
Catalytic oxidative dehydrogenation of malic acid to oxaloacetic acid†
Karine
De Oliveira Vigier  a
and
François
Jérôme
*a
a
and
François
Jérôme
*a
a
and
François
Jérôme
*a
Abstract
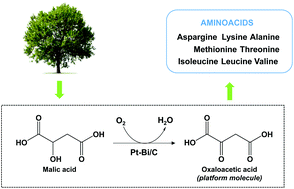
Here we report the oxidative dehydrogenation of malic acid to oxaloacetic acid, a key precursor in the fabrication of amino acids, over Pt–Bi/C catalysts. Under optimized conditions, we discovered that OAA was selectively produced with up to 60% conversion (i.e. 60% yield). The recurrent unwanted decarboxylation of OAA to pyruvic acid was circumvented by successfully conducting the catalytic reaction at 25 °C. A comparison with the classical Fenton oxidation reaction is discussed.
- This article is part of the themed collections: International Symposium on Green Chemistry 2019 and 2019 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

