
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards cleaner PolarClean: efficient synthesis and extended applications of the polar aprotic solvent methyl 5-(dimethylamino)-2-methyl-5-oxopentanoate†
Levente
Cseri
 *a and
Gyorgy
Szekely
*ab
*a and
Gyorgy
Szekely
*ab
aSchool of Chemical Engineering and Analytical Science, University of Manchester, The Mill, Sackville Street, Manchester, M1 3BB, UK. E-mail: levente.cseri@manchester.ac.uk; gyorgy.szekely@manchester.ac.uk; Tel: +44 (0)1613062662
bAdvanced Membranes & Porous Materials Center, Physical Science Engineering Division (PSE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: gyorgy.szekely@kaust.edu.sa; Tel: +966128082769
First published on 12th July 2019
Abstract
As a result of recent efforts in green solvent selection, methyl 5-(dimethylamino)-2-methyl-5-oxopentanoate, sold under the brand name Rhodiasolv PolarClean, has received considerable scientific and industrial attention as a possible non-toxic replacement for common polar aprotic solvents. However, the multicomponent nature and multi-step synthesis of this solvent remains an obstacle for its more widespread use and niche applications. In this work, a retrosynthetic approach was taken to identify novel shorter synthetic routes in alignment with green chemistry principles. High purity methyl 5-(dimethylamino)-2-methyl-5-oxopentanoate was obtained in novel single-step reactions via two different base-catalysed Michael additions from readily available building blocks. The more advanced synthetic route shows great potential owing to the swift (30 min), solvent-free reaction and catalytic amounts of base (<6.5 mol%). Green metrics analysis, including Atom Economy, Complete E factor, Carbon Intensity and hazard analysis found the new synthesis to be more sustainable than the patented routes. Application of this green solvent was demonstrated for the first time for O- and N-arylation in SNAr reaction with solvent recovery with similar or superior yields compared to other green solvents. Moreover, broad opportunities for this green solvent in membrane science were identified, where the use of conventional, toxic polar aprotic solvents in large quantities is unavoidable. Important practical solvent properties and parameters such as dielectric constant, solubility parameters, solvent miscibility and NMR residual shifts have been determined to facilitate the uptake of methyl 5-(dimethylamino)-2-methyl-5-oxopentanoate as a green solvent.
Introduction
The alarming environmental concerns of the past decade have resulted in a series of ambitious sustainability plans established by international organisations and companies.1 These efforts have manifested themselves in stricter regulatory environment and soaring research interest in sustainable technologies and green chemistry. Solvents have always been of central interest in green chemistry simply because they account for the largest proportion of total mass used in chemical manufacturing.2 To mitigate the environmental, health and safety concerns associated with the use of organic solvents, a series of green solvents have been developed and reported in the literature. These green solvents usually can be characterised by lower acute and chronic toxicity, decreased environmental impact and fewer safety issues compared to their conventional counterparts. Furthermore, they are often renewables-based and thus more attractive from a sustainability point of view. However, green alternatives are not equally available for all solvent classes. While the landscape of green alcohols or low-polarity esters is highly populated, there are only a few examples of green solvents with polar aprotic characteristics as shown in Fig. 1. The latest GSK's solvent sustainability guide features 154 solvents with 53 of them earning a green (recommended) flag.3 However, none of the 14 assessed dipolar aprotic solvents was ranked in this category.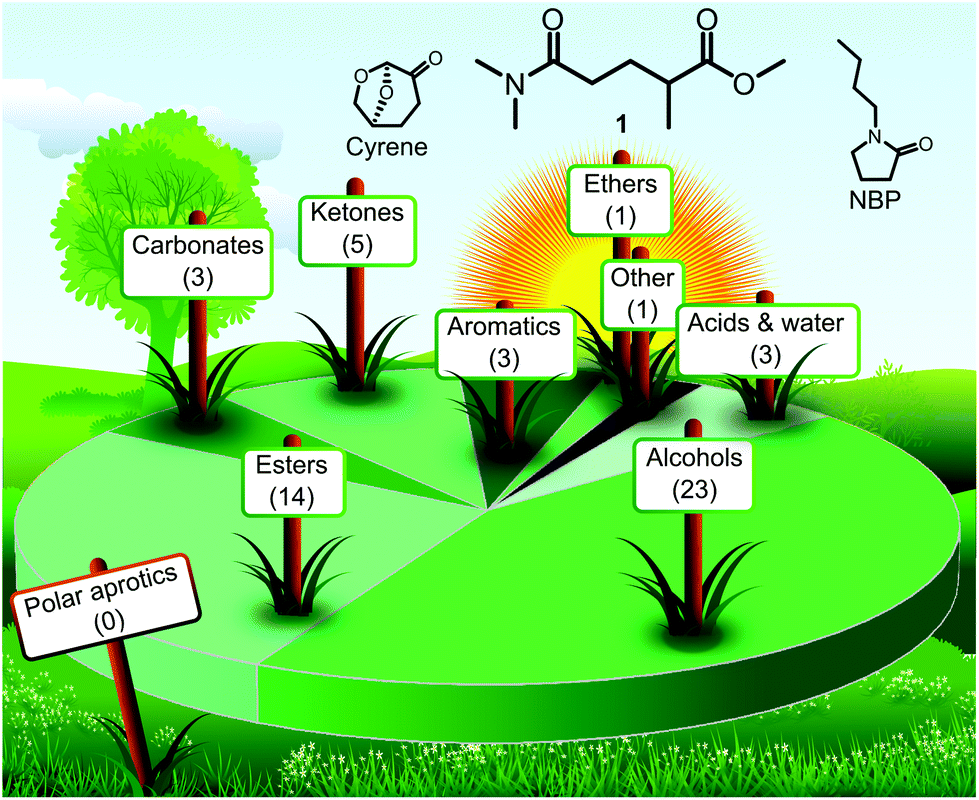 | ||
| Fig. 1 Landscape of solvent types among the 53 solvents marked with green composite colour in GSK's solvent sustainability guide3 with the structures of new, potentially green polar aprotic solvents over the horizon: PolarClean (1), cyrene and N-butylpyrrolidone (NBP). | ||
Polar aprotic solvents are extremely useful for their solvation characteristics and promotion of a variety of chemical reactions. The need for green alternatives in this solvent class has been repeatedly articulated due to the reproductive toxicity of the conventional members, N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMAc) and N-methylpyrrolidone (NMP).4 The few promising green alternatives for polar aprotic solvents include γ-valerolactone (GVL),5 Cyrene,6 propylene carbonate (PC),7N-butylpyrrolidone (NBP)8 and methyl 5-(dimethylamino)-2-methyl-5-oxopentanoate (1).9 While some of these solvents are only available commercially on a gram scale, 1 is the main component of the solvent PolarClean, which is produced at the ton scale by Solvay. A toxicological study of 1 found low acute toxicity (no observed adverse effect at 1000 mg kg−1 day−1 rat) and no evidence of carcinogenity, genotoxicity or mutagenicity.10 The eco-friendly nature and high solvency of 1 enabled applications of PolarClean in crop formulation,11 membrane technology12–15 and chemical synthesis.16–18
A significant effort is invested into the development of greener synthetic routes for green solvents.19 There are several major obstacles to overcome for producing a greener PolarClean. First, this solvent is a yellowish, multicomponent mixture. The patented production procedure of PolarClean reports three synthetic routes (A1–3 in Scheme 1) leading to 1.20 All three routes start from 2-methylglutaronitrile – produced by hydrocyanation of butadiene – which contains around 11% dinitrile isomers. 2-Methylglutaronitrile undergoes hydrolysis to produce 2-methylglutaric acid. In routes A1 and A2, 2-methylglutaric acid is cyclised in the next step to 2-methylglutaric anhydride. The esterification and amidation of the two carbonyl groups in different orders ultimately lead to 1 by these routes. The dinitrile isomers present in the starting material react identically with the main compound and are therefore present in the product. Moreover, the ring opening of the cyclic anhydride can occur in two positions resulting in the formation of methyl 5-(dimethylamino)-4-methyl-5-oxopentanoate. Thus, the 1 content of the products from routes A1 and A2 are rather low, around 37% and 69%, respectively. Route A3 leads to 1 from 2-methylglutaric acid via subsequent diesterification and amidation. For this route, the final isomeric composition is not reported. Second, its synthesis cannot be considered efficient from a green chemistry point of view. It consists of multiple synthetic and purification steps and entails the generation of considerable amounts of waste. Consequently, there is a need for a selective and green synthesis of 1 to boost its applications in the fine chemical industry as a green alternative for polar aprotic solvents. In this work, new synthetic strategies leading to the selective production of 1 were developed. The syntheses were evaluated against the current routes, using green metric analysis. The produced solvents were explored for membrane science via polymer dissolution and miscibility tests, and in nucleophilic aromatic substitution (SNAr) as reaction medium, against other green polar aprotic solvent candidates. Moreover, practically relevant physical and spectroscopic analysis, as well as NMR chemical shifts in different deuterated solvents, are reported to promote the application of 1 even from the early stages of research and development.
