
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGVL pulping facilitates nanocellulose production from woody biomass
Mingjie
Chen
 a,
Qianli
Ma
b,
J. Y.
Zhu
c,
David
Martin Alonso
d and
Troy
Runge
*a
a,
Qianli
Ma
b,
J. Y.
Zhu
c,
David
Martin Alonso
d and
Troy
Runge
*a
aDepartment of Biological System Engineering, University of Wisconsin-Madison, Madison, WI 53726, USA. E-mail: trunge@wisc.edu
bState Key Lab Pulp and Paper Engineering, South China University of Technology, 381 Wushan Road, Guangzhou 510630, China
cUSDA Forest Service, Forest Products Laboratory, Madison, WI 53726, USA
dGlucan Biorenewables LLC, 505 South Rosa Road. Suite 112, Madison, WI 53719, USA
First published on 2nd September 2019
Abstract
Nanocellulose is conventionally produced from woody biomass including wood, crops and forest/agricultural residues, by top-down methods. Due to biomass recalcitrance, pretreatment, with a subsequent bleaching process, is mandatory to break down the resistance of the composite for nanocellulose extraction. In the present study, gamma-valerolactone (GVL) pulping without further bleaching/purification was used for producing cellulose nanomaterials from wood. GVL pulp and bleached kraft pulp from Aspen were comparatively studied as starting materials for (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) cellulose nanofibril (TCNF) preparation. The TEMPO oxidation process and properties of the as-prepared TCNF solutions and films were studied to investigate the advantages of GVL pulp for TCNF production. There was no difference between nanocellulose prepared from GVL pulp and that from bleached kraft pulp. But, with the GVL pulping process, no subsequent bleaching process was required while the properties of the as-prepared nanocellulose were preserved.
1. Introduction
Nanocellulosic materials have interesting mechanical and biological properties that have created significant interest for medical and material applications.1–4 Nanocellulose is conventionally produced from woody biomass, wood or forest/agricultural residues, by intensive processes involving enzymatic/chemical/physical operations with typically numerous steps.5 Woody biomass is a natural composite of lignin, cellulose and hemicellulose6 that has a structure naturally resistant to degradation and separation into individual components.7 As a result, breaking down the resistance of the composite is an important prior step to extract nanocellulose8 from woody biomass which is usually accomplished with a pretreatment/pulping step.Purified cellulose from traditional pulping processes is typically used as feedstock to produce nanocellulose. However, extraction of purified cellulose by traditional kraft pulping is expensive and requires large amounts of capital equipment; other methods have been developed for nanocellulose extraction. For example, Zhu's group extracted nanocellulose by an aqueous p-TsOH hydrotrope treatment of wood at low temperatures (≤80 °C) followed by mechanical fibrillation.9–11 In this process, lignin is deposited onto the as-prepared nanocellulose, resulting in hydrophobic nanocellulose films with dark colour.9 Li et al. extracted nanocellulose from wood flour by ethanol organosolv pretreatment followed by alkaline hydrogen peroxide treatment and ultrasonic nanofibrillation.12 However, an alkaline hydrogen peroxide bleaching process was required to obtain cellulose with high purity.12 Lee et al. extracted cellulose nanocrystals from electron-beam irradiation pretreated pulp.13 However, subsequently an alkaline solution was applied for isolation purpose,13 which will make the process more complex. A major issue of existing biomass pretreatment/pulping methodologies is that pulps, generally, with various contents of lignin and/or hemicellulose and dark colours are obtained.
Although nanocellulose could be extracted from unbleached pulps, with lignin and/or hemicellulose,10 the presence of lignin and hemicellulose has significant effects on the nanocellulose extraction and the properties of the resulting nanocellulose.14 The dark colours from lignin would be passed to the resultant nanocellulose,11 which will limit its application in various fields, such as cosmetics. Rojo et al. showed that the presence of lignin results in an increase in hydrophobicity of nanocellulose films and reduces the oxygen permeability of nanocellulose films by up to 200-fold.15 Horseman et al. showed that lignin containing cellulose nanofibrils show lower thermal stability, mechanical properties and water uptake ability than reference cellulose nanofibrils.16 Meng et al. showed that the removal of xylan facilitates (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) oxidation of pulps and results in a nanocellulose solution with high light transmittance.17 In summary, it is crucial to use purified cellulose as a starting material to obtain nanocellulose with desired properties and limited energy/chemical exhaust of the process.
A bleaching process after pretreatment/pulping is generally applied to obtain cellulose of high purity and light/white colour in production of nanocellulose from woody biomass, with typically employing hydrogen peroxide and chlorine dioxide.18,19 However, such bleaching processes are costly from both economic and environmental standpoints.20,21
Here, we come to a question whether it is possible to eliminate the bleaching process without any significant effects on the nanocellulose extraction from woody biomass, while maintaining the properties of highly purified nanocellulose. Previous studies have produced nanocellulose without bleaching,10,11,16,17,22 but these studies have not produced nanocellulose with similar qualities as bleached materials. This has not been accomplished because fractionating cellulose to high purity is a crucial step in nanocellulose extraction from woody biomass. It is difficult to achieve the desirable properties of nanocellulose similar to those of nanocellulose produced from bleached kraft pulps, as it would require a process that removes almost all of the lignin while not degrading the cellulose. Fortunately, our recent studies showed that lignin content in the pulp could be tuned by a gamma-valerolactone (GVL) pulping process, while most hemicelluloses are removed, resulting in cellulose of high purity and a light colour. These results led us to hypothesize that these pulps could be used to produce nanocellulose of equal quality to that produced with bleached kraft pulp but without the costly bleaching process or modification of the obtained highly purified nanocellulose.23
The use of the GVL pulping process has several environmental advantages over traditional processes. First, the process allows the removal of bleaching processes that typically use chlorine-based chemicals and produce significant aqueous pollutants.21 Second, GVL is a biobased solvent that can be produced from biomass and possibly integrated into the pulping process as described by Alonso et al.23 Additionally, GVL allows for lower temperature pulping processes which further reduces the environmental impacts and has a lower vapour pressure than most other solvents considered for biomass pulping.23
Herein, to elucidate that GVL pulping is a suitable pretreatment process for nanocellulose extraction from wood, GVL pulp and bleached kraft pulp from Aspen wood chips were comparatively studied as starting materials for TEMPO cellulose nanofibril (TCNF) preparation. The TEMPO oxidation process and properties of the as-prepared TCNF solutions and TCNF films were studied to analyze the advantages of GVL pulp for TCNF production.
