martínez, M. J. Martínez and M. Alcalde, Appl. Environ. Microbiol., 2012, 78, 1370–1384 CrossRef CAS PubMed.
- I. Mateljak, E. Monza, M. F. Lucas, V. Guallar, O. Aleksejeva, R. Ludwig, D. Leech, S. Shleev and M. Alcalde, ACS Catal., 2019, 9, 4561–4572 CrossRef CAS.
- A. Kunamneni, S. Camarero, C. García-Burgos, F. J. Plou, A. Ballesteros and M. Alcalde, Microb. Cell Fact., 2008, 7, 1–17 CrossRef PubMed.
- P. Alessandra, P. Cinzia, G. Paola, F. Vincenza and G. Sannia, Bioeng. Bugs, 2010, 1, 252–262 Search PubMed.
- D. M. Mate, D. Gonzalez-Perez, M. Falk, R. Kittl, M. Pita, A. L. De Lacey, R. Ludwig, S. Shleev and M. Alcalde, Chem. Biol., 2013, 20, 223–231 CrossRef CAS PubMed.
- V. Frappier, M. Chartier and R. J. Najmanovich, Nucleic Acids Res., 2015, 43, W395–W400 CrossRef CAS PubMed.
- K. Kataoka, S. Hirota, Y. Maeda, H. Kogi, N. Shinohara, M. Sekimoto and T. Sakurai, Biochemistry, 2011, 50, 558–565 CrossRef CAS PubMed.
- E. Monza, M. F. Lucas, S. Camarero, L. C. Alejaldre, A. T. Martínez and V. Guallar, J. Phys. Chem. Lett., 2015, 6, 1447–1453 CrossRef CAS PubMed.
- G. Santiago, F. De Salas, M. F. Lucas, E. Monza, S. Acebes, Á. T. Martinez, S. Camarero and V. Guallar, ACS Catal., 2016, 6, 5415–5423 CrossRef CAS.
- I. Pardo and S. Camarero, Cell. Mol. Life Sci., 2015, 72, 897–910 CrossRef CAS PubMed.
- M. M. Gromiha, M. C. Pathak, K. Saraboji, E. A. Ortlund and E. A. Gaucher, Proteins: Struct., Funct., Bioinf., 2013, 81, 715–721 CrossRef CAS PubMed.
- M. Alcalde, M. Zumarraga, J. Polaina, A. Ballesteros and F. Plou, Comb. Chem. High Throughput Screening, 2006, 9, 719–727 CrossRef CAS PubMed.
- I. Mateljak, A. Rice, K. Yang, T. Tron and M. Alcalde, ACS Synth. Biol., 2019, 8, 833–843 CrossRef CAS PubMed.
- D. M. Mate, D. Gonzalez-Perez, R. Kittl, R. Ludwig and M. Alcalde, BMC Biotechnol., 2013, 13, 38 CrossRef CAS PubMed.
- F. S. Hartner, C. Ruth, D. Langenegger, S. N. Johnson, P. Hyka, G. P. Lin-Cereghino, J. Lin-Cereghino, K. Kovar, J. M. Cregg and A. Glieder, Nucleic Acids Res., 2008, 36, 1–15 CrossRef PubMed.
- D. M. Soden, J. O'Callaghan and A. D. W. Dobson, Microbiology, 2002, 148, 4003–4014 CrossRef CAS PubMed.
- L. Otterbein, E. Record, S. Longhi, M. Asther and S. Moukha, Eur. J. Biochem., 2000, 267, 1619–1625 CrossRef CAS PubMed.
- M. Orlikowska, M. de J. Rostro-Alanis, A. Bujacz, C. Hernández-Luna, R. Rubio, R. Parra and G. Bujacz, Int. J. Biol. Macromol., 2018, 107, 1629–1640 CrossRef CAS PubMed.
- N. J. Christensen and K. P. Kepp, PLoS One, 2013, 8 DOI:10.1371/journal.pone.0061985.
- K. Chattopadhyay and S. Mazumdar, Biochemistry, 2000, 39, 263–270 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- L. Julió Plana, A. D. Nadra, D. A. Estrin, F. J. Luque and L. Capece, J. Chem. Inf. Model., 2019, 59, 441–452 CrossRef PubMed.
- L. Giver, A. Gershenson, P. O. Freskgard and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12809–12813 CrossRef CAS PubMed.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS PubMed.
- M. Andberg, N. Hakulinen, S. Auer, M. Saloheimo, A. Koivula, J. Rouvinen and K. Kruus, FEBS J., 2009, 276, 6285–6300 CrossRef CAS PubMed.
- N. Hakulinen, L.-L. Kiiskinen, K. Kruus, M. Saloheimo, A. Paananen, A. Koivula and J. Rouvinen, Nat. Struct. Biol., 2002, 9, 601–605 CAS.
- M. Gelo-Pujic, H. H. Kim, N. G. Butlin and G. T. R. Palmore, Appl. Environ. Microbiol., 1999, 65, 5515–5521 CAS.
- G. Bleve, C. Lezzi, S. Spagnolo, G. Tasco, M. Tufariello, R. Casadio, G. Mita, P. Rampino and F. Grieco, Protein Eng., Des. Sel., 2013, 26, 1–13 CrossRef CAS PubMed.
- M. Hu, X. Zhou, Y. Shi, J. Lin, M. Irfan and Y. Tao, Appl. Biochem. Biotechnol., 2014, 174, 2007–2017 CrossRef CAS PubMed.
- F. Autore, C. Del Vecchio, F. Fraternali, P. Giardina, G. Sannia and V. Faraco, Enzyme Microb. Technol., 2009, 45, 507–513 CrossRef CAS.
- W. Liu, J. Kumar, S. Tripathy, K. J. Senecal and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 71–78 CrossRef CAS.
- F. J. O. Neill, R. J. Greenwood and J. S. Knapp.
- J. C. Michaelson, Endeavour, 1993, 17, 121–126 CrossRef CAS.
- A. C. Sousa, M. F. M. M. Piedade, L. O. Martins and M. P. Robalo, Green Chem., 2016, 18, 6063–6070 RSC.
- F. Mao and P. Tang, Ranliao Yu Ranse, 2010, 47, 1–3 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc02475a |
| This journal is © The Royal Society of Chemistry 2019 |