
Persulfate-mediated synthesis of polyfunctionalized benzenes in water via the benzannulation of alkynes and α,β-unsaturated compounds†

Abstract
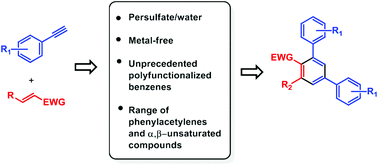
A persulfate-promoted metal-free route in water toward the synthesis of unprecedented polyfunctionalized benzenes is reported. In our approach, the targeted products are delivered in high to moderate yields from phenylacetylenes and α,β-unsaturated compounds via the benzannulation reaction. This method has a good scope and gives access to functionalized benzene rings that offer a wealth of opportunities for further functionalization.
 Please wait while we load your content...
Please wait while we load your content...