2. Results and discussion
2.1. Pulp preparation
Previous studies have shown that the presence of lignin and hemicellulose in pulp would consume excess NaClO and NaOH during TCNF preparation and also have a significant effect on the properties of the produced TCNF.17,24–26 Therefore, feedstocks of high cellulose purity are desired in the TEMPO oxidation processes of TCNF preparation. We previously demonstrated that GVL pulping is an advanced biomass fraction process suitable for producing dissolving pulps that makes use of a green solvent and lower temperatures than conventional processes.23 Herein, GVL pulp (Fig. 1) composed of 1.5% Klason lignin, 95.8% glucan and 2.7% pentosan was prepared from Aspen wood chips by GVL pulping at 125 °C for 3 hours using a 70/30 GVL/water mass ratio and 0.1 M sulfuric acid as the catalyst. | ||
| Fig. 1 GVL and bleached kraft pulp prepared from Aspen. | ||
The effect of reaction parameters on the resultant pulp was investigated and previously discussed.23 The results of that study indicated a narrow range of reaction parameters that would allow for efficient separation of cellulose, hemicellulose, and lignin without excessive degradation. This study used those optimal reaction conditions but further investigated the effect of residual lignin content of the resultant pulp altered by the washing process, which was decreased from 10.4% to 1.5% through repeated washing with 70/30 GVL/water as shown in Table 1. We tried producing nanocellulose with each of the four pulps but only the low lignin pulp (1.5 wt% lignin) from the 3 times washed GVL sample produced an acceptable nanocellulose dispersion, with the higher lignin content samples resulting in hazy yellow nanocellulose, indicating the importance of almost complete removal of lignin in the initial feedstock. Based on these experiments, the 3× washed GVL pulp sample was chosen for the full analysis and comparison to typical bleached kraft pulp.
| GVL washing | ×0 times | ×1 time | ×2 times | ×3 times |
|---|---|---|---|---|
| Lignin content of GVL pulp | 10.4% ± 0.2% | 7.8% ± 0.1% | 2.6% ± 0.3% | 1.5% ± 0.2% |
Delignification by GVL pulping was due to the promotion of acid catalysed cleavage of β-O-4 linkages by GVL and the excellent solubility of resultant lignin in GVL/water. The cleavage of β-O-4 linkages was confirmed by HSQC studies. As shown in Fig. 2, the relative content of β-O-4 linkages dramatically decreased from 86% to 29%, while the relative content of β–β and β-5 linkages increased from 8% and 6% to 55% and 16%, respectively. Also, the aromatic signals of both G and S units (Fig. 2) of the GVL lignin are shown to be extensively overlapped due to the acid catalysed depolymerization and subsequent condensation27,28 of lignin. These results confirmed the acid catalysed cleavage of β-O-4 linkages by the GVL pulping process, which subsequently resulted in delignification of the Aspen wood.
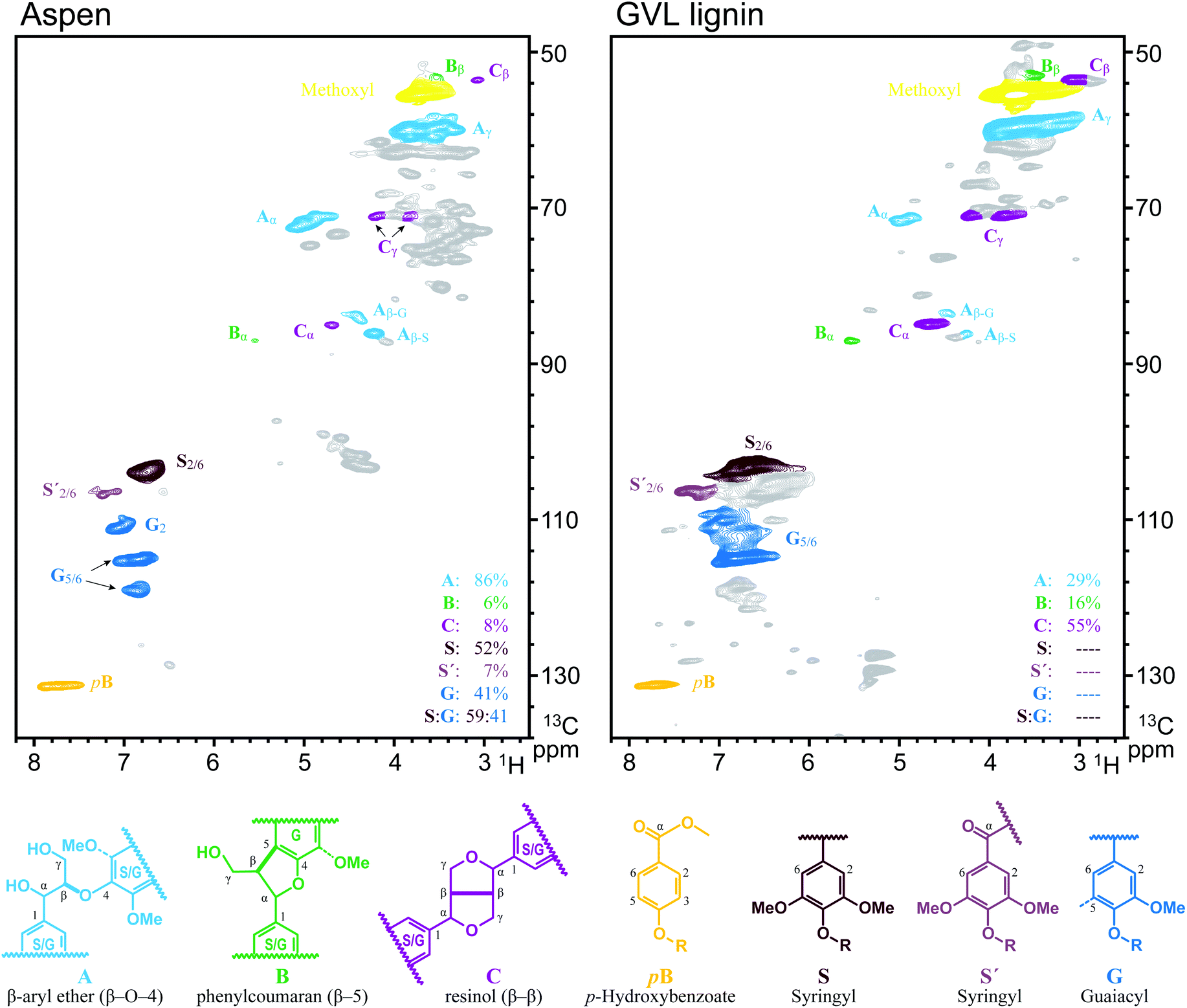 | ||
| Fig. 2 HSQC studies of lignin. | ||
For comparison, bleached kraft pulp (Fig. 1) composed of 0.5% Klason lignin, 77.5% glucan and 22.0% pentosan was prepared at an active alkali dosage of 18 wt% with a sulfidity of 30%, a liquid/solid ratio of 5, an H factor of 1200, and the D(EP)D bleaching sequence according to ref. 29. Both of these two pulps were then used for TCNF production. The GVL pulp had a light colour (Fig. 1) due to extensive delignification by the GVL pulping process. Also, compared to the bleached kraft pulp, the GVL pulp showed higher purity of glucan (95.8%, Fig. 1), which is primarily attributed to cellulose. These results suggested that the GVL pulp could be an ideal feedstock for TCNF preparation due to its low lignin and hemicellulose content, resulting in a high purity cellulose composition.
