
Reaction engineering implications of cellulose crystallinity and water-promoted recrystallization†

Abstract
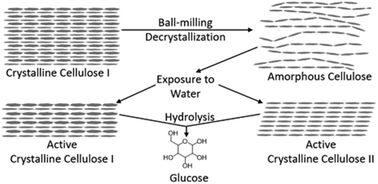
Mechanical decrystallization and water-promoted recrystallization of cellulose were studied to understand the effects of cellulose crystallinity on reaction engineering models of its acid-catalyzed hydrolysis. Microcrystalline cellulose was ball-milled for different periods of time, which decreased its crystallinity and increased the glucose yield obtained from acid hydrolysis treatment. Crystallinity increased after acid hydrolysis treatment, which has previously been explained in terms of rapid hydrolysis of amorphous cellulose, despite conflicting evidence of solvent promoted recrystallization. To elucidate the mechanism, decrystallized samples were subjected to various non-hydrolyzing treatments involving water exposure. Interestingly, all non-hydrolyzing hydrothermal treatments resulted in recovery of crystallinity, including a treatment consisting of heat-up and quenching that was selected as a way to estimate the crystallinity at the onset of hydrolysis. Therefore, the proposed mechanism involving rapid hydrolysis of amorphous cellulose must be incomplete, since the recrystallization rate of amorphous cellulose is greater than the hydrolysis rate. Several techniques (solid-state nuclear magnetic resonance, X-ray diffraction, and Raman spectroscopy) were used to establish that water contact promotes conversion of amorphous cellulose to a mixture of crystalline cellulose I and cellulose II. Crystallite size may also be reduced by the decrystallization-recrystallization treatment. Ethanolysis was used to confirm that the reactivity of the cellulose I/cellulose II mixture is distinct from that of truly amorphous cellulose. These results strongly point to a revised, more realistic model of hydrolysis of mechanically decrystallized cellulose, involving recrystallization and hydrolysis of the cellulose I/cellulose II mixture.
- This article is part of the themed collection: 2019 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