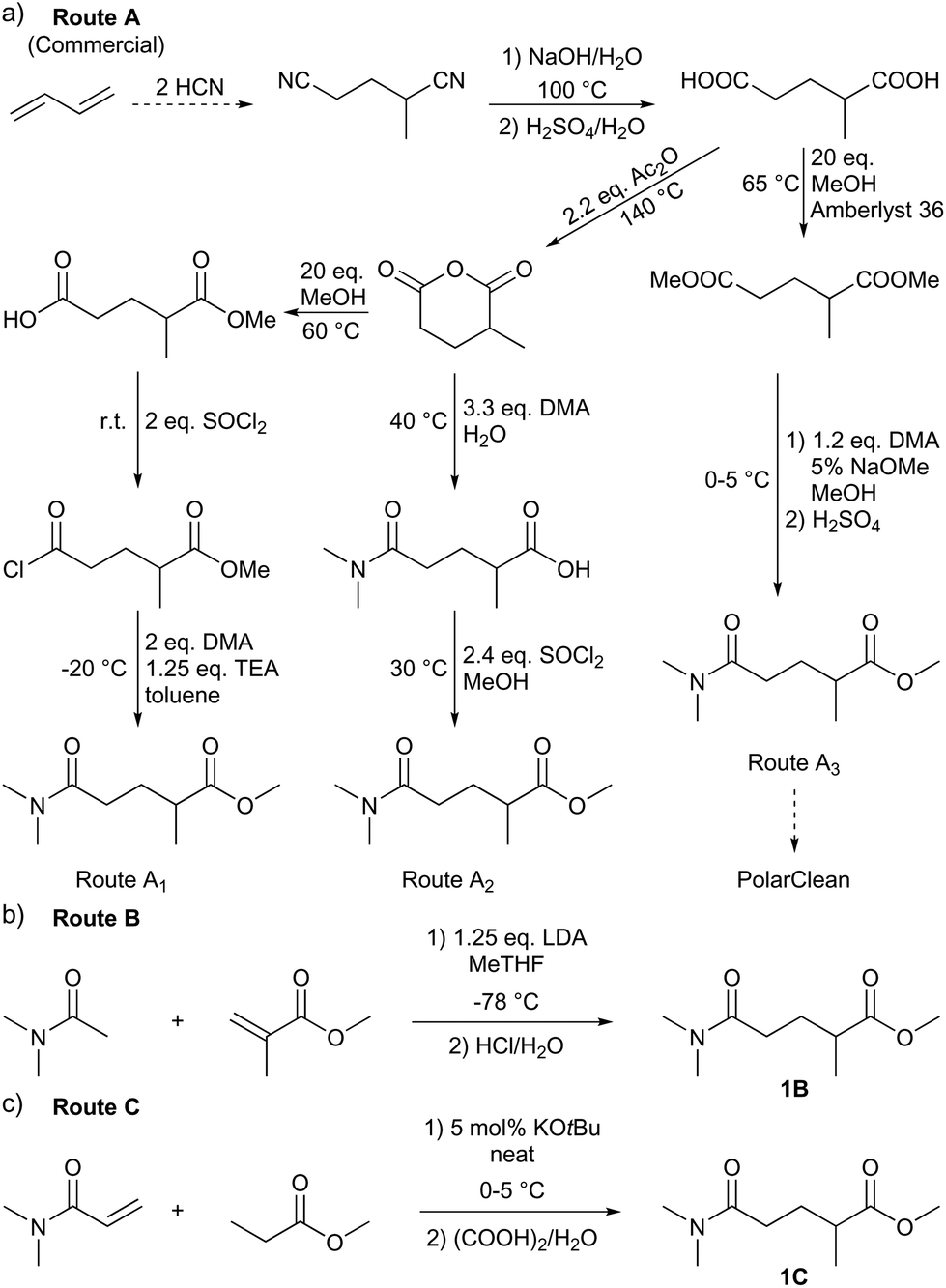 | ||
| Scheme 1 Synthetic strategies leading to methyl 5-dimethylamino-2-methyl-5-oxopentanoate (1) reported in the literature (a) and in this work (b and c). | ||
Results and discussion
Synthesis
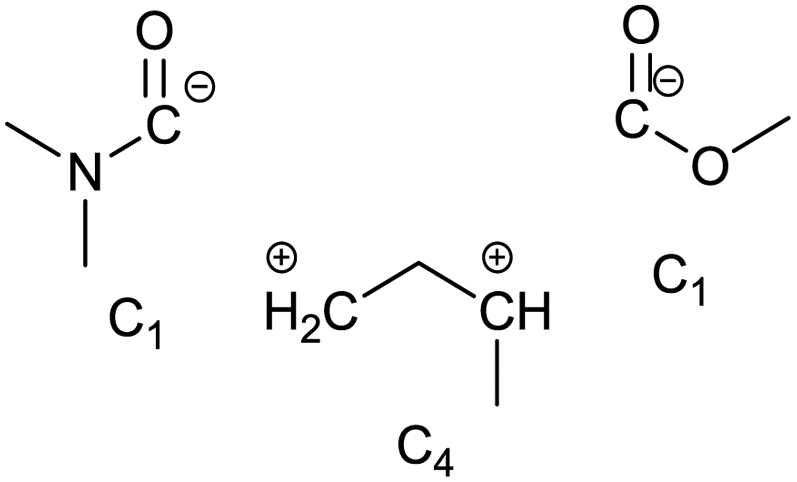
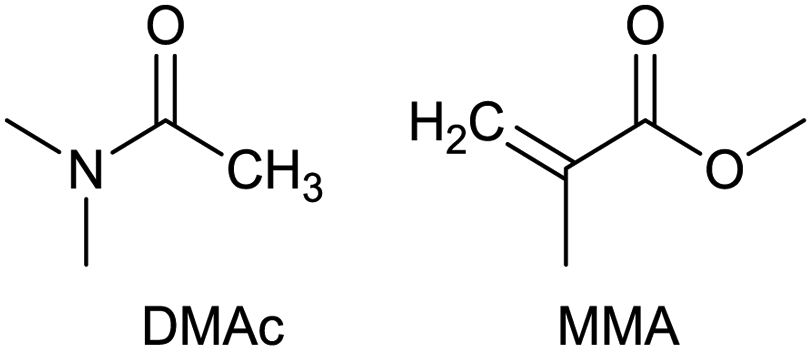
To identify new, potentially more efficient synthetic strategies leading to 1, a retrosynthetic approach was taken (Table 1). In the commercial synthesis (route A, Scheme 1) the carbon backbone of 1 (2-methylglutarate) is constructed in a C4 + C1 + C1 manner. Alternatively, C4 + C2 (route B, Scheme 1) or C3 + C3 (route C, Scheme 1) strategies could be considered. Owing to the C–H acidic character of the α position in carbonyl derivatives, Michael addition can be an obvious choice for the C–C bond formation in both approaches. In the early stage development of this work, the C4 + C2 strategy was favoured since both building blocks, methyl methacrylate (MMA) and N,N-dimethylacetamide (DMAc) are inexpensive, readily available, large volume chemicals. Although current industrial production of both building blocks rely on fossil feedstock, renewable-based strategies in the future will plausibly be derived from bio-based itaconic acid and acetic acid.21,22|
|
||
|---|---|---|
| Route | Synthons | Synthetic equivalents |
| A |
|
|
| B |
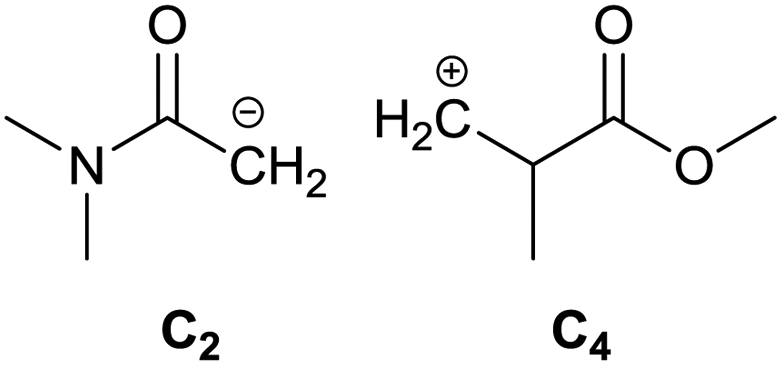
|
|
| C |
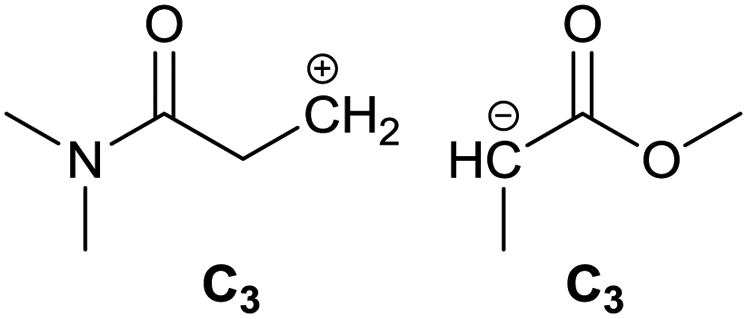
|
|
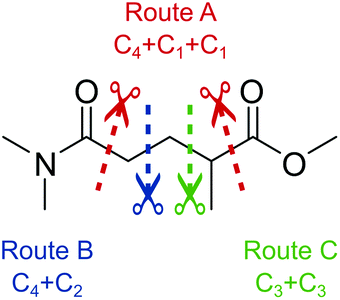
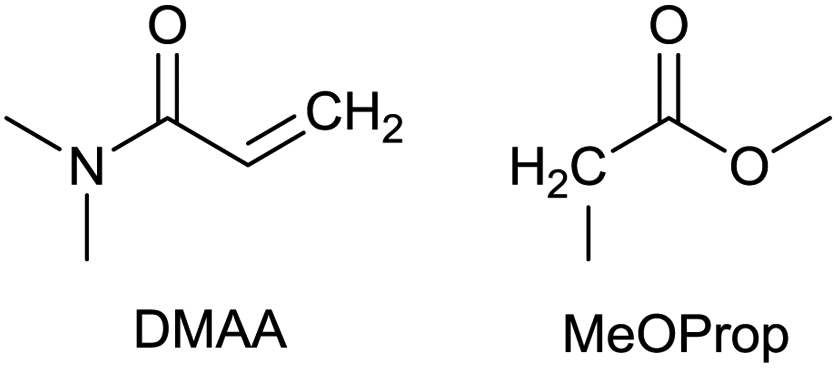
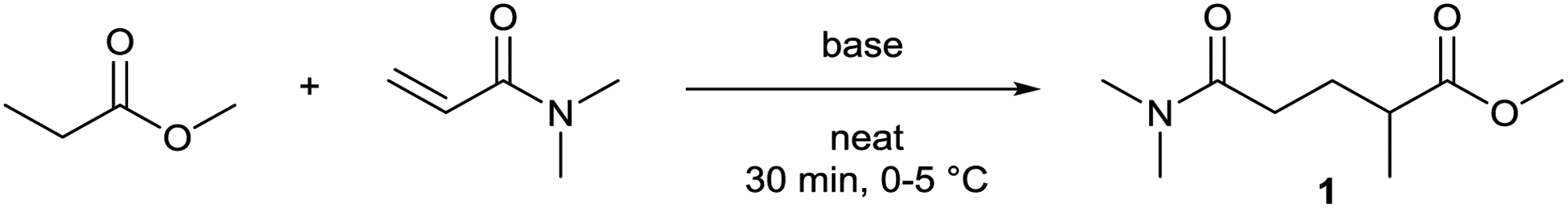
The Michael addition begins with the deprotonation of the C–H acidic reagent, in this case DMAc. The pKa value of DMAc is around 29.4;23 therefore a very strong base, such as lithium diisopropylamide (LDA; pKa of conjugated acid: 35.724) is required. Michael reactions of the Li-enolate form of DMAc with other α,β-unsaturated carbonyl compounds have been reported previously.25 In line with