2.2. Oxidation of the pulp by the TEMPO process
The as-prepared GVL pulp was used for TCNF preparation by oxidation at the C6 position (Fig. 3A) with TEMPO, sodium bromide, and sodium hypochlorite solution. The oxidation reaction was confirmed by FTIR studies and HSQC studies. As shown in Fig. 3B, new peaks at 1600 cm−1 and 1408 cm−1 were observed for the TCNF samples, both from GVL pulp and bleached kraft pulp. These two peaks are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and C–O bond stretching vibrations from carboxylate groups (–COO−), respectively.30,31 The presence of these peaks in TCNF samples suggests the successful oxidation of the pulp by the TEMPO process. The peaks at 3275 cm−1 and 710 cm−1 are attributed to the cellulose Iβ allomorph.32 The peak at 897 cm−1 is attributed to C1–H of β-(1,4) glycosidic linkage of cellulose.33 These peaks, due to cellulose Iβ and β-(1,4) glycosidic linkage, were preserved in the TCNF samples, indicating that the oxidation proceeded at the surface of the cellulose crystal,24,34 leaving the bulk of the cellulose crystal intact.
O bond and C–O bond stretching vibrations from carboxylate groups (–COO−), respectively.30,31 The presence of these peaks in TCNF samples suggests the successful oxidation of the pulp by the TEMPO process. The peaks at 3275 cm−1 and 710 cm−1 are attributed to the cellulose Iβ allomorph.32 The peak at 897 cm−1 is attributed to C1–H of β-(1,4) glycosidic linkage of cellulose.33 These peaks, due to cellulose Iβ and β-(1,4) glycosidic linkage, were preserved in the TCNF samples, indicating that the oxidation proceeded at the surface of the cellulose crystal,24,34 leaving the bulk of the cellulose crystal intact.
 | ||
| Fig. 3 Oxidation of pulp by the TEMPO process. | ||
To further validate the oxidation at C6 of cellulose, gel-state HSQC studies were performed. Chemical signal assignments are according to ref. 35. A new peak at 77.8/3.66 ppm was observed for both TCNFs derived from GVL pulp and kraft pulp (Fig. 3D and E). This peak is attributed to C5′/H5′ associated with C6 carboxylate groups (Fig. 3), suggesting the oxidation at the C6 position of the cellulose glucose unit. Two peaks at 77.8/3.66 ppm and 76.6/33.3 ppm, attributed to C5′/H5′ and C5/H5 of oxidized and non-oxidized cellulose, respectively, were observed for the TCNF samples. This result indicates the partial oxidation of primary alcohol into carboxylate at the C6 position of glucose units, consistent with the results from FTIR studies showing that oxidation occurred at the surface of cellulose crystals, leaving the bulk of the cellulose crystal intact.
A new peak at 96.7/4.48 ppm, assigned to C1/H1 of the reducing end glucose unit of cellulose, was observed for both of the TCNF samples. The presence of the HSQC signal of the reducing end glucose unit for TCNF samples is, probably, due to depolymerization of cellulose during the TEMPO process,36,37 which increases the concentration of the reducing end glucose unit to a detectable level.
To demonstrate the depolymerization of cellulose during the TEMPO process, the average viscometric DP of the pulps and oxidized pulp was determined according to ASTM D4243-16. The DPs of GVL pulp and kraft pulp were 472 and 724, respectively. The DPs of oxidized GVL pulp and oxidized kraft pulp prepared with a NaClO dosage of 4.8 mmol g−1 were 241 and 276, respectively. It was expected that the oxidized pulps have a lower DP than the original pulp, since depolymerization of cellulose by the TEMPO process has been extensively proven.24,37 It was interesting that the oxidized GVL pulp has a DP (241) close to that of oxidized kraft pulp (276), even though the DP of kraft pulp (724) is noticeably higher than that of GVL pulp (472). The results of the FTIR, HSQC, and DP studies suggest that there is no detectable significant chemical differences between GVL pulp and bleached kraft pulp in application of the TEMPO nanocellulose production process.
The as-prepared GVL pulp was demonstrated to be an excellent candidate for TCNF production by analyzing the yield, NaOH consumption of the TEMPO process, and carboxylate content of the oxidized pulp. As shown in Table 2, the yield of pulp oxidation by the TEMPO process was ∼100%, regardless of the NaClO dosage and origin of pulp. There were even samples with yield higher than 100%, which was attributed to the system error of the determination method (by weight after oven drying). The NaOH consumption of the bleached kraft was in the range of 0.37–1.60 mmol g−1 with a NaClO dosage of 1.2–4.8 mmol g−1, while the carboxylate group content was 0.38–1.11 mmol g−1. In the case of the GVL pulp, the NaOH consumption and carboxylate group content were 0.34–1.57 mmol g−1 and 0.34–1.17 mmol g−1, respectively. These results demonstrate that the GVL pulp and bleached kraft pulp behaved similarly during the TEMPO oxidation process.
