
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtending the design space in solvent extraction – from supercritical fluids to pressurized liquids using carbon dioxide, ethanol, ethyl lactate, and water in a wide range of proportions†
Veronika
Pilařová‡
 a,
Said
Al Hamimi‡
b,
Larissa P.
Cunico
b,
Lucie
Nováková
a and
Charlotta
Turner
*b
a,
Said
Al Hamimi‡
b,
Larissa P.
Cunico
b,
Lucie
Nováková
a and
Charlotta
Turner
*b
aCharles University, Department of Analytical Chemistry, Faculty of Pharmacy in Hradec Králové, Heyrovského 1203, CZ-500 05 Hradec Králové, Czech Republic
bLund University, Department of Chemistry, Centre for Analysis and Synthesis, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: charlotta.turner@chem.lu.se
First published on 11th September 2019
Abstract
Plants and food byproducts contain high-value bioactive compounds like carotenoids, tocopherols, flavonoids, and other phenolic compounds that can potentially be extracted prior to usage of the lower-ranked biomass, all in line with a circular bioeconomy. Extraction is typically performed by leaching using an organic solvent, or alternatively with more advanced techniques such as supercritical fluid extraction (SFE), pressurized liquid extraction (PLE), microwave assisted extraction (MAE) or ultrasound assisted extraction (UAE). With the exception of SFE, mass transfer is enhanced and solvent consumption is minimised by the use of elevated temperature, which has a negative impact on the extraction yield and structural preservation for thermally labile bioactive compounds. Hence, in this study we aim to explore the use of CO2 in extraction solvent mixtures as a viscosity-lowering entrainer. Furthermore, we aim to disregard the instrumental boundaries between SFE and PLE by extending the design space for extraction method development, using solvent mixtures of pressurized carbon dioxide, water, and either ethanol or ethyl lactate, covering a uniquely large area of the solvent's relative permittivity. Such a strategy will inherently enable extraction method optimisation for target compounds differing a lot in polarity, here demonstrated for β-carotene, α-tocopherol, and quercetin in sea buckthorn berry pomace. Our results show that by the use of experimental design, optimum extraction conditions were found for each of the three compounds: supercritical CO2 with 10 vol% of ethanol at 300 bar and 80 °C for β-carotene, liquid CO2/ethanol/water (50/44/6, v/v/v) at 210 bar and 60 °C for α-tocopherol, and liquid CO2/ethyl lactate/water (33/54/13, v/v/v) at 200 bar and 62 °C for quercetin. Extraction rates were fast in all three cases, giving complete extractions within less than 20 min at the highest flow rate explored, 3 mL min−1. The developed extraction methods gave significantly higher extraction yields than published optimised extraction methods based on PLE. Moreover, the optimized methods for the less polar compounds, β-carotene, and α-tocopherol, enabled a significantly lower consumption of organic solvents (30–52%), which makes them greener in comparison with the more conventional PLE extraction methods.
Introduction
Renewable resources such as plants, microalgae, seaweeds, and food industry by-products contain high-value biologically active compounds and constitute an under-utilized source of compounds of value in food and fine chemical production. Traditionally, well-established techniques such as maceration, distillation, and Soxhlet extraction are often employed to obtain bioactive compound extracts, despite the fact these methods are time-consuming, involve the use of a large amount of toxic organic solvents, and often provide relatively low yields of the bioactive compounds. Due to these drawbacks, alternative more efficient extraction methods that utilize green solvents have been explored over the years, including microwave assisted extraction (MAE), ultrasound assisted extraction (UAE), pressurised liquid extraction (PLE), supercritical fluid extraction (SFE), and recently also carbon dioxide-expanded liquid extraction (CXLE).1–4SFE is usually based on the use of supercritical carbon dioxide (scCO2) at pressure and temperature over the critical point, i.e. 74 bar and 31 °C,5 but in practice between 200 and 400 bar and 40 and 80 °C, respectively. Under such conditions, the fluid acquires physicochemical properties such as liquid-like density and gas-like viscosity, giving fast mass transfer and high solubility of non-polar compounds.6–8 However, neat scCO2 does not have sufficient solvent strength to extract polar compounds. For this reason, the addition of 1–15 vol% of the organic modifier, also called co-solvent, is often used to increase the relative permittivity of the fluid. Methanol and ethanol are preferred as co-solvents mixed with scCO2, but also water and ethyl acetate have been used in binary and ternary mixtures to increase the relative permittivity of the extraction solvent and thus the solubility of the compounds to be extracted.8–11
Recently, it has been demonstrated that instead of adding a co-solvent to scCO2, liquid CO2 can be added to an organic solvent, thereby obtaining a solvent of even higher relative permittivity than in the SFE case above (Fig. 1). Such a solvent is commonly called a CO2-expanded liquid (CXL), although other descriptions have been used, such as gas expanded liquid, enhanced fluidity liquid, and binary fluid.12,13 CXLs have similar density to neat organic solvents, however with the benefit of lower viscosity, and tunable relative permittivity.14,15 In comparison with SFE, significantly lower pressures are used, i.e. equipment is simpler to use and of lower-cost. From our point of view, CXLE is an extraction technique that provides enhanced mass transfer without necessarily using elevated temperature,16 which on the contrary is the case for most solvent-based extraction techniques, including PLE, MAE, and UAE. For thermally labile compounds, for instance flavonoids extractable from plants and foods, elevated temperatures cause relatively rapid degradation, especially in the presence of oxygen.17,18 In another situation, elevated temperature might be desired to enhance the desorption of the target-compound from the sample.
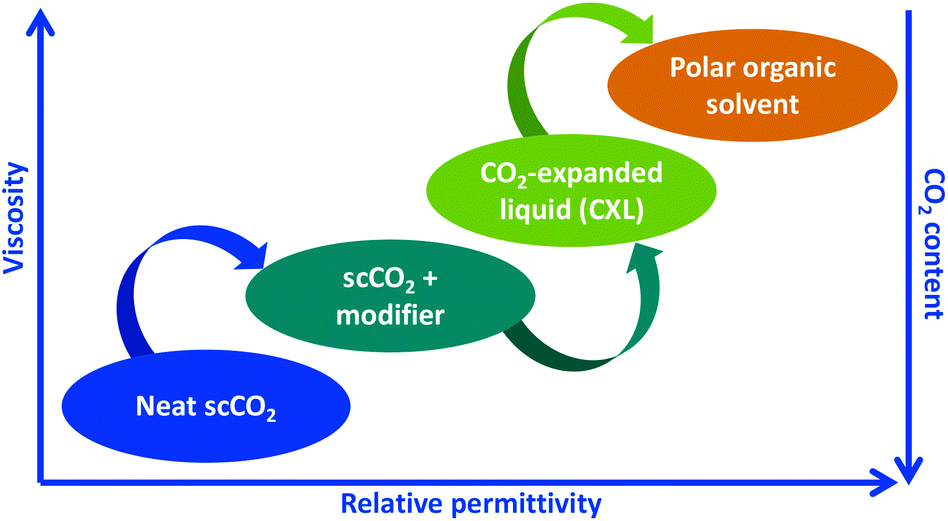 | ||
| Fig. 1 Scheme of physicochemical properties of the green solvent mixture composed of 0 to 100% CO2; from neat scCO2, to organic solvent-modified scCO2, CXL, and finally neat polar organic solvent, for instance ethanol. Redrawn from ref. 15. | ||
So far, SFE, CXLE, and PLE are considered as three completely different extraction techniques. Commercial instruments for SFE and PLE are very different, e.g. a PLE system cannot be used with liquid CO2 as a solvent, and an SFE system might not necessarily support the use of 100% organic solvent. For CXLE there are no commercial instruments, but in some cases a commercial SFE system can be used. In our opinion, it would be of value if mixtures of liquid (or supercritical) CO2 and green solvents such as ethanol, water and ethyl lactate could be used in a wide range of mixture ratios, using one single piece of equipment and one method optimisation. This would however require knowledge of phase equilibria for different mixtures, in order to ensure that the used mixtures are present as a single liquid phase.