our expectations, the reaction proceeded swiftly at −78 °C and served 1B after acidic work-up and vacuum distillation. The synthesis provided 1 in a single step as opposed to the 4–6 step commercial routes. Consequently, the 8th principle of green chemistry has been addressed.26 However, the stoichiometric amount of the pyrophoric and highly reactive LDA poses a safety hazard, while the −78 °C reaction temperature requires substantial energy. Furthermore, the purification including the acidic work-up and extraction generates both aqueous and organic waste. These issues ultimately originate from the inherently weak C–H acidic character of DMAc. The C3 + C3 strategy allows the utilisation of the higher C–H acidity of esters (pKa: ∼25). Similarly to DMAc and MMA, the building blocks methyl propionate (MeOProp) and dimethylacrylamide (DMAA) are also readily available and they can potentially be obtained through renewable based production.21,22 In route C, potassium tert-butoxide (KOtBu) was used as a base. In polar solvents, such as DMSO or HMPA, KOtBu has enhanced basicity, which allows the reversible deprotonation of weak C–H acids.27 To emulate this polar environment through the high concentration of DMAA and to minimise waste, no solvent was used, in line with the 5th principle of green chemistry.26 The Michael addition proceeded rapidly in the presence of a catalytic amount of KOtBu and thus the 9th principle of green chemistry has been addressed.26 100% conversion was achieved in less than 2.5 minutes upon addition of the base at the laboratory scale. During the swift exothermic reaction, a considerable amount of heat was evolved which necessitated the cooling of the reaction mixture. Nonetheless, 0–5 °C proved to be sufficient in contrast with the extreme −78 °C for route B. The reaction was quenched upon the addition of saturated aqueous oxalic acid, a less corrosive alternative for HCl,28 and the pure 1C was obtained by vacuum distillation.