| Samples | NaClO dosage to pulp (mmol g−1) | Yield of oxidized pulp (%) | Carboxylate content of oxidized pulp (mmol g−1) | NaOH consumption (mmol g−1) |
|---|---|---|---|---|
| Oven dried bleached kraft pulp | 1.2 | 101.5 ± 0.1 | 0.38 ± 0.01 | 0.37 ± 0.03 |
| 2.4 | 99.8 ± 0.3 | 0.83 ± 0.01 | 0.99 ± 0.09 | |
| 4.8 | 101.8 ± 0.7 | 1.11 ± 0.07 | 1.60 ± 0.07 | |
| Over dried GVL pulp | 1.2 | 102.3 ± 1.0 | 0.34 ± 0.01 | 0.34 ± 0.07 |
| 2.4 | 99.0 ± 0.1 | 0.70 ± 0.03 | 0.89 ± 0.18 | |
| 4.8 | 101.2 ± 1.1 | 1.17 ± 0.03 | 1.57 ± 0.10 | |
| Freeze dried GVL pulp | 4.8 | 101.6 ± 1.0 | 1.09 ± 0.01 | 1.60 ± 0.09 |
| Never dried GVL pulp | 4.8 | 101.0 ± 1.1 | 1.15 ± 0.03 | 1.74 ± 0.14 |
The reason why GVL and kraft pulps behave similarly during TEMPO oxidation is that the oxidation proceeds at the surface of the cellulose crystal, but not the bulk of the cellulose crystal, as elucidated above. Though the DP of the GVL pulp was noticeably lower than that of the kraft pulp, FTIR results showed that both pulps displayed a cellulose Iβ crystalline structure. The cellulose Iβ polymorph is a well-known native cellulose polymorph,38 which suggests that both GVL and kraft pulping preserve the native cellulose crystallinity of Aspen wood chips. Thus, the cellulose of both GVL and kraft pulps should have similar crystal surfaces regardless of the difference in their DPs, which contributes to the similar behaviours of GVL and kraft pulps during the TEMPO oxidation process.
Pulps were conventionally dried under different conditions, such as oven drying and freeze drying. Since drying is known to have a tremendous impact on the properties of pulp39–42 drying methods were investigated. Thus, oven dried, freeze dried and never dried GVL pulps were applied to the TEMPO oxidation process. The yield, NaOH consumption, and carboxylate group content of all these pulps were measured and found to be similar, suggesting that the drying processes had no effect on the oxidation of GVL pulp.
2.3. Properties of TCNFs in solution
Nanocellulose was prepared from oxidized GVL pulp (1.17 mmol g−1 carboxylate groups) and bleached kraft pulp (1.11 mmol g−1 carboxylate groups) by a homogenization process. Photos of the as-prepared TCNF solution in water (0.1 wt%) are presented in Fig. 4a. Both the GVL and kraft TNCF solutions were shown to be highly transparent due to extensive delignification and the high purity of polysaccharides of the pulps. No visible difference was observed between the GVL TCNF and kraft TCNF solutions because of the similar chemical structure, DP and crystallinity of the oxidized pulps. The sizes of TCNFs were further studied by TEM (Fig. 4b). The diameters of GVL and kraft TCNFs measured by TEM were in the range of 8–26 nm and 6–26 nm, respectively. The diameter distribution was fitted by the Lorentz function. The peaks of the Lorentz fitted curve were found to be similar at 12.6 nm and 12.0 nm for GVL TCNFs and kraft TCNFs, respectively. Additionally, these diameters are in the TCNF typical diameter range of TEMPO nanocellulose reported by previous studies.24,43–45 The data show that the TCNFs from both GVL and kraft pulps have a similar diameter/diameter distribution, likely because of homogenization, a process that delaminates the cellulose microfiber and liberates the cellulose crystal as nanofibrils.5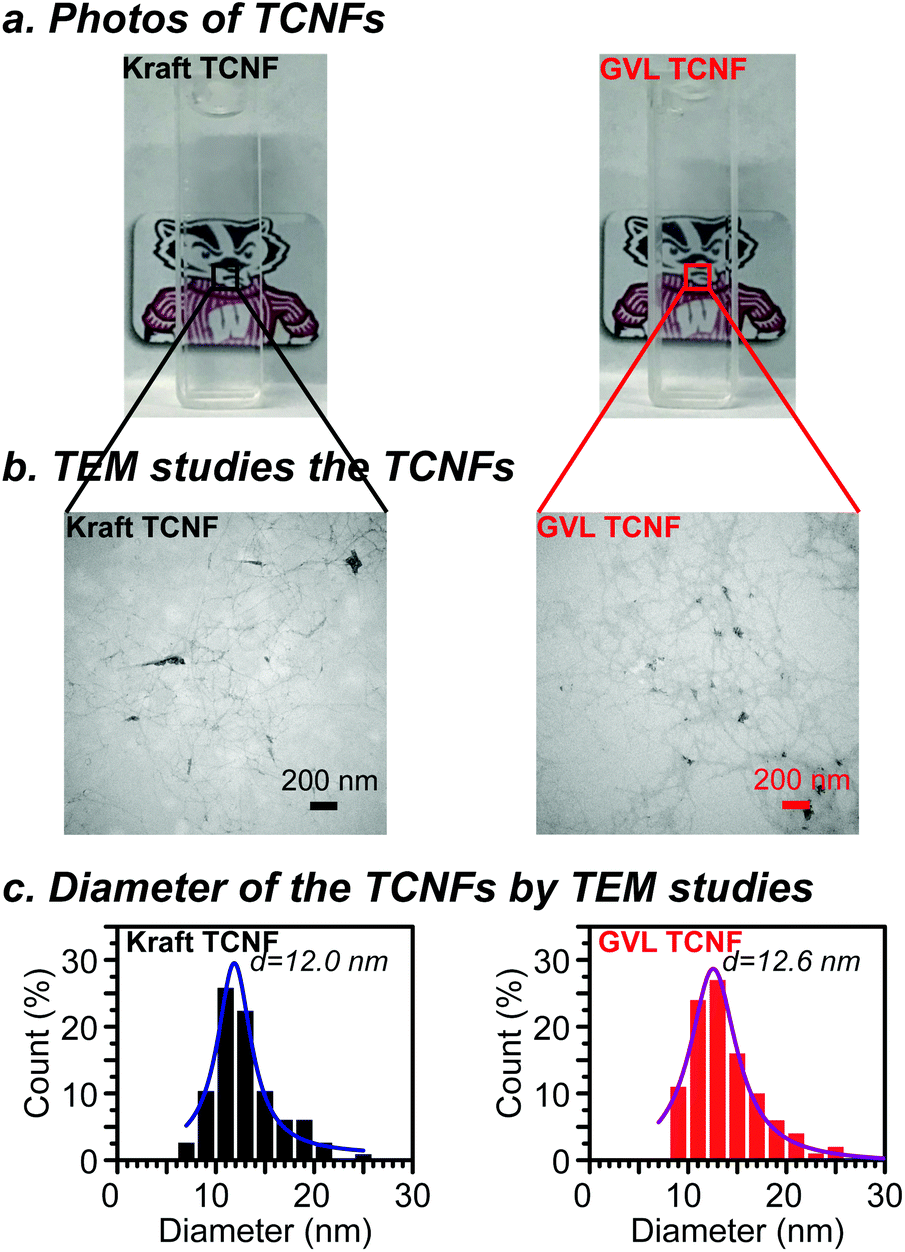 | ||
| Fig. 4 Photos, TEM results and diameters of the TCNF in solution. | ||
The transmittances of 0.1 wt% TCNF solution at 600 nm were 87% and 88% for GVL TCNFs and bleached kraft TCNFs, respectively, similar to those of previous reports.46,47 The UV-vis spectra of GVL and kraft TCNFs almost completely overlapped in the whole range of 190–900 nm, indicating similar optical properties of TCNF materials produced from the GVL and kraft pulp. The transmittance of the TNCF solution in the UV-vis spectrum is a result of both light absorption and scattering which could correspond to the chemical structure and the size of TCNFs, respectively.48 As discussed above, the GVL and kraft TCNFs have a similar chemical structure and diameter. Therefore, it is reasonable that the GVL TCNF solution has optical properties similar to those of the kraft TCNF solution.