The aim of this study is to explore the use of 0 to 90% addition of liquid/supercritical CO2 to green solvent mixtures (“co-solvent”) of ethanol, ethyl lactate and water, for the extraction of three types of bioactive compounds with largely different physicochemical properties, β-carotene, α-tocopherol, and quercetin (Table 1). The sample used for this purpose is sea buckthorn berry pomace, which is a by-product recovered from juice production. In fact, industrial berry pomace is a large untapped source of fibre, has a wide range of antioxidants,19–22 and can be added to food and bakery products to improve nutritional and sensory properties.19 Sea buckthorn berries contain biologically important compounds including ascorbic acid, tocopherols, carotenoids, polyunsaturated fatty acids, flavonoids, and other phenolic compounds, which offer health benefits like antioxidant activity, cardioprotective, immunomodulatory, and anti-cancer activity, hepatoprotective activity, and anti-stress activity.23–26 Hence, this study not only provides for the first time a holistic approach merging SFE, PLE, and CXLE in green solvent extraction, but also shows the potential of using sea buckthorn berry pomace as a high-value renewable source of β-carotene, α-tocopherol, and quercetin.
| Compound | Structure | MW (g mol−1) | Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
|---|---|---|---|
| β-Carotene |
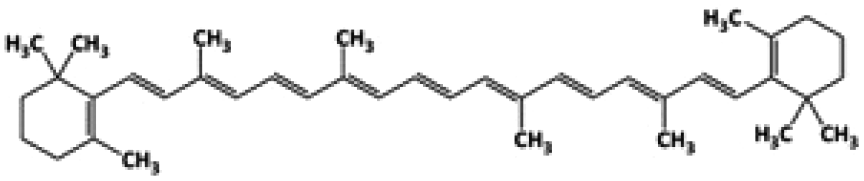
|
536.89 | 13.5 |
| α-Tocopherol |
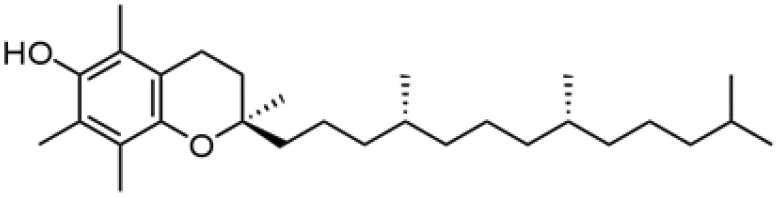
|
430.71 | 10.5 |
| Quercetin |
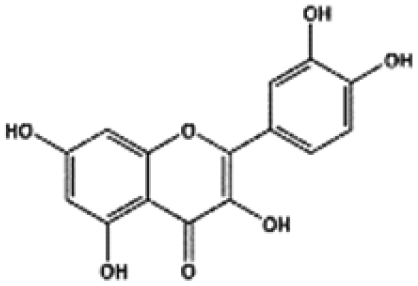
|
302.04 | 2.0 |
Results and discussion
Selection of appropriate green extraction solvents based on Hansen solubility parameters
In extraction, solubility of target compound(s) in the extraction solvent is important to ensure high recovery in a short time. In this study, we aimed to screen and develop green extraction methods based on compressed CO2 mixed with “green” co-solvents such as water, ethanol and ethyl lactate. The aim was also to explore a uniquely wide range of compositions of these solvents (0 to 90 vol% CO2 and 0 to 20 vol% water added to either ethanol or ethyl lactate), as well as a wide range of pressure (100–300 bar) combined with relatively moderate temperatures (40–80 °C), thereby disregarding typical instrumental boundaries between SFE and PLE. In order to assess the suitability of the green solvents investigated here, Hansen solubility parameters (HSP) were initially calculated for pure solvents and mixtures, along with the studied compounds (β-carotene, α-tocopherol, and quercetin). See Table 2 for dispersive, polar and hydrogen bonding interaction of the compounds and solvents investigated in this study. It is clear that CO2, ethanol, ethyl lactate and water demonstrate a wide range of different polar and hydrogen bonding interactions, and different mixtures of these solvents should enable high solubility of the investigated compounds. Note that HSP are merely a theoretical tool, which describes intermolecular interactions based on the group contribution method. They are not by any means an exact method for determining the ideal solvent mixture to dissolve a compound. Furthermore, when conducting extractions from real complex samples (such as sea buckthorn berry pomace as in this study), other aspects should be taken into account, like sample matrix effects and mass transfer limitations. For instance, the addition of CO2 to a polar organic solvent does not only decrease the relative permittivity of the solvent, it also lowers the viscosity and thereby enhances the mass transfer. Furthermore, the addition of water to the solvent does not only alter the solvent strength, but also enhances the solvent's ability to desorb compounds from the sample matrix in cases where the compounds are strongly adsorbed by polar/hydrogen bonding. What can be concluded from Table 2 is that the selected solvents for this study make sense, since they together embrace the wide range of solubility parameters calculated for the target compounds.| Compound | δ D (MPa1/2) | δ P (MPa1/2) | δ H (MPa1/2) | Conditions |
|---|---|---|---|---|
| β-Carotene | 17.4 | 0.8 | 1.7 | Ambient |
| α-Tocopherol | 15 | 1.4 | 2.8 | Ambient |
| Quercetin | 23 | 15.8 | 25 | Ambient |
| ScCO2 | 10.7 | 4.5 | 4.8 | 300 bar, 60 °C |
| Liquid CO2 | 10.5 | 4.4 | 4.9 | 100 bar, 25 °C |
| EL | 16.0 | 7.6 | 12.5 | Ambient |
| EtOH | 15.8 | 8.8 | 19.4 | Ambient |
| Water | 15.5 | 16.0 | 42.3 | Ambient |
| CO2 + EtOH | 9.8 | 4.6 | 5.7 | CO2/EtOH (90/10, v/v), 300 bar, 80 °C |
| CO2 + EtOH + water | 12.1 | 6.7 | 12.7 | CO2/EtOH/water (50/44/6, v/v/v), 210 bar, 60 °C |
| CO2 + EL + water | 13.1 | 7.3 | 13.2 | CO2/EL/water (33/54/13, v/v/v), 200 bar, 62 °C |
Phase diagrams for binary and ternary green solvent mixtures
Once the selection of green solvents has been made, the question to be asked is which proportions of these mixtures can be used in the extraction system. In this study, a continuous flow (dynamic) extraction system is used, in which solvent mixtures are produced continuously in a mixer (T-shaped joint). It is important that the obtained mixtures are homogeneous and occurring as single-phase liquids, which was ensured by using a coil of 200 cm after the T-joint. However, when CO2 is one of the components of a binary or ternary system, the mixture might be found as a single liquid phase or as two to three phases (one vapor phase and one to two liquid phases), all of which depends on the nature of the liquid solvent(s), temperature, pressure, and the molar fraction of CO2. Jessop and Subramaniam classified solvents based on their ability to dissolve CO2.13 Water is classified as class I, since it has insufficient ability to dissolve CO2, whereas ethanol and ethyl lactate are class II solvents, because of their ability to dissolve large amounts of CO2. There are many studies in which phase diagrams for the binary system of CO2 and ethanol and the ternary system of CO2, ethanol and water have been experimentally determined.27–29 However, in the literature there is only one study conducted to investigate the phase behaviour of CO2 and ethyl lactate mixtures,30 but unfortunately, there are no studies on the ternary mixture of CO2, ethyl lactate and water.In this study, the extraction solvent used is found as a pure organic solvent (ethanol or ethyl lactate), a binary system (CO2/ethanol, CO2/ethyl lactate, ethanol/water or ethyl lactate/water) or a ternary system (CO2/ethanol/water or CO2/ethyl lactate/water). To illustrate the phase diagram of the extraction system under each condition, the molar fractions were calculated using the mass of each component delivered from each pump, see the Experimental section. Table S1 (ESI†) shows examples of the calculations for mixtures of water, CO2, and ethanol at different pressures and ratios of CO2 to solvent containing water. Previous studies30,31 showed that the binary systems of CO2 with ethanol or ethyl lactate at 100 bar and above in the range of 30–60 °C are found as a single liquid phase at any molar fraction of CO2. The data obtained by Lim et al.27 were used to illustrate the phase diagram of the ternary system of CO2, ethanol and water at 60 °C and 142 bar (Fig. 2). The explored extraction solvent compositions used in this study (0–90 vol% CO2 added to ethanol, which in turn contained 0–20 vol% of water) show that all the conditions were found as a single liquid phase except when the water was present at 20 vol% in the ethanol and the content of CO2 in the solvent was 45 vol%. For this composition, at 60 °C and 142 bar, the liquid phase co-exists with a vapor phase. Such conditions should be avoided for instruments that employ mixing in-line upstream of the extraction vessel. In the case of ethyl lactate, there is no existing study on the phase diagram of the ternary system of CO2, ethyl lactate and water. Therefore, we are not certain about the state of the investigated conditions. A future study on phase equilibria of CO2 + water + ethyl lactate would certainly be valuable.