Quality assessment
The purity and impurity profile of 1B and 1C was compared with commercial PolarClean from two different batches denoted with X and Y (Table 2). 1C is transparent, while the commercial 1B, PolarCleanX and PolarCleanY have a yellow colour. Solvents and impurities with colours are highly undesirable in the pharmaceutical industry, where discolouration of the product could result in failed batches, even if the analytical purity meets the requirements.29 The UV-Vis spectroscopy based ASTM D1209 Standard Test Method was used to quantify the colour difference.30 Both batches, PolarClean X and Y, showed a medium yellow colour with values of 233 ± 1 and 203 ± 1 on the Pt–Co scale, respectively. The colouration of 1B is stronger with a value higher than the upper end point of the Pt–Co scale (>500). On the other hand, 1C is virtually transparent, as proved by its low Pt–Co colour value (22 ± 1).|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample | GC-MS composition (%) | Yellow colourationa | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| a Platinum–Cobalt color, Test Method D1209.30 | |||||||||
| PolarCleanX | 85.7 | 8.9 | 4.3 | 1.1 | — | — | — | — | 233 ± 1 |
| PolarCleanY | 83.8 | 10.0 | 5.0 | 1.2 | — | — | — | — | 203 ± 1 |
| Ref. 9 | 95.1 | — | Trace | Trace | — | — | 4.6 | — | <10031 |
| 1B | 90.1 | — | — | — | 8.9 | 1.0 | — | — | >500 |
| 1C | >99 | — | — | — | — | — | — | — | 22 ± 1 |

Table 2 shows the quantitative analysis of the solvent components performed by GC-MS. PolarCleanX and PolarCleanY have similar impurity profiles and purities (85.7% and 83.8%, respectively). For both solvents, the main impurity is the regioisomer methyl 4-(dimethylamino)-2-ethyl-4-oxobutanoate (2), while 2,N,N,N′,N′-pentamethylglutaramide (3) and dimethyl 2-methylglutarate (4) are also present in low quantities. The ratio of regioisomers was found to be around 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (1:2) in both cases, which is close to the ratio of 2-methylglutaronitrile and 2-ethylsuccinonitrile reported for the starting material.31 The impurity profile and the absence of methyl 5-(dimethylamino)-4-methyl-5-oxopentanoate regioisomer suggest that the commercially available PolarClean is produced via the patented route A3. However, significantly different purity and impurity profile was reported earlier for commercial PolarClean.9 The impurities present in that case indicate that those solvent batches were produced through a different route (route A1 or A2), which consolidates the demand for a more consistent synthetic procedure. The GC-MS chromatogram shows higher purity of 1B at around 90%, while diphenylbutane isomers contribute the remaining 10%. 5 (1,4-diphenylbutane) and 6 (1,3-diphenylbutane) originate from the lithium diisopropylamide solution, which become enriched in the product as their boiling points are close to that of 1. The presence of aromatic hydrocarbons can also explain the strong yellow colour developed by 1B. As a consequence of this impurity, 1B forms an oil-in-water-type emulsion when diluted with water that can affect and limit its range of applications. This impurity and discolouration can be avoided by using lithium diisoproylamide from a styrene–free synthetic route. 1C has the highest purity (above 99%) among the analysed solvents. Although the formation of by-product 4,N,N,N′,N′-pentamethyl-4-(methoxycarbonyl)pimelamide (8) was observed due to the Michael addition of 1 to DMAA; its substantially higher boiling point enabled its complete retention during vacuum distillation. The GC-MS results confirmed that a solvent with higher purity than the commercial PolarClean can be obtained via the newly proposed synthetic route, which could open new opportunities for its exploitation in fine chemical production.
:1 (1:2) in both cases, which is close to the ratio of 2-methylglutaronitrile and 2-ethylsuccinonitrile reported for the starting material.31 The impurity profile and the absence of methyl 5-(dimethylamino)-4-methyl-5-oxopentanoate regioisomer suggest that the commercially available PolarClean is produced via the patented route A3. However, significantly different purity and impurity profile was reported earlier for commercial PolarClean.9 The impurities present in that case indicate that those solvent batches were produced through a different route (route A1 or A2), which consolidates the demand for a more consistent synthetic procedure. The GC-MS chromatogram shows higher purity of 1B at around 90%, while diphenylbutane isomers contribute the remaining 10%. 5 (1,4-diphenylbutane) and 6 (1,3-diphenylbutane) originate from the lithium diisopropylamide solution, which become enriched in the product as their boiling points are close to that of 1. The presence of aromatic hydrocarbons can also explain the strong yellow colour developed by 1B. As a consequence of this impurity, 1B forms an oil-in-water-type emulsion when diluted with water that can affect and limit its range of applications. This impurity and discolouration can be avoided by using lithium diisoproylamide from a styrene–free synthetic route. 1C has the highest purity (above 99%) among the analysed solvents. Although the formation of by-product 4,N,N,N′,N′-pentamethyl-4-(methoxycarbonyl)pimelamide (8) was observed due to the Michael addition of 1 to DMAA; its substantially higher boiling point enabled its complete retention during vacuum distillation. The GC-MS results confirmed that a solvent with higher purity than the commercial PolarClean can be obtained via the newly proposed synthetic route, which could open new opportunities for its exploitation in fine chemical production.
Green metrics analysis
Green metrics analysis was used to compare the patented synthetic procedures with those proposed in this work (Fig. 2). The key process performance indicators are the number of steps, complexity and ideality. All three previously reported routes have a complexity of 3, meaning that there are three construction steps involved. However, the idealities are different, namely 60%, 75% and 100% for routes A1, A2 and A3, respectively. This indicates a gradual improvement among the patented routes by eliminating the concession steps. Both routes B and C in this work demonstrate an improvement in the key process performance indicators having a complexity of 1 and an ideality of 100% that confirms the simplicity and straightforward nature of the proposed synthetic methodologies.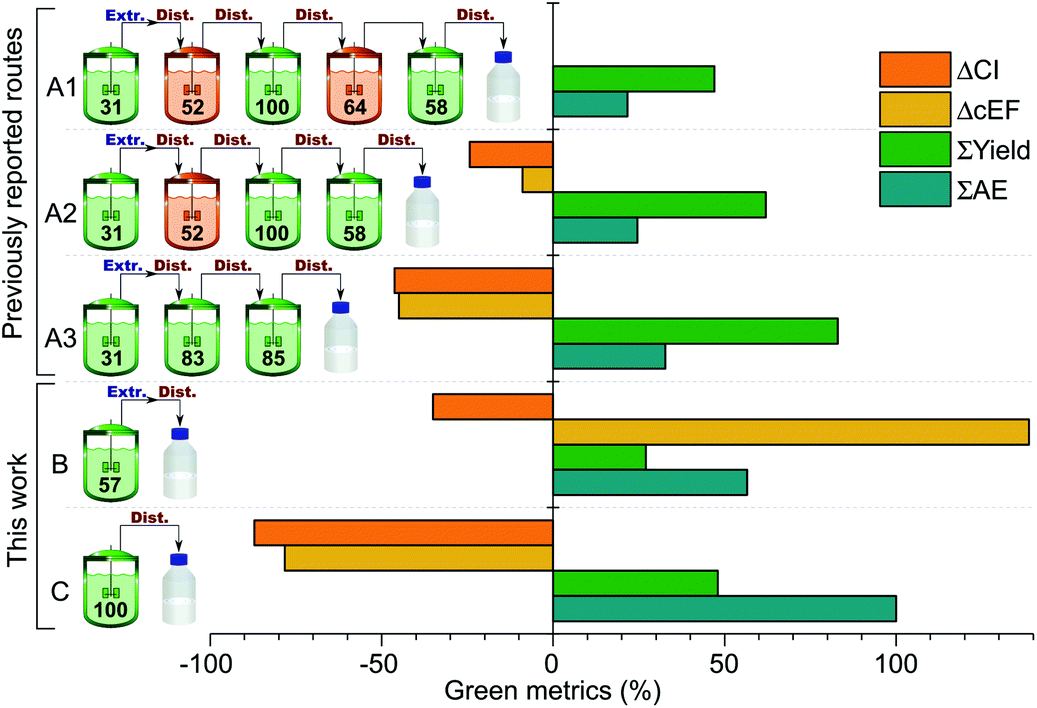 | ||
| Fig. 2 Green metrics comparison of the synthetic routes leading to 1. The green and orange reactors symbolise construction steps and concession steps, respectively. The percentage values in the reactors show the Atom Economy (AE) of that synthetic step. ΔCI and ΔcEF are the relative differences in the estimated CI and the cEF compared to the longest reported route (CI = 222.9 kg kg−1 and cEF = 25.8). ΣYield and ΣAE are the overall yield and cumulated AE, respectively. Extr.: extraction; Dist.: distillation. | ||
The Atom Economy (AE) values of the individual steps in routes A1–3 cover a broad range, from 31% to 100%. The overall AE in these cases falls within the range of 21–33%, indicating that even if the use of exact stoichiometric quantities of starting materials and a chemical yield of 100% is assumed, less than one third of the mass of starting materials is incorporated into the product. In contrast, the single step in route B has a significantly higher AE of 57%, while the only step in route C has an ideal AE of 100%. However, the synthetically less challenging steps in the patented routes result in good overall yields, ranging from 47% for A1 to 83% for A3, while the overall yields for route B and C are 27% and 48%, respectively.