The rheological properties of TCNFs (1 wt% gel) were studied with a TA ARES rheometer using a steady rate sweep model (Fig. 5). Both the GVL and kraft TCNFs showed strong shear thinning behaviour, similar to reported results.49 The shear shinning behaviour of the TCNF solution was due to the nanofibril alignment under shearing forces. Importantly, the GVL TCNF has almost exactly the same viscosity behaviours as the kraft TCNF. The rheological behavior of the TCNF solution is a macro-level phenomenon of the motions of cellulose nanofibrils under shearing forces. At the micro-level, the surface chemistry and shape of cellulose nanofibrils are the two major contributions to the rheological behavior.50 It has been shown that the GVL TCNF and kraft TCNF have a similar chemical structure and size, which results in the similar rheological behavior of the two TCNF solutions. All these results indicate that the GVL pulp is a suitable starting material for nanocellulose production and produces nanocellulose with similar properties to the typical feedstock bleached kraft pulp.
 | ||
| Fig. 5 UV-vis spectrum and rheological analysis of TCNF solutions. | ||
2.4. Properties of TCNF films
The crystalline structure and mechanical and thermal properties of the GVL TCNF film were studied to understand the impact of using GVL pulp as a feedstock for nanocellulose production. As shown in Fig. 6a, both the pulps and TCNFs, either from the GVL process or the kraft process, showed a typical cellulose I pattern with diffraction peaks at 16.0° and 22.5°. This was expected as the nanocellulose production process used does not change the crystalline allomorph of cellulose,51 consistent with FTIR results. Though the crystalline index of the GVL pulp (71%) was different from that of the kraft pulp (67%), the crystalline index of GVL TCNFs was similar to that of kraft TCNFs, suggesting that the GVL pulp is suitable for TCNF production without modifying the resulting nanocellulose's crystalline structure.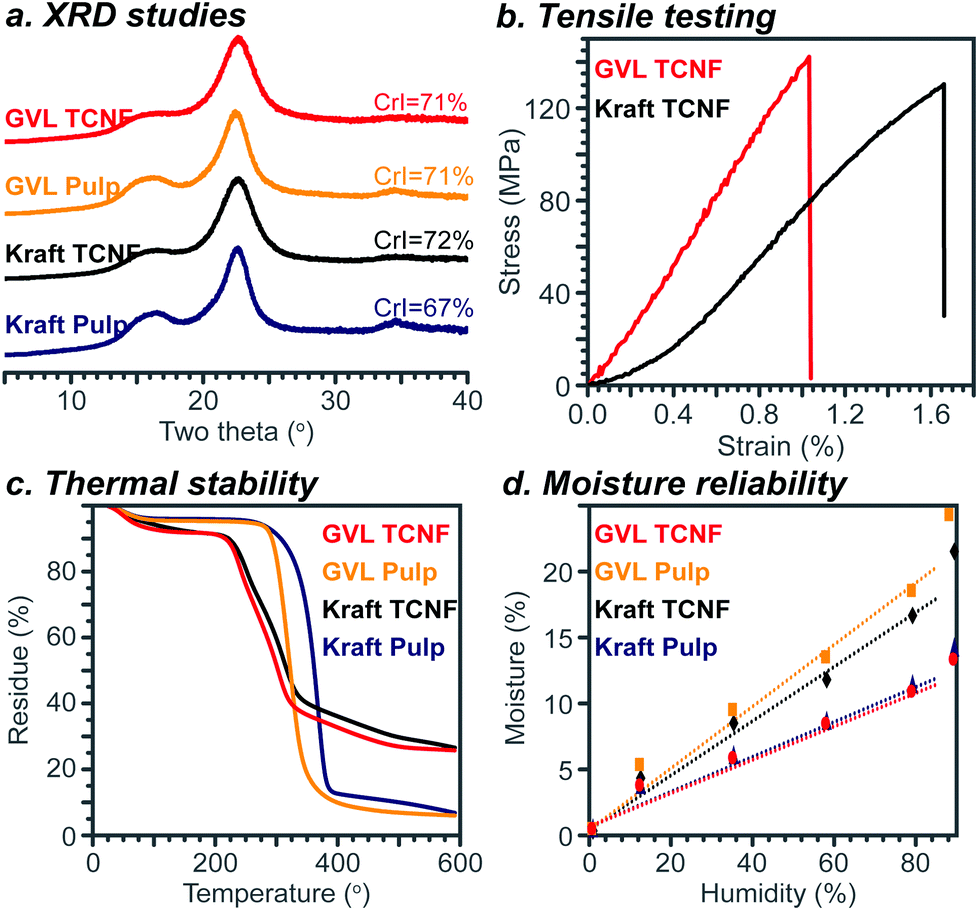 | ||
| Fig. 6 Structure and properties of TCNF films. In this figure, the legend description Kraft indicates bleached kraft pulp. | ||
The mechanical properties of TCNFs were measured according to ASTM D638-14. The tensile strength, elongation at break, and Young's modulus were 142 ± 34 MPa, 1.03 ± 0.29%, and 14.3 ± 0.7 GPa for the GVL TCNF film, respectively, while those were 131 ± 30 MPa, 1.66 ± 0.37%, and 10.3 ± 0.9 GPa for the kraft TCNF film, respectively. Though the tensile strength and Young's modulus of GVL TCNFs were slightly higher than those of bleached kraft TCNFs, these values were similar enough to suggest no significant difference in the mechanical properties between GVL and bleached kraft TCNF films. The similar mechanical properties of GVL and kraft TCNFs could be attributed to a comprehensive result of the similar DP, carboxylate content, crystalline structure and size of the two materials.