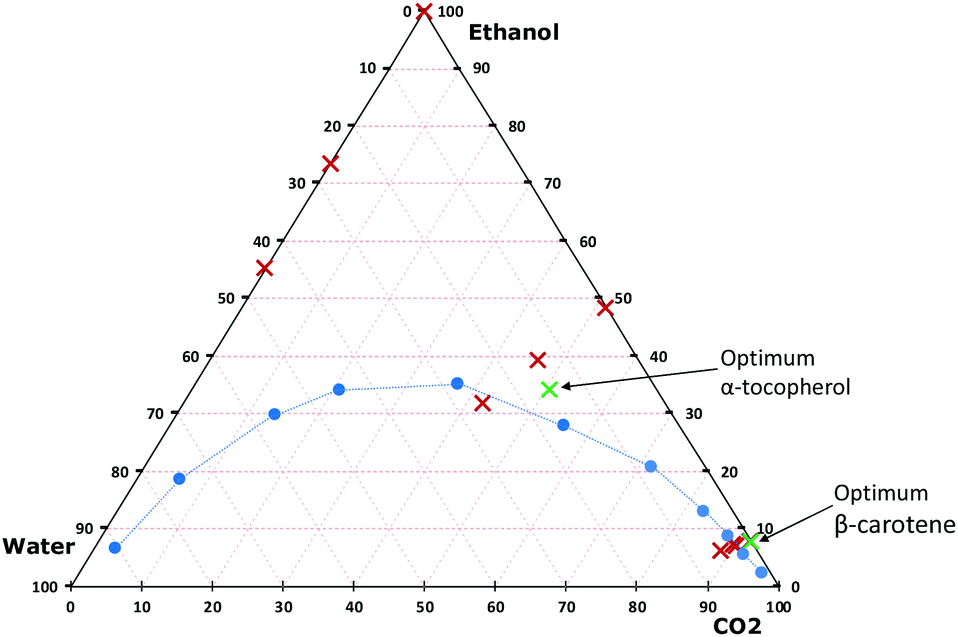 | ||
| Fig. 2 Phase diagram of a ternary system composed of CO2, water, and ethanol. The blue circles mark the boundary between a single liquid phase and two phases (vapor/liquid) at 60 °C and 142 bar.27 The red crosses present the investigated conditions (plotted for 200 bar) and green crosses are the optimal conditions for the extraction of β-carotene and α-tocopherol from sea buckthorn berry pomace. Note that the pressure and temperature conditions are not the same as for the plotted equilibrium data. | ||
Optimization of the extraction conditions
To understand the impact of temperature, pressure, water content in the organic solvent (ethanol or ethyl lactate), and the volumetric ratio between CO2 and the solvent, on the extracted amount of β-carotene, α-tocopherol, and quercetin, two face centred central composite designs were created for ethanol and ethyl lactate, individually. The range of the examined variables was selected to ensure that optimal extraction conditions can be found for each of the target compounds, considering their largely different physicochemical properties. All recovered extract solutions were analysed by either HPLC/DAD or ultrahigh performance supercritical fluid chromatography (UHPSFC/DAD), and the obtained peak areas for each compound under different extraction conditions were treated as the response in the Design of Experiment (DOE) models in order to find the optimal conditions for the target compounds.Optimum conditions for β-carotene
The obtained results show that the use of ethyl lactate as an extraction solvent for β-carotene gave an invalid model (Fig. S1, ESI†) with R2 = 0.3 that describes the model fit and Q2 = <0.1 that estimates the future prediction precision. Any model that has a value for Q2 of less than 0.25 is considered an invalid model due to the lack of prediction power. We have no explanation for this, but hypothetically, this might be explained by complex interactions between the factors which cannot be represented as a correlation between them. On the other hand, when ethanol was used as a solvent, the fitting of the model gave valid values (R2 = 0.83 and Q2 = 0.71), as also demonstrated in the plots of observed versus predicted values (Fig. S2, ESI†). The coefficient plot for β-carotene using ethanol mixtures as the extraction solvent in Fig. S3 (ESI†) shows that the presence of water in ethanol decreased significantly the extracted amount of β-carotene. This is expected since water is a very polar solvent and β-carotene is a very nonpolar compound as shown in Table 2 with HSPs. A higher CO2 ratio has a positive effect on the extracted amount of β-carotene. Such results were reported previously by Baysal et al. who examined co-solvent (ethanol) percentage in CO2 between 5 and 15 vol% for maximum yield of β-carotene from tomato.32 They found that a higher amount of ethanol decreased the extractable amount of β-carotene. Contour plots (Fig. S4, ESI†) confirm the observation from the coefficient plot, of which the most interesting contour plot showing CO2 ratio vs. water content of the co-solvent (ethanol) is shown in Fig. 3.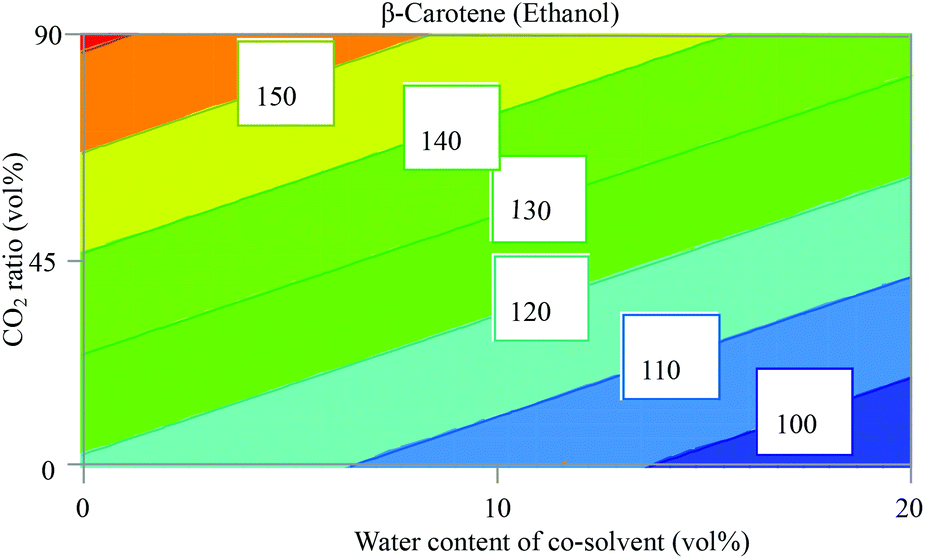 | ||
| Fig. 3 Contour plot from a face centred central composite designs (FC-CCD) showing the direct and interactions’ influence of the CO2 ratio (vol%) and water content in ethanol (vol%) on the extracted amount of β-carotene (peak area absorbance (mAU)). Temperature was 60 °C and pressure 200 bar. | ||
An optimizer function based on a simplex algorithm with a non-linear desirability function was applied to find the conditions where the highest response is existing. It turned out that scCO2 containing 10 vol% ethanol (with no water added) at 300 bar and 80 °C was the optimum extraction condition for β-carotene in sea buckthorn berry pomace.