The AEs, the yields and the amount of auxiliary chemicals (such as solvents and catalysts) and water used in the synthesis all have an effect on the Complete E factor (cEF) of a process. The cEF values of the different routes are presented relative to route A1 in Fig. 2. Route A2 and A3 have about 9% and 45% lower cEFs, i.e. waste generated per mass unit of PolarClean produced. On the other hand, route B has a higher cEF owing to the large amount of solvent used in the extraction step. The key reagent LDA was supplied as a solution, which also has an adverse effect on cEF. Route C in contrast represents a drop in cEF by almost 80% relative to route A1, due to the solvent-free synthesis and catalytic amount of base used.
Although cEF is an important measure of the waste generation associated with a chemical process, it does not take into account other important process metrics such as the energy consumption. To achieve an overall view of the sustainability of the different synthetic routes, carbon intensity (CI) was estimated for comparison. The obtained CI values given in kg CO2 eq. per kg of product provide an estimation of the CO2 emissions associated with the laboratory scale synthesis. CI values are presented relative to route A1 in Fig. 2. Similarly to cEF, routes A2 and A3 have lower CIs than A1 due to the fewer synthetic and purification steps and the lower waste generation. Route B has a CI around 35% lower than A1 despite the much higher cEF, which can be attributed to the substantially lower energy consumption of the one-step synthesis and single vacuum distillation. Meanwhile, the relative CI associated with route C is around −87%, representing an almost tenfold decrease in CO2 emissions compared to the patented A1 route.
The breakdown of contributors to the CI of different routes is shown in Fig. 3. In all cases, the two main contributors are the energy consumption associated with the heating and the produced chemical waste, accounting for 71–83% of the totalvalue. In all patented routes, the heat energy constitutes more than half of the total CI. However, this contribution is reduced to below 40% for route B and C due to the fewer synthetic and purification steps.
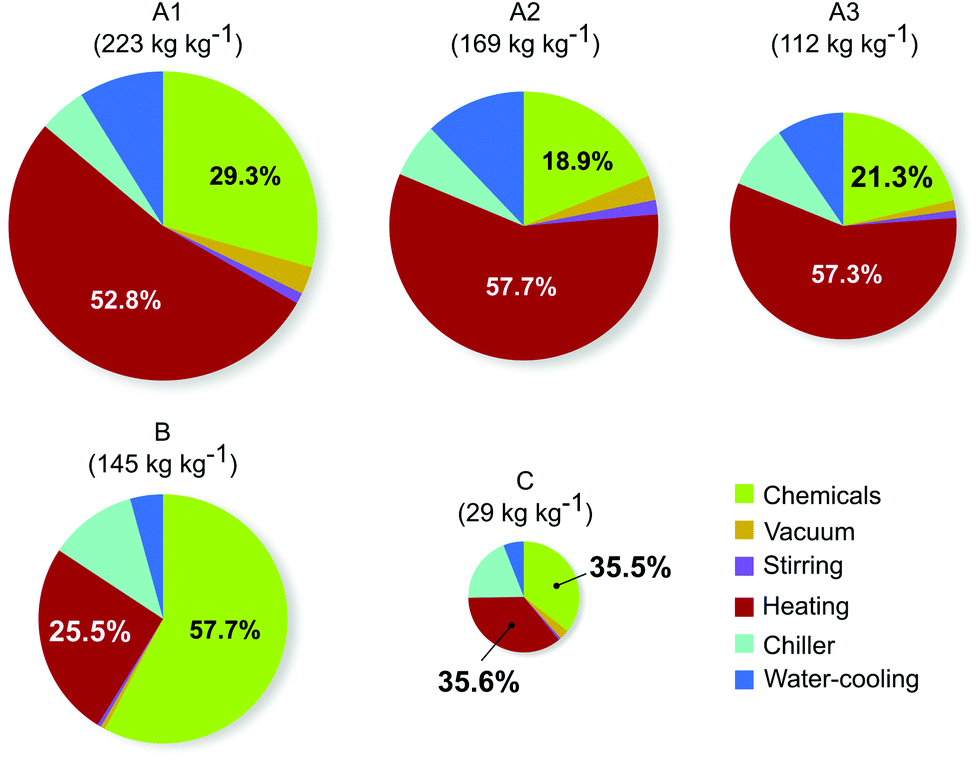 | ||
| Fig. 3 Overall carbon intensities (CI) and their breakdown to the different contributors for synthetic routes A–C leading to 1. The areas of the pie charts correspond to the total CI. | ||
The health and safety risks associated with the chemicals used in the different synthetic routes are summarised in Fig. 4. Instead of focusing on the risks of one specific chemical or another, the figure attempts to give an easily comprehensible, intuitive overview of the chemical hazards through the Globally Harmonized System of Classification and Labelling of Chemicals (GHS) hazard pictograms. The area of the pictograms shown in the figure is proportional to the amount of chemicals (including reagents, catalysts, solvents, isolated intermediates and product) used for the synthesis of unit mass of product via that route. The patented routes exhibit more or less similar chemical hazard profiles but in route A3 there are no unknown chemical hazards originating from the isolated intermediates. Route C demonstrates a clear improvement in the amount and nature of hazardous chemicals. Significantly less (one third to one fifteenth in mass) toxic and corrosive chemicals are used compared to the patented routes, and the need for hazardous chemicals is completely eliminated. Although more chemicals are used in route B in total, none of them are classified as toxic.
 | ||
| Fig. 4 Chemical hazards associated with the different synthetic routes. The area of a pictogram is proportional to the amount of corresponding chemicals used. The question mark denotes chemicals with unknown hazards. | ||
Reaction optimisation
Among the investigated synthetic routes, route C proved to be the most promising in terms of synthetic design, purity and green metrics analysis. The main drawback is the generation of considerable amounts of the double Michael adduct, 8, which can be easily crystallised from the distillation residue of 1C with as high as >99% purity. Although using excess amounts of MeOProp mitigates the by-product formation as it shown in Table 3, it compromises the cEF and CI of the synthesis and more base catalyst is needed to achieve 100% conversion of DMAA. If the high-purity 8 by-product could also be utilised, the overall economy of the proposed synthesis would improve, and the cEF and CI would further decrease by 14% (0.8) and 9% (4 kg kg−1), respectively. For instance, diamides similar in structure to 8 have applications as diuretic agents.32|
|
|||||
|---|---|---|---|---|---|
| Entry | Base [pKa] | Amount of base (mol%) |
n
MeOProp:nDMAA |
Isolated yield of 1 (%) | Relative amounts of 1:8a |
| a Based on the relative peak areas determined by HPLC. b P4-phosphazene. | |||||
| 1 | KOtBu [19.2] | 2.5 | 1:1 |
17 | 30:70 |
| 2 | KOtBu [19.2] | 3 | 2:1 |
32 | 46:54 |
| 3 | KOtBu [19.2] | 5 | 5:1 |
48 | 65:35 |
| 4 | KOtBu [19.2] | 6.5 | 10:1 |
62 | 78:22 |
| 5 | P4-phos.b [42.1] | 2.5 | 1:1 |
14 | 25:75 |
| 6 | P4-phos.b [42.1] | 3 | 2:1 |
22 | 35:65 |
| 7 | BEMP [27.6] | 3 | 2:1 |
0 | — |
| 8 | MTBD [25.4] | 10 | 2:1 |
0 | — |
| 9 | TMG [13.6] | 10 | 2:1 |
0 | — |
| 10 | DBU [12.5] | 10 | 2:1 |
0 | — |
Nonetheless, we hypothesise that the formation of 8 could be eliminated via the development of efficient and selective Brønsted base catalysts. Strong, commercially available non-ionic bases were explored for route C to replace KOtBu. The catalyst candidates were chosen from the bases marked with yellow or green composite flags in GSK's base selection guide.28 The P4-phosphazene base exhibited similar activity and selectivity as KOtBu. However, there was no conversion with weaker organic bases such as P1-phosphazene (BEMP), methyltriazabicyclodecene (MTBD), tetramethylguanidine (TMG) or diazabicycloundecene (DBU).