Thermal stability is another important property of nanocellulose. The TGA curves of pulps and TCNFs are shown in Fig. 6c. The weight loss before 150 °C was attributed to water evaporation.52 The moisture content was 4.1%, 4.6%, 7.8%, and 8.2% for the bleached kraft pulp, GVL pulp, kraft TCNF, and GVL TCNF, respectively. The dependence of the moisture content of the TCNF films and pulps on humidity is shown in Fig. 6d. The results show that the GVL and kraft TCNF have a similar moisture content due to the similar chemical structure. Also, the moisture contents of TCNFs were higher than those of the pulps. The onset thermal degradation temperatures were 323 °C, 292 °C, 225 °C, and 222 °C for the bleached kraft pulp, GVL pulp, kraft TCNF, and GVL TCNF, respectively. It is reasonable that the TCNFs have a higher moisture content and lower thermal stability than pulps, due to the low DP of TCNFs and introduction of the carboxylate group which is highly hydrophilic and thermally unstable.53–55 Importantly, the GVL TCNF showed similar thermal stability to both the bleached kraft TCNF and reported TEMPO nanocellulose because of the similar chemical structure of the materials.56
2.5. A comparative discussion
The biomass composition and structure makes it difficult to produce nanocellulose directly from untreated woody biomass;7 pretreatments are used to overcome this recalcitrance.8 Some biomass pretreatment processes result in pulps with high lignin and/or hemicellulose content which alter the nanocellulose extraction processes and the properties of extracted nanocellulose.57,58 Pulping processes that do remove significant lignin and hemicellulose require a subsequent bleaching process to allow for high quality, opaque nanocellulose production.12,59 However this additional process is capital intensive, requires additional costs of chemicals and energy, and causes additional pollution. However, using our GVL pulping process, high purity cellulose pulp with a white colour was obtained and found to be ideal for use in TCNF production. The appearance of the as-prepared GVL TCNF was similar to that of the kraft TCNF, and the UV-vis spectral studies showed that the GVL TCNF solution was highly transparent, similar to those of kraft TCNF and reported studies. Further studies by FTIR, NMR, XRD, TEM, the steady-rate rheological test, the tensile test and TGA analysis showed that there was no observed difference in the chemical mechanism of TCNF preparation, size, and rheological, mechanical and thermal properties between GVL TCNFs and kraft TCNFs. These results showed that GVL pulp is a suitable source for nanocellulose extraction without altering the existing nanocellulose extraction processes and the properties of the resulting nanocellulose. The GVL pulp was obtained by a one-step GVL pulping process without any subsequent bleaching or purification process, which provides a significant advantage to the typical kraft pulping process for nanocellulose extraction from woody biomass.3. Conclusions
Herein, GVL pulping was applied in biomass pretreatment for nanocellulose extraction from wood. GVL pulp with a high purity of cellulose up to 95.8% was obtained without subsequent bleaching/purification processes for nanocellulose extraction. There was no significant difference between nanocellulose samples prepared from GVL pulp and those from bleached kraft pulp in terms of the chemical mechanism and chemical consumption of the TEMPO oxidation process and the properties of nanocellulose solutions and films. From these results, the GVL pulping process was shown to be an excellent pretreatment process for nanocellulose extraction from woody biomass requiring no additional bleaching step.4. Experimental
4.1. Materials
Aspen chips were produced from big-tooth aspen trees (Populus grandidentata) harvested from Dane County, WI, USA, that were approximately 30 year old. GVL (>98% purity) was purchased Shenzhen Nexconn Pharmatechs Ltd (Shenzhen, China). NaOH, certified ACS pellets, and sodium bromide were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Concentrated sulfuric acid, TEMPO, Na2S, DMSO-d6 and pyridine-d5 were purchased from Sigma–Aldrich (St Louis, MO, USA). Cupriethylenediamine solution (CED, 1 m) was purchased from Fisher Scientific (Fair Lawn, NJ, USA). Chlorine dioxide (ClO2) was produced from acidified sodium chlorite. Sugars (arabinose, galactose, glucose, mannose, and xylose, all 99 + %) were purchased from Acros Organics (Geel, Belgium). NaClO was purchased from RICCA Chemical.4.2. GVL pulping
GVL pulp was prepared from Aspen chips according to an optimized process previously reported.23 Briefly, 100 g of Aspen chips and 400 g of a liquid solution of GVL/water 7/3 by weight and 0.1 M sulfuric acid were added to a 1 L stainless steel rotating digester and heated at 125 °C for 3 hours. After the reaction was completed, the solid was collected by filtration and used as is or washed with 300 g fresh GVL/water 7/3 solution once, twice, or thrice to further remove degraded lignin as shown in Fig. 1. Each pulp sample was thoroughly washed with DI water.4.3. Kraft pulping and D(EP)D bleaching
Bleached kraft pulp was prepared from Aspen chips according to ref. 29. Briefly, 100 g of Aspen chips were loaded into a 1 L stainless steel rotating digester and cooked at 165 °C with an active alkali dosage of 18 wt% with a sulfidity of 30%, a liquid/solid ratio of 5, and an H factor of 1200. Details of the D(EP)D bleaching sequence were described previously.294.4. TCNF extraction