Optimum conditions for α-tocopherol
Both DOEs with ethanol and ethyl lactate gave valid models for α-tocopherol (Fig. S1†). However, the model with ethanol showed higher values of R2 and Q2 compared to that of ethyl lactate. In addition, the response values from the experiments with ethanol are greater than the ones using ethyl lactate, which implies that higher amounts of α-tocopherol were extracted with ethanol. Thus, the DOE with ethanol was selected and addressed in this discussion. The fitting summary of the model has values of R2 = 0.87 and Q2 = 0.79, which reveals a valid method that has the power to predict the optimal extraction conditions within the investigated experimental range. The coefficient plot in Fig. S3 (ESI†) for α-tocopherol shows that only water content in ethanol has a positive significant effect on the extracted amount. This might be attributed to the slightly higher δP of α-tocopherol compared to CO2. This also can be due to the interactions of water with other variables to enhance the solubility of α-tocopherol in the mixture of the solvents. However, most reported studies in α-tocopherol extraction showed that addition of a co-solvent such as ethanol increased the extraction recovery compared to neat CO2. Dubbs et al. studied the solubility of α-tocopherol in a mixture of ethanol/water and found that 70% ethanol achieved the highest solubility.33 The contour plots in Fig. S5 (ESI†) are non-linear, and reveal strong interactions between the different variables. The plots also indicate that the optimum conditions are found in the middle point of the range for each variable. Fig. 4 shows that the maximum extractable amount is found in the middle of the design. The presence of water up to 12 vol% in ethanol (ca. 6 vol% in the ternary solvent mixture) enhances the recovery; however a further increase in water content in ethanol increases the polarity of the solvent, which might affect the solubility of α-tocopherol negatively. By applying an optimizer function targeting the maximum amount of α-tocopherol, the optimal conditions were found at 210 bar, 60 °C, and the composition CO2/EtOH/water (50/44/6, v/v/v).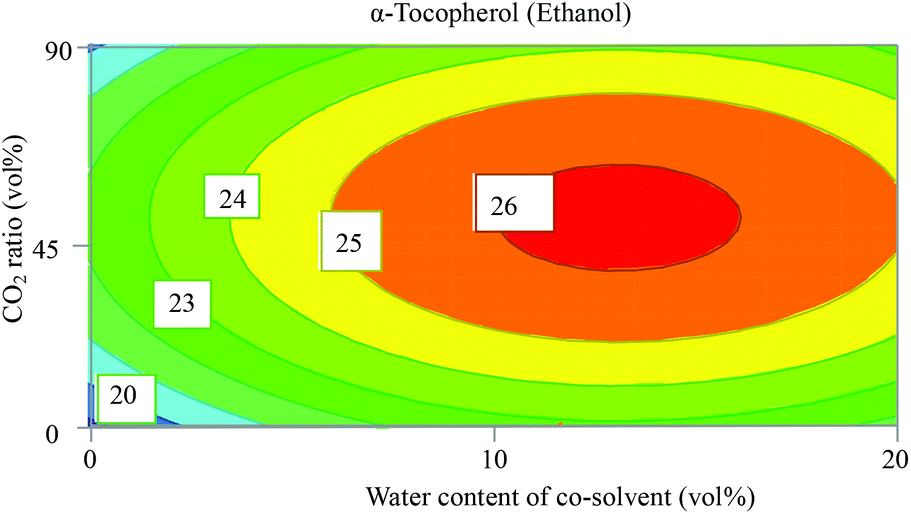 | ||
| Fig. 4 Contour plot from a face centred central composite designs (FC-CCD) showing the direct and interactions’ influence of the CO2 ratio (vol%) and water content in ethanol (vol%) on the extracted amount of α-tocopherol (peak area absorbance (mAU)). Temperature was 60 °C and pressure 200 bar. | ||
Optimum conditions for quercetin
Quercetin differs from β-carotene and α-tocopherol since it belongs to the class of phenolic compounds which are relatively polar compounds due to the presence of hydroxyl groups. This is clear from the high values of δP and δH in Table 2. Intuitively, scCO2 is not the best choice to extract quercetin although there are studies on this topic.34,35 In the study conducted by Xiao Ruan,35 the best conditions to extract quercetin from Taxus chinensis (Chinese yew) were acquired with CO2/ethanol (18/82, v/v) at 46 °C and 250 bar, and extraction time exceeding two hours. Obviously, the presence of a polar solvent is essential to increase the solubility and extractability of quercetin. In our study, both DOEs with ethanol and ethyl lactate gave valid models for the extraction of quercetin, with R2 and Q2 of around 0.9 and 0.8, respectively (Fig. S1†). The observed versus predicted value plot in Fig. S2 (ESI†) shows a high degree of linearity for both designs with ethanol and ethyl lactate. However, ethyl lactate gave higher responses (extracted amount) in comparison with ethanol. Hence, ethyl lactate was selected to be investigated further in this study. Marta Lores et al.36 studied the potential to use ethyl lactate to extract polyphenols including quercetin at high temperature using the PLE technique, as compared to extraction with the same solvent under ambient conditions. They found that at higher temperature the extracted amount of polyphenols increased considerably compared to under ambient conditions. This supports the fact that a higher temperature increases the dissociation from the sample matrix and the mass transfer in the solvent lowers the relative permittivity, which altogether enhances the solubility, extractability, and extraction rate. In this study, instead of increasing the temperature to enhance mass transfer, CO2 was added to the organic phase to decrease the viscosity. Addition of CO2 to ethyl lactate enhanced the extractability of quercetin more than increasing the temperature as can be seen from the plot of CO2 ratio vs. water content (Fig. 5), water content vs. temperature (Fig. S6, ESI†), and in the coefficient plot (Fig. S3, ESI†).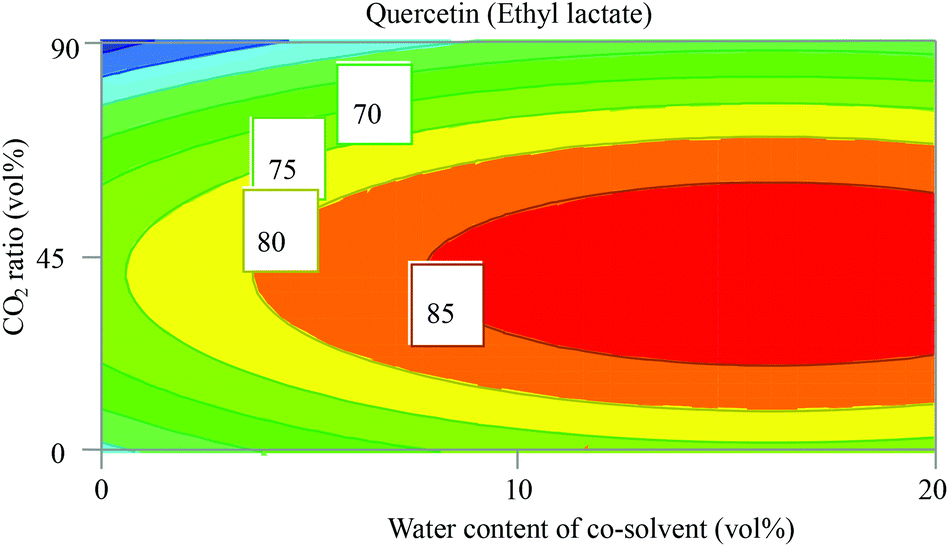 | ||
| Fig. 5 Contour plot from a face centred central composite designs (FC-CCD) showing the direct and interactions’ influence of the co-solvent/CO2 ratio and water content in ethyl lactate on the extracted amount of quercetin (peak area absorbance (mAU)). Temperature was 60 °C and pressure 200 bar. | ||
Addition of 30 vol% CO2 to ethyl lactate containing 10 vol% water increased the response from 70 to >85 (mAU). In contrast, increasing the temperature from 40 to 60 °C at 10 vol% water in the co-solvent increased the response to almost 80 (mAU). The coefficient plot in Fig. S3 (ESI†) for quercetin using ethyl lactate solvent mixtures reveals that increasing the water content increased the extracted amount of quercetin, while the increase in the CO2 ratio had a significantly negative impact. The contour plots in Fig. S6 (ESI†) show that the maximum extracted amount of quercetin is found at the highest water content of the co-solvent (20 vol%) and in the middle of the ranges of temperature, pressure, and CO2 ratio. The optimum extraction conditions for quercetin were found at 200 bar, 62 °C and the composition CO2/ethyl lactate/water (33/54/13, v/v/v).