A slight variation of the MeOProp building block could prevent the addition of a second DMAA molecule and subsequently eliminate the formation of 8. Methyl isobutyrate readily reacts with DMAA in the presence of a catalytic amount of KOtBu in solvent-free conditions to provide 9, an analogue of 1 containing an extra methyl group (Scheme 2). In light of the facile production, 9 could be an attractive green solvent if it proves to have similar non-toxic and non-harmful characteristics to 1.
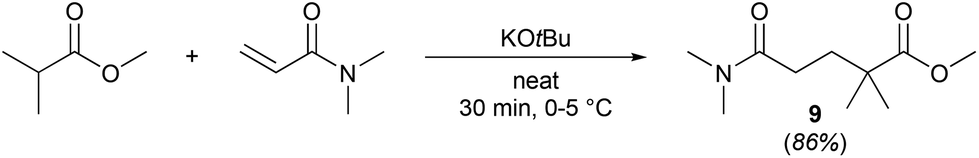 | ||
| Scheme 2 Simple and efficient synthesis of methyl 5-(dimethylamino)-2,2-dimethyl-5-oxopentanoate (9), a close structural analogue of 1. | ||
Extending the application of PolarClean
Nucleophilic aromatic substitution (SNAr) is among the most commonly used chemical transformations in polar aprotic solvents. SNAr reactions are widely used in different fields of chemical synthesis including pharmaceuticals, fine chemicals and polymer syntheses. To evaluate the performance of 1 as a reaction medium and to compare it with other alternative green solvents, O-arylation with SNAr mechanism was selected as a test reaction (Table 4). (4-Phenoxyaryl) aryl sulfones have importance due to their applications in polymer science and also as antiviral agents, advanced optical materials or microelectronic materials.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Solvent dielectric constant | Isolated yield (%) | HPLC purity (%) |
| a The product crystallised upon the addition of water and it was collected by filtration unless otherwise stated. b The product was purified by extraction and subsequent preparative HPLC. | ||||
| 1 | 1C | 28.3 | 91 | 98 |
| 2 | PolarCleanX | 29.9 | 92 | 95 |
| 3 | GVL | 36.5 | 86 | 92 |
| 4 | Cyrene | 37.3 | 11b | 99 |
| 5 | PC | 65.5 | 1b | 37 |
| 6 | DMAc/toluene33 | 37.8/2.4 | 70 | N/A |
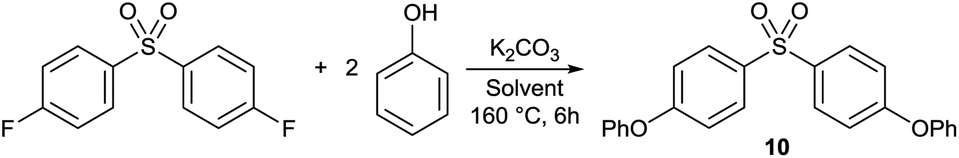
Yields as high as 92% and 91% were achieved for bis(4-phenoxyphenyl) sulfone (10) in PolarCleanX and 1C, respectively. The product crystallised from the reaction mixture and it was isolated by filtration. No by-product of the reaction mixture was observed by HPLC analysis. These results emphasize the chemical stability of 1 and its good solvent properties for SNAr reaction. Among other green solvents, reaction in GVL provided the best performance with a high yield of 86%. The reaction in Cyrene and PC resulted in a complex mixture of compounds indicating the reactive nature of these solvents under SNAr conditions (Fig. S61 and S63, ESI†). The product could not be crystallised, it could only be obtained in low yields after subsequent extraction and preparative HPLC purification steps. The instability of Cyrene in basic media,34 and the use of PC as an alkylating agent for phenols have been reported.35
Solvent recovery experiments were performed to study the feasibility of recycling of 1C after the reaction. Vacuum distillation proved to be a straightforward way to recover 1C with high purity (>99%) from the aqueous mother liquor (Vwater:V1C = 10:3). For the less energy intensive extraction, the aqueous mother liquor was washed with toluene to remove apolar impurities including any residual product or substrate. 1C was retained in the aqueous phase due to its polar nature, however its polarity was sufficiently low to be extracted with EtOAc from the aqueous phase. 1C was obtained with high purity (>99%) after drawing off EtOAc from the extract.
The dielectric constant of 1 has been experimentally determined to be 28.3 ± 3.5, which places it between acetone (20.7) and DMF (36.7) on the polarity scale. The actual value has shown a slight variation depending on the synthetic method and thus the impurity profile, but no direct correlation between the dielectric constants and the yield was found.
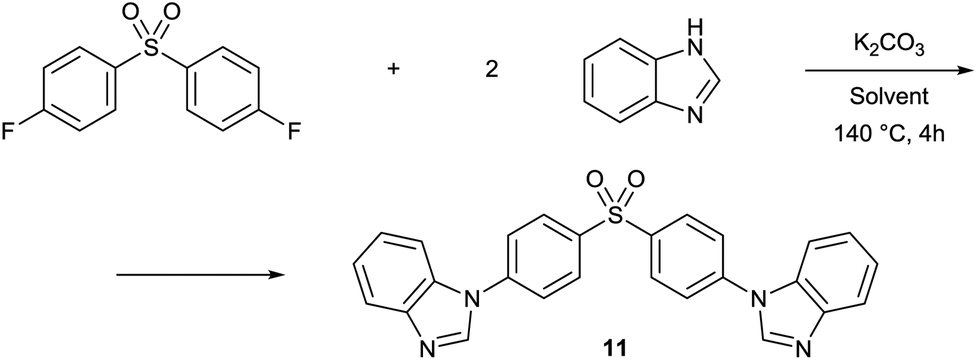
The same set of solvents have been tested for N-arylation of benzimidazole with bis(4-fluorophenyl) sulfone (Table 5). Similarly to the O-arylation, PolarCleanX, 1C and GVL provided the product with good isolated yields ranging 77–87%, while Cyrene and PC did not prove to be inert under the reaction conditions. The test reaction results indicate good stability and solvency of 1 even under the harsh conditions of SNAr reaction, suggesting a broader range of applicability as a reaction medium compared to some other green polar aprotic solvent candidates.
Polar aprotic solvents are essential for polymer synthesis and polymer membrane fabrication.36 The results of the SNAr reactions provide a good indication on the applicability of 1 in polymer synthesis. To study the potential of 1 for membrane fabrication, polymer solubility tests were performed and compared with other aprotic green solvent candidates (Table 6).
| Polymer | Solvent | ||||||
|---|---|---|---|---|---|---|---|
| 1C | PolarcleanX | GVL | Cyrene | PC | MeTHF | ISDME | |
| PBI | + | + | − | − | − | − | − |
| PSf | +++ | +++ | +++ | +++ | − | +++ | +++ |
| PI | + | + | ES | +++ | ES | − | + |
| PAN | + | + | + | + | +++ | − | + |
| PVDF | +++ | +++ | +++ | − | − | − | − |
| PLA | − | − | + | +++ | ES | + | +++ |
| PIM-1 | + | + | + | + | − | +++ | +++ |
| PVA | − | − | − | − | − | − | − |
| PIM-COOH | +++ | +++ | +++ | +++ | + | +++ | +++ |
| PDMBI-I | − | − | − | − | − | − | − |
Both 1C and PolarCleanX exhibited similar solvency. In agreement with previously reported membrane fabrication procedures,13,14 polysulfone (PSf) and polyvinylidene fluorid (PVDF) were soluble in PolarCleanX as well as in 1C. Polybenzimidazole (PBI), polyimide (PI), polyacrylnitrile (PAN)and PIM-1 were dissolved only partially in these solvents. Therefore, 1 is not applicable as a solvent for membrane fabrication from these polymers, although it may find an application as an activating solvent.38 Furthermore the hydrolysed form of PIM-1, PIM-COOH, exhibited good solubility in both PolarCleanX and 1C alongside the majority of tested green polar aprotic solvents. Polyvinylalcohol (PVA) and the anion exchange polymer PDMBI-I (permethylated derivative of PBI) were generally insoluble in all tested solvents.