Pulp (2 g) was suspended and oxidized at the C6 position with TEMPO (25 mg), sodium bromide (250 mg), sodium hypochlorite (NaClO, 1.2–4.8 mmol g−1 to pulp) solution according to ref. 36 and 60. The solution of NaClO was added drop by drop (1 mL min−1) at room temperature to initiate the TEMPO-mediated oxidation. The suspension was stirred, and the pH was maintained at 10.0 ± 0.5 by continuous addition of 0.5 M NaOH solution. The oxidation was considered complete when the NaClO was consumed and pH was measured to be constant. Upon completion, excess ethanol was added to quench the reaction and the pulp was separated by filtration and thoroughly washed with DI water. The washed pulp was diluted to make a 0.5 wt% suspension which was then sheared with a Polytron PT 10–35 GT homogenizer at a maximum speed for 5 min. The obtained gel was diluted into 0.1 wt% solution and centrifugated at 4000 rpm to remove large particles. The resulting solution was then concentrated to 1 wt% gel solution by microfiltration.4.5. Nanocellulose film preparation
Nanocellulose films were prepared as described by Ma et al.11 TCNF films were made using a 1 wt% solution with a filtration system (YT30 142HW, Millipore Corporation, Bedford, MA, USA). The filtration membrane (JVWP14225, Millipore Corporation, USA) had a reported pore size of 0.45 μm supported by filter paper (P2, Fisher Scientific, USA). The filtration tank was pressurized to 3.45 bar. TCNF films were formed after 30 min of dewatering. The TCNF films were dried by placing them between two steel plates with a 23 kg weight applied to provide pressure at room temperature for 72 h.4.6. Moisture determination
Moisture content of the TCNF and pulp samples was determined by conditioning the 110 °C oven dried sample in a desiccator with saturated salt solution for 24 hours. The saturated salt solutions used for humidity control were LiCl (11%), MgCl2 (33%), Mg(NO3)2 (55%), NaCl (75%) and KCl (85%) solutions. The moisture content was calculated as follows:| Moisture% = (m − m0)/m0 × 100%. |
4.7. Characterization
Component analysis of the pulps was according to the NREL method.61 FTIR spectra were collected in the wavenumber range of 400–4000 cm−1 with an FTIR spectrophotometer (Spectrum 100, PerkinElmer Inc., Norwalk, CT) equipped with a universal attenuated total reflectance probe. Gel-state HSQC spectroscopy was performed according to ref. 35. Pulp was ball-milled for 4 h and then dispersed into DMSO-d6/pyridine-d5 (4/1 v/v) to form a gel solution. HSQC spectra were acquired on a Bruker Biospin (Billerica, MA) AVANCE 700 MHz spectrometer fitted with a cryogenically cooled 5 mm TXI gradient probe with inverse geometry (proton coils closest to the sample). For the lignin samples, DMSO-d6 was applied as the HSQC solvent. The viscometric DP of pulp was determined according to ASTM D 4243-16. UV-Vis spectra were collected with a Shimadzu UV-1800 UV spectrophotometer (Canby, OR, USA) from 190 nm to 900 nm at 0.5 nm resolution. TEM study was performed with a JEOL 1200EX TEMSCAN (JEOL USA) operated at 80 kV. The nanocellulose was deposited onto a copper grid and observed directly with TEM after drying at room temperature. A steady-rate rheological test was performed with an advanced rheometric expansion system (ARES, TA Instruments, USA) equipped with parallel plates of 25 mm in diameter. The shear data were collected from shear rate tests ranging from 0.001 to 200 s−1 at room temperature (20 °C). XRD patterns were collected with a Bruker D8 Advance X-ray diffractometer equipped with Ni-filtered Cu Kα1 radiation (λ = 0.15418 nm) at room temperature. The scattering angle range was 5°−45° 2θ at a scanning speed of 8° min−1 and a 2θ step interval of 0.02°. Tensile testing was performed with and an electromechanical universal test system (MTS Criterion Model 43) according to ASTM D882-12. The grip length was set to 25 mm. Test specimens were cut to 15 mm width × 70 mm length. The rate of grip separation was set to 4 mm min−1. Thermal stability of nanocellulose was analyzed with a TA Q500 thermal analyzer (TA Instrument, USA). The samples (approximately 10 mg) were heated from room temperature to 600 °C at a heating rate of 10 °C min−1 under a N2 atmosphere.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the United States Department of Agriculture National Institute of Food and Agriculture, under ID number WIS02002, and the NSF Small Business Innovation Research program under ID number 1632394. The authors also wish to thank Carl Houtman and Roland Gleisner of the USDA Forest Products Lab (Madison, Wis.) for their assistance with this research.References
- C. Chen and L. Hu, Acc. Chem. Res., 2018, 51, 3154–3165 CrossRef CAS PubMed.
- F. Sun, W. Liu, Z. Dong and Y. Deng, Chem. Eng. J., 2017, 330, 774–782 CrossRef CAS.
- A. Dufresne, Mater. Today, 2013, 16, 220–227 CrossRef CAS.
- D. Klemm, E. D. Cranston, D. Fischer, M. Gama, S. A. Kedzior, D. Kralisch, F. Kramer, T. Kondo, T. Lindström, S. Nietzsche, K. Petzold-Welcke and F. Rauchfuß, Mater. Today, 2018, 21, 720–748 CrossRef CAS.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- M.-J. Chen, R.-M. Li, X.-Q. Zhang, J. Feng, J. Feng, C.-F. Liu and Q.-S. Shi, ACS Sustainable Chem. Eng., 2017, 5, 360–366 CrossRef CAS.
- T. Auxenfans, C. Terryn and G. Paës, Sci. Rep., 2017, 7, 8838 CrossRef PubMed.
- M. Rajinipriya, M. Nagalakshmaiah, M. Robert and S. Elkoun, ACS Sustainable Chem. Eng., 2018, 6, 2807–2828 CrossRef CAS.
- J. Dou, H. Bian, D. J. Yelle, M. Ago, K. Vajanto, T. Vuorinen and J. Zhu, Ind. Crops Prod., 2019, 129, 15–23 CrossRef CAS.
- H. Bian, L. Chen, R. Gleisner, H. Dai and J. Y. Zhu, Green Chem., 2017, 19, 3370–3379 RSC.
- Q. Ma, J. Zhu, R. Gleisner, R. Yang and J. Y. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 14480–14489 CrossRef CAS.
- Y. Li, Y. Liu, W. Chen, Q. Wang, Y. Liu, J. Li and H. Yu, Green Chem., 2016, 18, 1010–1018 RSC.
- M. Lee, M. H. Heo, H. Lee, H.-H. Lee, H. Jeong, Y.-W. Kim and J. Shin, Green Chem., 2018, 20, 2596–2610 RSC.