Extraction kinetics
To complete the optimization of the extraction process for the target compounds, the flow rate was investigated at three levels; 1, 2 and 3 mL min−1 under the optimum conditions for each compound. Variation of the flow rate provides information about the best flow rate to complete the extraction in a short time. In addition, it can be used to investigate whether the extraction process is limited by solubility or by mass transfer. In this study, fractions were collected at defined time periods for the three different flow rates for each analyte to reveal the extraction profile and kinetics as shown in Fig. 6. It is clear that for all compounds, during the first 15 min the extracted amounts increased with the flow rate (Fig. 6A). When the extracted amounts instead are plotted versus solvent volumes used (Fig. 6B), the curves clearly overlap during the first part of the extraction. This indicates that the extraction process is limited by solubility rather than by mass transfer. This observation reveals that the compounds have been quickly transferred to the surface of the sample matrix but dissolution is limited by solubility. Therefore, running the extraction at a higher flow rate will increase the extracted amount per time unit. Hence, running the extraction at the highest possible flow rate during this initial extraction period (0–15 min) is recommended to shorten the extraction time. After 15 min, the extracted amount per time unit is less, indicating that the extraction rate is limited by the mass transfer of the compounds inside the sample matrix. It may also indicate that there remain only small amounts of extractable compounds in the sample, meaning that the concentration difference between the sample and solvent is minimal. Overall, the plots in Fig. 6 confirm that the extraction rates of the investigated compounds are controlled by solubility, explained by the low viscosity and fast mass transfer obtained with the optimal solvent mixtures. A high flow rate not only reduces the extraction time, it also minimises the risk of degradation especially for unstable compounds like the compounds studied here. Therefore, extraction at 3 ml min−1 for 20 min was considered the optimal extraction conditions for the flow rate and extraction time for the investigated compounds.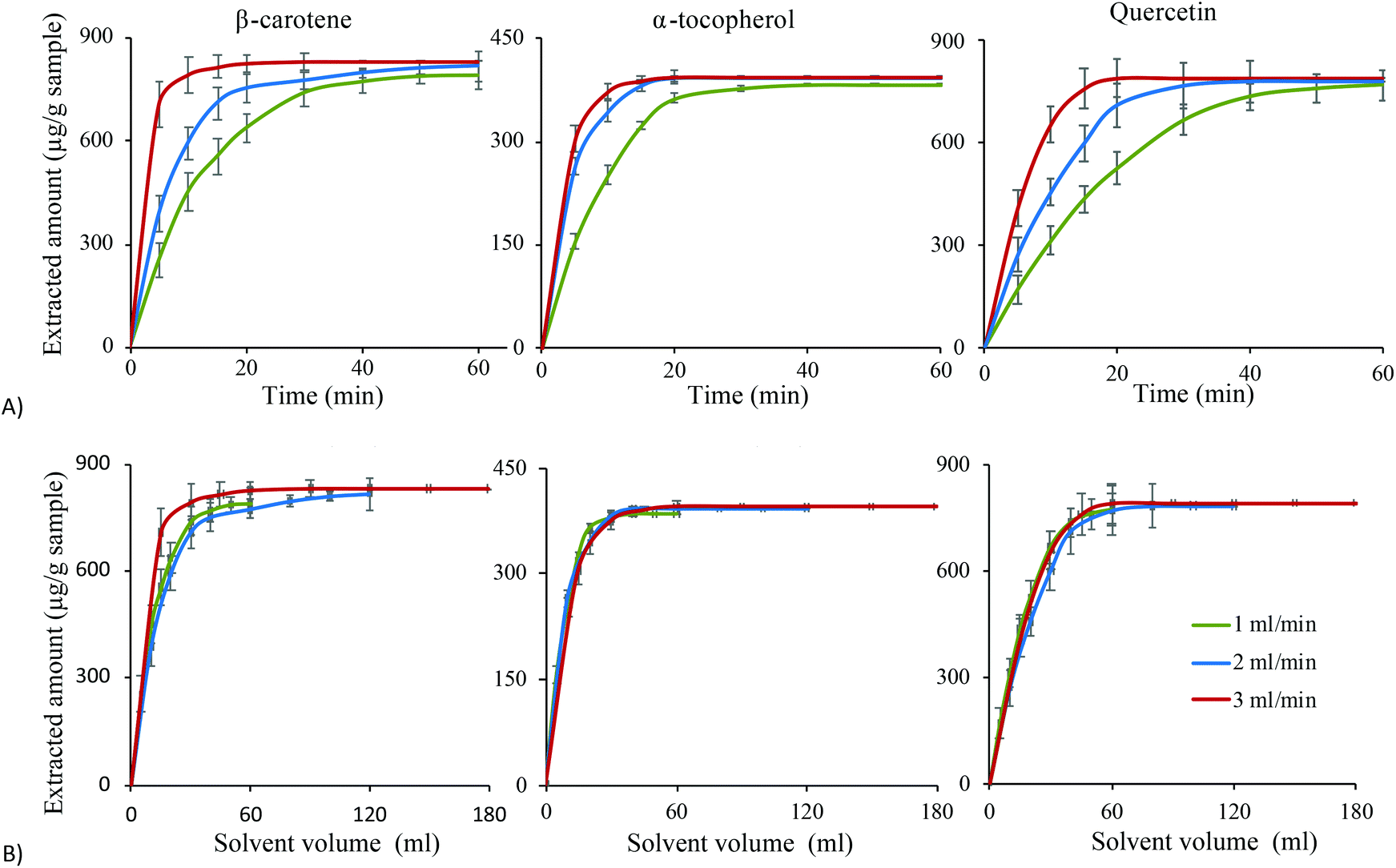 | ||
| Fig. 6 Extracted amount of β-carotene, α-tocopherol, and quercetin (μg g−1 of sea buckthorn pomace sample) versus (A) extraction time, and (B) solvent volume, at three different flow rates using the optimum extraction conditions for each compound. | ||
Comparison with other extraction methods
In the literature, there are many methods developed to extract the investigated compounds using techniques based on SFE and PLE. The previously published SFE methods of selected compounds enable the extraction from various vegetable and fruit matrices. For quercetin, the SFE conditions are usually set up as follows: CO2/ethanol (10–85%, v/v) mixture as the extraction solvent, pressure 70–400 bar, and temperature 35–60 °C. In all cases, the extraction took more than 2 hours (120–160 min).4,35,37–39 For α-tocopherol and β-carotene, a wide range of different extraction conditions have been used in SFE, including pressures (100–500 bar) and temperatures (20–80 °C). In this case, different organic solvents (alcohols, oils, propane, chloroform) have been used as a co-solvent to extract these lipophilic compounds. The extraction time varied from 30 min to 5 hours depending on the extraction conditions.40–44 In this study however, we selected published carefully optimised extraction methods based on PLE to conduct a comparison with the herein developed methods. The solvent to sample ratios were kept similar to enable a somewhat fair comparison between the methods. The results are illustrated in Fig. 7, which reveals that the developed methods are more efficient in the extraction of the investigated compounds. This could be explained by higher solubility as well as faster mass transfer. For instance, the developed method for quercetin increased the extracted amount by 40% compared to the PLE method in the literature. This significant difference could also be explained by less thermal degradation during the extraction.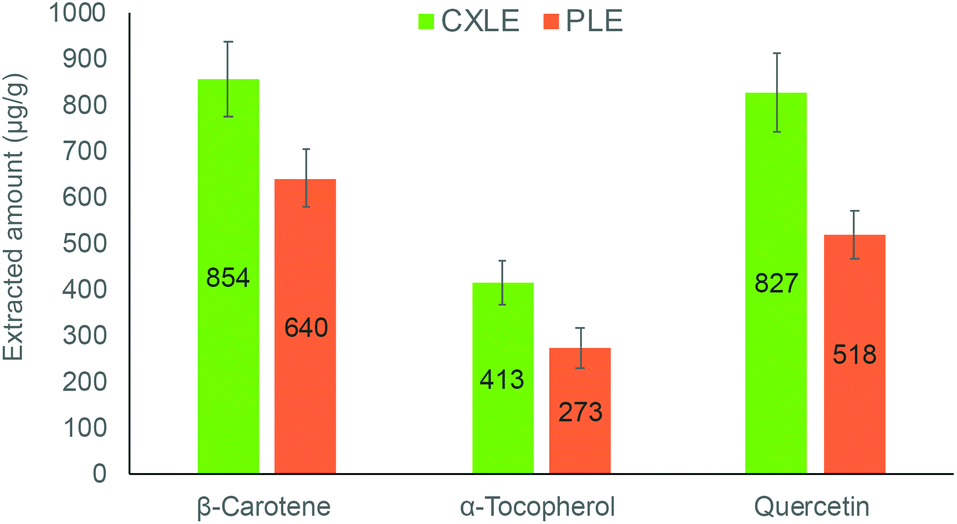 | ||
| Fig. 7 Comparison of the extracted amount of compounds from sea buckthorn berry pomace using the optimised CXLE methods as compared to PLE methods found in the literature49–51 and applied here. | ||
In addition, the developed methods are less organic solvent-demanding compared to the conventional methods. For example, the developed method for β-carotene requires only 6 mL of ethanol while the PLE method consumes more than 20 mL of ethanol to complete the extraction. The method for α-tocopherol requires toxic solvents like n-isopropanol and methanol. On the other hand, the developed extraction method for quercetin is longer and requires a higher amount of organic solvent when compared to the PLE method. However, the developed method operates at much lower temperature and that is more suitable for sensitive compounds like a flavonoid such as quercetin. Table 3 summarizes the comparison between the conditions and extracted amounts of developed methods and the method from the literature.
| Quercetin | α-Tocopherol | β-Carotene | ||||
|---|---|---|---|---|---|---|
| PLE | CXLE | PLE | CXLE | PLE | CXLE | |
| Extraction solvent | H2O/EtOH/formic acid, 94/5/1, v/v/v | CO2/EL/H2O 33/54/13, v/v/v | MeOH/2-propanol 50/50, v/v | CO2/EtOH/H2O 50/44/6, v/v/v | EtOH | CO2/EtOH 90/10, v/v |
| Temperature [°C] | 120 | 62 | 50 | 60 | 60 | 80 |