Because solubility data is indispensable for solvent selection, prediction tools are on the rise. Solubility parameters have been established in the literature to predict polymer solubility in various solvents; for instance the Hildebrand Solubility Parameter (δ) relates the solvency behaviour of a solvent to its heat of vaporisation. The δ value for PolarClean was calculated to be 18.3 MPa0.5 (8.94 cal0.5 cm−1.5) based on experimental vapour pressure data.9 Estimation of δ provided a value of 22.5 MPa0.5 (11.0 cal0.5 cm−1.5) for 1 based on a functional group-contribution method.39 The more commonly used Hansen solubility parameters were estimated using a similar functional group-contribution method.40 Values of 16.6, 13.4 and 9.5 were obtained for the dispersion (δd), polar (δp) and hydrogen bonding (δh) parameters. The miscibility of 1 with 33 traditional and green organic solvents plus water was tested (summarised in Table S18, ESI†). 1B, 1C and PolarCleanX all proved to be miscible with the vast majority of the tested solvents with the highly apolar n-heptane, n-hexane and cyclohexane being the exceptions. These miscibility data are of high importance in solvent selection for multicomponent reaction media, extraction, membrane phase inversion and thin film composite membrane fabrication.
Table 7 shows the resistance of commercially available organic solvent nanofiltration (OSN) membranes in 1 and other green solvents. The results have correlation with the polymer solubility tests shown in Table 6. Membranes made of polymers, which were insoluble in a particular solvent, were expected to show good stability in it and vice versa. The in-house prepared PBI membrane was unstable in 1 but showed good resistance to the other tested solvents. The good solvent properties of 1 caused disintegration of NF030705 and Nadir NP010 in 1, which were stable in some other green solvents. On the contrary, both GMT-oNF-1 and Duramem 900 showed good stability in 1, allowing them to be used for OSN.
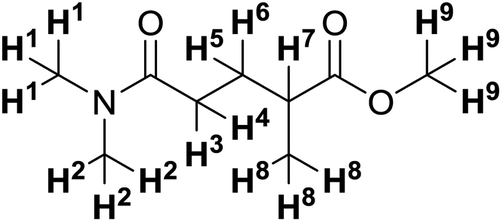
Many relatively new green solvents are side-lined in laboratories due to the lack of experimental data and practical experience working with them. Trace NMR peaks – commonly from solvents – can be tricky to identify although their assignation is essential for reliable data interpretation. Residual NMR peak compilations are available for common solvents41,42 and a recent extension for green solvents has been specifically aimed to advance green chemistry initiatives.43 The trace residual NMR shifts of 1 have been identified in 10 different NMR solvents to provide a reference for chemists (Tables 8 and 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Solvent | 1H Chemical shift (ppm) | |||||
| H1–2 | H3–4 | H5–6 | H7 | H8 | H9 | |
| a Overlapping with solvent signal. b Overlapping with water signal. | ||||||
| CDCl3 | 2.99 (s) | 2.40–2.25 (m) | 2.00–1.90 (m) | 2.54 (h) | 1.19 (d) | 3.68 (s) |
| 2.94 (s) | 1.87–1.78 (m) | |||||
| CD3CN | 2.94 (s) | 2.28 (t) | 1.83 (dq) | 2.49 (dp) | 1.12 (d) | 3.62 (s) |
| 2.83 (s) | 1.66 (dq) | |||||
| CD3OD | 3.05 (s) | 2.39 (t) | 1.90 (dq) | 2.53 (h) | 1.17 (d) | 3.67 (s) |
| 2.92 (s) | 1.73 (dq) | |||||
| (CD3)2SO | 2.93 (s) | 2.26 (t) | 1.76 (dq) | ∼2.47a | 1.08 (d) | 3.59 (s) |
| 2.79 (s) | 1.58 (dq) | |||||
| C4D8O | 2.94 (s) | 2.27 (t) | 1.85 (dq) | ∼2.50b | 1.12 (d) | ∼3.59a |
| 2.83 (s) | ∼1.70a | |||||
| C7D8 | 2.61 (s) | 1.94–2.03 (m) | 2.01–1.92 (m) | 2.48 (h) | 1.07 (d) | 3.34 (s) |
| 2.22 (s) | 1.90–1.78 (m) | |||||
| C6D6 | 2.62 (s) | 2.01 (t) | 2.12–2.03 (m) | 2.53 (h) | 1.08 (d) | 3.33 (s) |
| 2.15 (s) | 1.96–1.86 (m) | |||||
| D2O | 3.04 (s) | 2.40 (t) | 1.86 (dq) | 2.56 (h) | 1.14 (d) | 3.68 (s) |
| 2.89 (s) | 1.72 (dq) | |||||
| (CD3)2CO | 2.99 (s) | 2.31 (t) | 1.86 (dq) | 2.51 (h) | 1.12 (d) | 3.62 (s) |
| 2.83 (s) | 1.69 (dq) | |||||
| TFA | 3.27 (brs) | 2.76 (br) | 1.88 (br) | 2.64 (br) | 1.17 (br) | 3.73 (brs) |
| 3.19 (brs) | ||||||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Solvent | 13C Chemical shift (ppm) | ||||||||
| C1–2 | C3 | C4 | C5 | C6 | C7 | C8 | C9 | ||
| a Overlapping with solvent signal. b Cannot be detected under the applied experimental conditions. | |||||||||
| CDCl3 | 37.3 | 35.6 | 172.4 | 39.1 | 29.1 | 31.0 | 177.0 | 17.5 | 51.8 |
| CD3CN | 37.3 | 35.3 | 172.7 | 39.5 | 29.8 | 31.2 | 177.5 | 17.5 | 52.0 |
| CD3OD | 37.7 | 35.8 | 174.9 | 40.1 | 30.1 | 31.7 | 178.3 | 17.6 | 52.1 |
| (CD3)2SO | 36.6 | 34.8 | 171.3 | 38.0 | 28.6 | 29.9 | 176.1 | 16.9 | 51.4 |
| C4D8O | 36.9 | 35.1 | 171.7 | 39.6 | 29.9 | 31.2 | 176.7 | 17.7 | 51.4 |
| C7D8 | 36.0 | 34.7 | 170.9 | 39.0 | 29.3 | 30.7 | 176.2 | 17.5 | 50.9 |
| C6D6 | 36.1 | 34.9 | 171.1 | 39.1 | 29.4 | 30.8 | 176.4 | 17.6 | 51.1 |
| D2O | 37.4 | 35.4 | 175.3 | 38.8 | 28.4 | 30.4 | 179.0 | 16.2 | 52.2 |
| (CD3)2CO | 37.0 | 35.1 | 172.1 | 39.4 | ∼29.8a | 31.1 | 176.9 | 17.5 | 51.6 |
| TFA | —b | —b | 41.4 | 29.7 | 30.7 | —b | 17.6 | 55.3 | |

Conclusion
Two different one-step syntheses of methyl 5-(dimethylamino)-2-methyl-5-oxopentanoate (1) – the main component of the green solvent, PolarClean – have been developed via Michael additions using a retrosynthetic approach. The most advanced, solvent-free synthetic route was optimised in terms of reagent excess and base catalyst selection. An excess of methyl propionate was shown to have a positive effect on the selectivity and yield. KOtBu and organic, non-ionic phosphazene bases were found to be effective catalysts (<6.5 mol%). The produced solvents had excellent purities up to 99% and colourations down to 22 Pt–Co colour units in contrast to the respective values of 83–85% and 203–233 for commercial PolarClean. Green metrics analysis of the synthetic routes revealed that the novel synthesis could cut the Complete E factor and Carbon Intensity associated with the production of 1 by 59–78% and 76–87%, respectively, compared to patented procedures. The efficient synthetic design of the novel routes was also supported by their high Atom Economies of 57% and 100% in contrast to the 22–33% for the patented routes. The sustainability of the proposed synthesis was also demonstrated by the use of low-hazard chemicals employing a new, intuitive approach to quantify process health and safety.1 as a reaction medium was successfully applied for O- and N-arylations with SNAr mechanism for the first time to replace traditional polar aprotic solvents. 1 showed good stability under the harsh reaction conditions, and resulted in good yields of 77–91%, which are similar or better than green polar aprotic solvent candidates. 1 was recovered from the post-reaction mixtures in excellent purities (>99%) both with vacuum distillation and extraction. NMR chemical shifts in trace amounts were recorded in 10 different common NMR solvents. Applications of 1 in polymer and membrane science have been proposed. Solubility tests of 8 polymers and 4 commercial membranes revealed that 1 is (i) an excellent solvent for processing 3 of the polymers, and (ii) suitable as a filtration medium using two of the membranes. Hildebrand and Hansen solubility parameters were calculated. In conclusion, the novel green and straightforward synthesis, the new exemplary applications and the practical experimental characterisation of 1 presented in this work can pave the way for a replacement for common toxic polar aprotic solvents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Ms Hai Anh Le Phuong and Mr Fan Fei (both from The University of Manchester) for their assistance with UV-Vis spectroscopy and capacitance measurements, respectively.Notes and references
- S. Axon and D. James, Curr. Opin. Green Sustainable Chem., 2018, 13, 140–145 CrossRef.