| Pressure [bar] | 100 | 200 | 110 | 210 | 50 | 300 |
| Organic solvent usage [mL] | 10 | 32 | 50 | 26 | 20 | 6 |
| Extraction time [min] | 8 | 20 | 16 | 20 | 11 | 20 |
| Extracted amount [μg g−1] | 521 ± 57 | 828 ± 91 | 278 ± 43 | 412 ± 57 | 642 ± 68 | 854 ± 96 |
Experimental
Chemicals and reagents
Ultrapure CO2 in cylinders with a dip tube was provided by AGA industrial gases (Lidingö, Sweden). Ethanol 99.7% was ordered from Solveco (Rosenberg, Sweden). LC-HPLC grade methanol was provided by VWR (VWR Chemicals, France). Ethyl L-lactate, 99% and β-carotene, 99% were ordered from Alfa Aesar. Formic acid ≥95%, α-tocopherol ≥95.5%, and quercetin anhydrous ≥95% were purchased from Sigma-Aldrich (St Louis, MO). Water was purified using a Milli-Q purification system (Millipore, Billerica, MA). Stock solutions for the standards were prepared at concentrations of 1.00 mg mL−1 and then diluted with EtoH or ethyl lactate to different concentrations to establish the calibration curves.Sea buckthorn samples
Sea buckthorn pomace was collected from a juice processor on the day of pressing from Kelterei Walther GmbH, Arnsdorf, Germany. The pomace was freeze-dried at −24 °C for 72 h (Martin Christ Gefriertrocknungsanlagen GmbH, Osterode, Germany) and milled after lyophilization using a ZM 100 ultra-centrifugal mill, equipped with a 0.5 mm sieve (Retsch GmbH, Haan, Germany) at 14000 rpm. The milled samples were kept in sealed containers in a −20 °C freezer until the time of analysis.
Extraction instrument
An analytical SFE system (Waters MV-10, Milford, MA, USA) consisting of a fluid delivery module for pumping CO2 and the co-solvent, an oven for placing the extraction vessels, an automated back pressure regulator, a make-up pump, and a fraction collector module was used to conduct the experiments. The heads of the CO2 pump were cooled using a chiller operated at 4 °C. The sample (1.00 g) was mixed with glass beads and loaded into a 10 mL stainless steel extraction vessel. The flow was controlled as the volumetric ratio between CO2 and the co-solvent. The collected extracts were diluted to final volumes of either 10, 25 or 50 mL depending on the collected volume of the extract. After each extraction the system was flushed with a CO2/co-solvent mixture for 5 min followed by neat CO2. The collected extracts were stored in a −20° freezer until the time of analysis. The system was controlled by using ChromScope™ software (Waters, Milford, MA, USA).Hansen solubility parameters (HSP)
Hansen solubility parameters45 can be used to obtain the theoretical knowledge of the solubility of the compounds of interest (in this work: β-carotene, α-tocopherol, and quercetin) and the chosen solvents for the extraction of these compounds. According to this method, close values of HSP between the compound and the solvent would indicate high solubility. Since the original values of HSP from Hansen’s work45 were not available for β-carotene, α-tocopherol, and quercetin, the software HSP in Practice (HSPiP)46 was used to predict the HSP values. The temperature dependence of HSP was calculated by the equation of Jayasri and Yaseen.47 For the HSP calculations of CO2, the effect of pressure was considered and therefore the method of Williams et al.48 was used. In this work, the HSP for a mixture of solvents were obtained considering a linear relationship with the composition of the solvents in the mixture.Phase diagrams
To illustrate the phase diagram of the extraction solvent mixture under each condition, the molar fractions were calculated using the mass of each component delivered from each pump. The density of the liquids (water, ethanol, and ethyl lactate) at 25 °C was used to convert volumetric flow (mL min−1) to mass flow (g min−1). The density of CO2 was calculated at each pressure and 10 °C (pump head chiller) using the online software https://webbook.nist.gov/chemistry/fluid/.Design of experiments and method optimization
Two face centred central composite designs (FC-CCD) for ethanol and ethyl L-lactate with three centre points were created in MODDE 10.1 (Sartorius Stedim Biotech, Malmö, Sweden) and used to examine the effect of extraction temperature (40–80 °C), pressure (100–300 bar), water content in the co-solvent (0–20 vol%), and CO2 ratio of the co-solvent (0–90 vol%), on the extracted amount of β-carotene, α-tocopherol, and quercetin extracted from sea buckthorn pomace. The flow rate and extraction time were kept at 2.0 mL min−1 and 15 min, respectively. In total, 27 experiments for each design were performed in a random run order (Tables S2 and S3, ESI†). Multiple linear regression (MLR) was used to calculate the fitting model and response surface. The adequacy of the models was evaluated by the R2 and Q2 values (where R2 shows the model fit and Q2 shows an estimate of the future prediction precision), predicted vs. observed plot and coefficient plots. The optimum processing conditions for the maximum peak area for each target compound were obtained by using graphical and numerical analysis based on the criteria of the desirability function and the response surface plots.The flow rate and extraction time were studied separately using the optimal extraction conditions for each compound. The flow rate was varied between 1, 2, and 3 mL min−1 and the fractions were collected at defined time periods during a certain extraction time. Flow rates over 3 mL min−1 were not studied since these caused fluctuation in the back pressure, especially when ethyl lactate was used as a co-solvent. The data were acquired and plotted as the extracted amount in μg per g of sample versus time and solvent amount to understand the extraction kinetics.
Extraction with conventional methods
Three different conventional methods were selected to compare the extraction efficiency of the optimised methods for β-carotene, α-tocopherol, and quercetin using the PLE technique with a Dionex ASE-350 (Thermo Fisher, Germering, Germany). For β-carotene the selected method was optimized to extract β-carotene from the carrot by-product.49 Briefly, 1.00 g of the sample was extracted with pressurized hot ethanol in a 10 mL extraction cell. Extractions were performed at 60 °C, 100 bar, with 5 min pre-heating, and 2 min static extraction for two cycles. The sample was flushed with 60% volume of the extraction solvent. For α-tocopherol the method has been used to extract α-tocopherol from different foods like corn, mangoes and spinach.50 Briefly, 1.00 g of the sample was extracted with methanol:isopropanol (1:1 v/v) in an 10 mL extraction cell. Extractions were performed at 50 °C, 100 bar, with 5 min pre-heating, and 5 min static extraction for two cycles. The sample was flushed with 60% volume of extraction solvent. For quercetin the selected method was optimized to extract phenolic compounds from apple pomace.51 Briefly, 1.00 g of the sample was extracted with pressurized hot water containing 5% ethanol and 1% formic acid in a 10 mL extraction cell. Extractions were performed at 99 °C, 100 bar, with 5 min pre-heating, and 1 min static extraction for two cycles. The sample was flushed with 60% volume of extraction solvent. All the extracts were diluted to a final volume of 25 mL with the same solvent composition of extraction and kept at −20 °C until analysis.
HPLC-DAD analysis
The separation of β-carotene, α-tocopherol, and quercetin was carried out using an Agilent 1100 HPLC system with DAD detection. The samples were injected into an ODS-2 Hypersil analytical column (100 mm × 2.1 mm; 5.0 μm) and separated using gradient elution with 1% formic acid in water (eluent A) and 1% formic acid in MeOH (eluent B) with a flow rate of 0.6 mL min−1, and a temperature of 40 °C. The gradient started at 5% of eluent B, and increased to 60% over 15.0 min. Then, an isocratic step with 100% of eluent B followed until 25.0 minutes. At 26 minutes, the percentage of eluent B ramped to original conditions (5%). The total time of chromatographic separation including column equilibration was 30 minutes. Three different wavelengths were used for the compound detection, namely 295 nm for α-tocopherol, 370 nm for quercetin, and 450 nm for β-carotene. An example of a chromatogram is presented in Fig. S7 (ESI†).UHPSFC-DAD analysis
To verify the results from the HPLC-DAD system and to increase the selectivity and sensitivity, an ultrahigh performance supercritical fluid chromatography (UHPSFC-DAD) method was optimised for the separation of all compounds. The separation was carried out using a Waters Acquity UltraPerformance Convergence Chromatography (UPC2) system (Waters, Milford, MA, USA) with DAD detection at 295 nm for α-tocopherol, 365 nm for quercetin, and 445 nm for β-carotene. 2 μL of sample was injected into a HSS C18 SB analytical column (100 mm × 3.1 mm; 1.8 μm) from Waters (Milford, MA, USA) and separated using gradient elution with CO2 (eluent A) and MeOH containing 20 mM citric acid (eluent B) with a flow rate of 2.0 mL min−1. The gradient started at 2% of eluent B, increased to 10% over 5.0 minutes, and to 20% until 6.0 minutes. At 6.2 minutes, the percentage of eluent B ramped to original conditions (2%) for column equilibration. The total time of chromatographic separation was 8 minutes as shown in Fig. S8 (ESI†).Conclusions
In the present study, extraction efficiency of β-carotene, α-tocopherol, and quercetin from sea buckthorn pomace was screened in a wide range of physicochemically different solvents, including mixtures of liquid or supercritical CO2, water and ethanol or ethyl lactate. The most polar mixture investigated was CO2/ethanol/water (10/72/18, v/v/v), and the least polar was scCO2/ethanol (90/10, v/v). The optimization process showed that scCO2 with 10 vol% of ethanol at 300 bar and 80 °C were the best conditions to extract β-carotene. For α-tocopherol, the best conditions were CO2/ethanol/water (50/44/6, v/v/v) at 210 bar and 60 °C. Because of the higher polarity of quercetin, the addition of 20 vol% of water to ethyl lactate enhanced the recovery of quercetin. The best conditions were found with CO2/ethyl lactate/water (33/54/13, v/v/v) at 200 bar and 62 °C. The kinetics study showed that the extraction process for the three investigated compounds using their respective optimised solvent mixture was mainly controlled by solubility, meaning that at least for the first 20 min of the extraction, the highest possible flow rate should be used, i.e. at least 3 mL min−1. The quantitative analysis of investigated compounds extracted under the optimum conditions showed significantly higher amounts compared to published optimised methods in the literature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Anne-Marie Reißner from Technische Universität Dresden for providing the sample of sea buckthorn pomace. The authors gratefully acknowledge the financial support of the STARSS project (Reg. No. CZ.02.1.01/0.0/0.0/15_003/0000465) co-funded by ERDF, as well as the Swedish Research Council Formas (222-2014-1924 and 2016-00604).References
- T. Belwal, S. M. Ezzat, L. Rastrelli, I. D. Bhatt, M. Daglia, A. Baldi, H. P. Devkota, I. E. Orhan, J. K. Patra, G. Das, C. Anandharamakrishnan, L. Gomez-Gomez, S. F. Nabavi, S. M. Nabavi and A. G. Atanasov, TrAC, Trends Anal. Chem., 2018, 100, 82–102 CrossRef CAS.
- M. Cvjetko Bubalo, S. Vidović, I. Radojčić Redovniković and S. Jokić, Food Bioprod. Process., 2018, 109, 52–73 CrossRef CAS.
- M. Herrero and E. Ibañez, J. Supercrit. Fluids, 2018, 134, 252–259 CrossRef CAS.
- C. Rodríguez-Pérez, J. A. Mendiola, R. Quirantes-Piné, E. Ibáñez and A. Segura-Carretero, J. Supercrit. Fluids, 2016, 116, 90–100 CrossRef.