- L. Cseri, M. Razali, P. Pogany and G. Szekely, Organic Solvents in Sustainable Synthesis and Engineering, in Green Chemistry: An inclusive Approach, ed. B. Török and T. Dransfield, Elsevier, Oxford, 2017, ch. 3.15, pp. 513–553 Search PubMed.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS.
- J. S. Bello Forero, J. A. Hernández Muñoz, J. Jones Junior and F. M. da Silva, Curr. Org. Synth., 2016, 13, 834–846 CrossRef CAS.
- J. Sherwood, H. L. Parker, K. Moonen, T. J. Farmer and A. J. Hunt, Green Chem., 2016, 18, 3990–3996 RSC.
- A. Randová, L. Bartovská, P. Morávek, P. Matějka, M. Novotná, S. Matějková, E. Drioli, A. Figoli, M. Lanč and K. Friess, J. Mol. Liq., 2016, 224, 1163–1171 CrossRef.
- US Environmental Protection Agency, Methyl 5-(dimethylamino)-2-methyl-5- oxo pentanoate; Exemption from the Requirement of a Tolerance, Fed. Regist., 2013, 78, 32157–32161 Search PubMed.
- T. Vidal, V. Bramati, K. Murthy and B. Abribat, J. ASTM Int., 2011, 8, 1–8 Search PubMed.
- N. T. Hassankiadeh, Z. Cui, J. H. Kim, D. W. Shin, S. Y. Lee, A. Sanguineti, V. Arcella, Y. M. Lee and E. Drioli, J. Membr. Sci., 2015, 479, 204–212 CrossRef CAS.
- T. Marino, E. Blasi, S. Tornaghi, E. Di Nicolò and A. Figoli, J. Membr. Sci., 2018, 549, 192–204 CrossRef CAS.
- H. H. Wang, J. T. Jung, J. F. Kim, S. Kim, E. Drioli and Y. M. Lee, J. Membr. Sci., 2019, 574, 44–54 CrossRef CAS.
- X. Dong, A. Al-Jumaily and I. C. Escobar, Membranes, 2018, 8, 23 CrossRef.
- L. Luciani, E. Goff, D. Lanari, S. Santoro and L. Vaccaro, Green Chem., 2018, 20, 183–187 RSC.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Eur. Polym. J., 2015, 67, 409–417 CrossRef CAS.
- T. Lebarbé, A. S. More, P. S. Sane, E. Grau, C. Alfos and H. Cramail, Macromol. Rapid Commun., 2014, 35, 479–483 CrossRef.
- L. M. M. Mouterde, F. Allais and J. D. Stewart, Green Chem., 2018, 20, 5528–5532 RSC.
- O. Jentzer and M. Guglieri, Rhodia Operations, US Pat., 20140221211, 2014 Search PubMed.
- P. F. H. Harmsen, M. M. Hackmann and H. L. Bos, Biofuels, Bioprod. Biorefin., 2014, 8, 306–324 CrossRef CAS.
- P. N. R. Vennestrom, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef CAS.
- J. P. Richard, G. Williams, A. C. O'Donoghu and T. L. Amyes, J. Am. Chem. Soc., 2002, 124, 2957–2968 CrossRef CAS.
- R. R. Fraser and T. S. Mansour, J. Org. Chem., 1984, 49, 3442–3443 CrossRef CAS.
- N. Shinohara, J. Haga, T. Yamazaki, T. Kitazume and S. Nakamura, J. Org. Chem., 1995, 60, 4363–4374 CrossRef CAS.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Jankovic, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- D. Caine, Potassium tert-Butoxide, e-EROS Encycl. Reagents Org. Synth., 2006 DOI:10.1002/047084289X.rp198.pub2.
- R. K. Henderson, A. P. Hill, A. M. Redman and H. F. Sneddon, Green Chem., 2015, 17, 945–949 RSC.
- P. D. Oram and J. Strine, J. Pharm. Biomed. Anal., 2006, 40, 1021–1024 CrossRef CAS.
- ASTM Standard D1209, 2005 (2011), Color of Clear Liquids (Platinum-Cobalt Scale), ASTM International, West Conshohocken, PA, 2011, DOI:10.1520/D1209-05R11, http://www.astm.org.
- P. Leconte and C. Denis, Rhodia Operations, US Pat., 20090326260, 2009 Search PubMed.
- I. T. Barnish, J. C. Danilewicz, K. James, G. M. R. Samuels, N. K. Terrett, M. T. Williams and M. J. Wythes, Pfizer Inc., US Pat., 5030654, 1991 Search PubMed.
- D. Izuhara, H. Umeda, E. Amano and T. Kunita, Toray Industries Inc., US Pat., 9126908, 2015 Search PubMed.
- H. A. Le Phuong, L. Cseri, G. F. S. Whitehead, A. Garforth, P. Budd and G. Szekely, RSC Adv., 2017, 7, 53278–53289 RSC.
- T. Tabanelli, C. Giliberti, R. Mazzoni, R. Cucciniello and F. Cavani, Green Chem., 2019, 21, 329–338 RSC.
- P. Marchetti, M. F. Jimenez Solomon, G. Szekely and A. G. Livingston, Chem. Rev., 2014, 114, 10735–10806 CrossRef CAS.
- I. I. Ponomarev, I. I. Ponomarev, Y. A. Volkova, M. Yu. Zharinova and D. Yu. Razorenov, Mendeleev Commun., 2012, 22, 162–163 CrossRef CAS.
- M. F. Jimenez Solomon, Y. Bhole and A. G. Livingston, J. Membr. Sci., 2012, 423–424, 371–382 CrossRef CAS.
- E. Stefanis, L. Constantinou and C. Panayiotou, Ind. Eng. Chem. Res., 2004, 43, 6253–6261 CrossRef CAS.
- E. Stefanis and C. Panayiotou, Int. J. Thermophys., 2008, 29, 568–585 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- N. R. Babij, E. O. McCusker, G. T. Whiteker, B. Canturk, N. Choy, L. C. Creemer, C. V. De Amicis, N. M. Hewlett, P. L. Johnson, J. A. Knobelsdorf, F. Li, B. A. Lorsbach, B. M. Nugent, S. J. Ryan, M. R. Smith and Q. Yang, Org. Process Res. Dev., 2016, 20, 661–667 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods; synthetic procedures; green analysis data; instrumental procedures; solvent miscibility table, NMR, MS, FTIR spectra; GC and HPLC chromatograms. See DOI: 10.1039/c9gc01958h |
| This journal is © The Royal Society of Chemistry 2019 |