- A. D. Sanchez-Camargo, F. Parada-Alfonso, E. Ibanez and A. Cifuentes, J. Sep. Sci., 2017, 40, 213–227 CrossRef CAS PubMed.
- R. P. F. F. da Silva, T. A. P. Rocha-Santos and A. C. Duarte, TrAC, Trends Anal. Chem., 2016, 76, 40–51 CrossRef CAS.
- K. Y. Khaw, M. O. Parat, P. N. Shaw and J. R. Falconer, Molecules, 2017, 22, 1186–1207 CrossRef PubMed.
- J. Płotka-Wasylka, M. Rutkowska, K. Owczarek, M. Tobiszewski and J. Namieśnik, TrAC, Trends Anal. Chem., 2017, 91, 12–25 CrossRef.
- I. Brondz, B. Sedunov and N. Sivaraman, Int. J. Anal. Mass Spectrom. Chromatogr., 2017, 05, 17–39 CrossRef CAS.
- A. d. P. Sánchez-Camargo, E. Ibáñez, A. Cifuentes and M. Herrero, in Green Extraction Techniques - Principles, Advances and Applications, 2017, pp. 27–51, DOI:10.1016/bs.coac.2017.01.001.
- S. B. Hawthorne, Anal. Chem., 1990, 62, 633–642 CrossRef.
- C. Turner, Lipid Technol., 2015, 27, 275–277 CrossRef CAS.
- P. G. Jessop and B. Subramaniam, Chem. Rev., 2007, 107, 2666–2694 CrossRef CAS PubMed.
- L. P. Cunico and C. Turner, in The Application of Green Solvents in Separation Processes, 2017, pp. 155–214, DOI:10.1016/b978-0-12-805297-6.00007-3.
- M. Herrero, J. A. Mendiola and E. Ibáñez, Curr. Opin. Green Sustainable Chem., 2017, 5, 24–30 CrossRef.
- S. Al-Hamimi, A. Abellan Mayoral, L. P. Cunico and C. Turner, Anal. Chem., 2016, 88, 4336–4345 CrossRef CAS PubMed.
- A. Mustafa and C. Turner, Anal. Chim. Acta, 2011, 703, 8–18 CrossRef CAS PubMed.
- M. M. R. de Melo, I. Portugal, A. J. D. Silvestre and C. M. Silva, in The Application of Green Solvents in Separation Processes, 2017, pp. 325–348, DOI:10.1016/b978-0-12-805297-6.00011-5.
- A. Quiles, G. M. Campbell, S. Struck, H. Rohm and I. Hernando, Food Rev. Int., 2016, 34, 162–181 CrossRef.
- A. Quiles, E. Llorca, C. Schmidt, A.-M. Reißner, S. Struck, H. Rohm and I. Hernando, Int. J. Food Sci. Technol., 2018, 53, 1579–1587 CrossRef CAS.
- S. Struck, M. Plaza, C. Turner and H. Rohm, Int. J. Food Sci. Technol., 2016, 51, 1305–1318 CrossRef CAS.
- D. Quintin, P. Garcia-Gomez, M. Ayuso and A. M. Sanmartin, Trends Food Sci. Technol., 2019, 84, 19–21 CrossRef CAS.
- L. M. Bal, V. Meda, S. N. Naik and S. Satya, Food Res. Int., 2011, 44, 1718–1727 CrossRef CAS.
- R. Guo, X. Guo, T. Li, X. Fu and R. H. Liu, Food Chem., 2017, 221, 997–1003 CrossRef CAS PubMed.
- B. Olas, Food Chem. Toxicol., 2016, 97, 199–204 CrossRef CAS PubMed.
- G. Suryakumar and A. Gupta, J. Ethnopharmacol., 2011, 138, 268–278 CrossRef PubMed.
- J. S. Lim and Y. Y. Lee, J. Supercrit. Fluids, 1994, 7, 219–230 CrossRef CAS.
- I. Dalmolin, E. Skovroinski, A. Biasi, M. L. Corazza, C. Dariva and J. V. Oliveira, Fluid Phase Equilib., 2006, 245, 193–200 CrossRef CAS.
- N. E. Durling, O. J. Catchpole, S. J. Tallon and J. B. Grey, Fluid Phase Equilib., 2007, 252, 103–113 CrossRef CAS.
- D. Villanueva Bermejo, E. Ibáñez, R. P. Stateva and T. Fornari, J. Chem. Eng. Data, 2013, 58, 301–306 CrossRef CAS.
- C. Secuianu, V. Feroiu and D. Geană, J. Supercrit. Fluids, 2008, 47, 109–116 CrossRef CAS.
- T. Baysal, S. Ersus and D. A. J. Starmans, J. Agric. Food Chem., 2000, 48, 5507–5511 CrossRef CAS PubMed.
- M. D. Dubbs and R. B. Gupta, J. Chem. Eng. Data, 1998, 43, 590–591 CrossRef CAS.
- K. G. Martino and D. Guyer, J. Food Process Eng., 2004, 27, 17–28 CrossRef.
- X. Ruan, L. Y. Yan, X. X. Li, B. Liu, H. Zhang and Q. Wang, Molecules, 2014, 19, 17682–17696 CrossRef PubMed.
- M. Lores, M. Pajaro, M. Alvarez-Casas, J. Dominguez and C. Garcia-Jares, Talanta, 2015, 140, 134–142 CrossRef CAS PubMed.
- T. S. Tunna, M. Z. I. Sarker, K. Ghafoor, S. Ferdosh, J. M. Jaffri, F. Y. Al-Juhaimi, M. E. Ali, M. J. H. Akanda, M. S. Awal, Q. U. Ahmed and J. Selamat, Sep. Sci. Technol., 2017, 53, 243–260 CrossRef.
- M. Antunes-Ricardo, J. A. Gutiérrez-Uribe and D. Guajardo-Flores, J. Supercrit. Fluids, 2017, 119, 58–63 CrossRef CAS.
- J. Li, J. Zhang and M. Wang, Molecules, 2016, 21, 810–824 CrossRef PubMed.
- J. M. Prado, P. C. Veggi and M. A. A. Meireles, Curr. Anal. Chem., 2014, 10, 29–66 CrossRef CAS.
- M. de Andrade Lima, I. Kestekoglou, D. Charalampopoulos and A. Chatzifragkou, Molecules, 2019, 24, 466–480 CrossRef PubMed.
- R. K. Saini and Y.-S. Keum, Food Res. Int., 2016, 82, 59–70 CrossRef CAS.
- R. K. Saini and Y. S. Keum, Food Chem., 2018, 240, 90–103 CrossRef CAS PubMed.
- C. Fanali, G. D'Orazio, S. Fanali and A. Gentili, TrAC, Trends Anal. Chem., 2017, 87, 82–97 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, 2nd edn, 2012 Search PubMed.
- The Official Hansen Solubility Parameter Site, https://www.hansen-solubility.com/, (accessed June 2019).
- A. Jayasri and M. Yaseen, Nomograms for Solubility Parameters—A Review, 1980 Search PubMed.
- L. L. Williams, J. B. Rubin and H. W. Edwards, Ind. Eng. Chem. Res., 2004, 43, 4967–4972 CrossRef CAS.
- A. Mustafa, L. M. Trevino and C. Turner, Molecules, 2012, 17, 1809–1818 CrossRef CAS PubMed.
- P. Vinas, M. Bravo-Bravo, I. Lopez-Garcia, M. Pastor-Belda and M. Hernandez-Cordoba, Talanta, 2014, 119, 98–104 CrossRef CAS PubMed.
- M. Plaza, V. Abrahamsson and C. Turner, J. Agric. Food Chem., 2013, 61, 5500–5510 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc02140j |
| ‡ Shared first co-authorship. |
| This journal is © The Royal Society of Chemistry 2019 |