- A. Chaker, S. Alila, P. Mutjé, M. R. Vilar and S. Boufi, Cellulose, 2013, 20, 2863–2875 CrossRef CAS.
- E. Rojo, M. S. Peresin, W. W. Sampson, I. C. Hoeger, J. Vartiainen, J. Laine and O. J. Rojas, Green Chem., 2015, 17, 1853–1866 RSC.
- T. Horseman, M. Tajvidi, C. I. K. Diop and D. J. Gardner, Cellulose, 2017, 24, 2455–2468 CrossRef CAS.
- Q. Meng, H. Li, S. Fu and L. A. Lucia, React. Funct. Polym., 2014, 85, 142–150 CrossRef CAS.
- M. Smyth, A. García, C. Rader, E. J. Foster and J. Bras, Ind. Crops Prod., 2017, 108, 257–266 CrossRef CAS.
- H. Abushammala, I. Krossing and M.-P. Laborie, Carbohydr. Polym., 2015, 134, 609–616 CrossRef CAS PubMed.
- M. Chen, R. Li, T. Runge, J. Feng, S. Hu and Q.-S. Shi, Mater. Today Sustainability, 2019, 100008, DOI:10.1016/j.mtsust.2019.100008.
- T. Andersson, L. Förlin, J. Härdig and Å. Larsson, Can. J. Fish. Aquat. Sci., 1988, 45, 1525–1536 CrossRef CAS.
- H. Bian, L. Chen, H. Dai and J. Y. Zhu, Carbohydr. Polym., 2017, 167, 167–176 CrossRef CAS PubMed.
- D. M. Alonso, S. H. Hakim, S. Zhou, W. Won, O. Hosseinaei, J. Tao, V. Garcia-Negron, A. H. Motagamwala, M. A. Mellmer, K. Huang, C. J. Houtman, N. Labbé, D. P. Harper, C. T. Maravelias, T. Runge and J. A. Dumesic, Sci. Adv., 2017, 3, e1603301 CrossRef PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- Y. Okita, T. Saito and A. Isogai, Journal, 2009, 63, 529 CAS.
- T. Pääkkönen, K. Dimic-Misic, H. Orelma, R. Pönni, T. Vuorinen and T. Maloney, Cellulose, 2016, 23, 277–293 CrossRef.
- C. W. Lahive, P. J. Deuss, C. S. Lancefield, Z. Sun, D. B. Cordes, C. M. Young, F. Tran, A. M. Z. Slawin, J. G. de Vries, P. C. J. Kamer, N. J. Westwood and K. Barta, J. Am. Chem. Soc., 2016, 138, 8900–8911 CrossRef CAS PubMed.
- N. Li, Y. Li, C. G. Yoo, X. Yang, X. Lin, J. Ralph and X. Pan, Green Chem., 2018, 20, 4224–4235 RSC.
- S. Zhou, T. Runge, S. D. Karlen, J. Ralph, E. Gonzales-Vigil and S. D. Mansfield, ChemSusChem, 2017, 10, 3565–3573 CrossRef CAS PubMed.
- R. Grande, E. Trovatti, A. J. F. Carvalho and A. Gandini, J. Mater. Chem. A, 2017, 5, 13098–13103 RSC.
- C. Zhu, A. Soldatov and A. P. Mathew, Nanoscale, 2017, 9, 7419–7428 RSC.
- D. O. Carlsson, J. Lindh, M. Strømme and A. Mihranyan, Biomacromolecules, 2015, 16, 1643–1649 CrossRef CAS PubMed.
- W. Lan, C.-F. Liu, F.-X. Yue, R.-C. Sun and J. F. Kennedy, Carbohydr. Polym., 2011, 86, 672–677 CrossRef CAS.
- Y. Habibi, H. Chanzy and M. R. Vignon, Cellulose, 2006, 13, 679–687 CrossRef CAS.
- H. Kim and J. Ralph, RSC Adv., 2014, 4, 7549–7560 RSC.
- R. Shinoda, T. Saito, Y. Okita and A. Isogai, Biomacromolecules, 2012, 13, 842–849 CrossRef CAS PubMed.
- T. Saito, M. Hirota, N. Tamura and A. Isogai, J. Wood Sci., 2010, 56, 227–232 CrossRef CAS.
- V. L. Finkenstadt and R. P. Millane, Macromolecules, 1998, 31, 7776–7783 CrossRef CAS.
- K. Marta, P. Piotr, D. Marcin and B. Kamila, Ann. Warsaw Univ. Life Sci.–SGGW, For. Wood Technol., 2014, 86, 171–174 CAS.
- Y.-M. Chen, J.-Q. Wan, M.-Z. Huang, Y.-W. Ma, Y. Wang, H.-L. Lv and J. Yang, Carbohydr. Polym., 2011, 85, 759–764 CrossRef CAS.
- L. Fang and J. M. Catchmark, Cellulose, 2014, 21, 3951–3963 CrossRef CAS.
- H. Bian, L. Chen, H. Dai and J. Y. Zhu, Cellulose, 2017, 24, 4205–4216 CrossRef CAS.
- X. Cao, B. Ding, J. Yu and S. S. Al-Deyab, Carbohydr. Polym., 2012, 90, 1075–1080 CrossRef CAS PubMed.
- M. Rajinipriya, M. Nagalakshmaiah, M. Robert and S. Elkoun, Ind. Crops Prod., 2018, 118, 53–64 CrossRef CAS.
- F. Bettaieb, R. Khiari, A. Dufresne, M. F. Mhenni, J. L. Putaux and S. Boufi, Ind. Crops Prod., 2015, 72, 97–106 CrossRef CAS.
- Y. Chen, B. Geng, J. Ru, C. Tong, H. Liu and J. Chen, Cellulose, 2017, 24, 4831–4844 CrossRef CAS.
- J. Jiang, W. Ye, L. Liu, Z. Wang, Y. Fan, T. Saito and A. Isogai, Biomacromolecules, 2017, 18, 288–294 CrossRef CAS PubMed.
- L. Hu, G. Zheng, J. Yao, N. Liu, B. Weil, M. Eskilsson, E. Karabulut, Z. Ruan, S. Fan, J. T. Bloking, M. D. McGehee, L. Wågberg and Y. Cui, Energy Environ. Sci., 2013, 6, 513–518 RSC.
- H. Q. Lê, K. Dimic-Misic, L.-S. Johansson, T. Maloney and H. Sixta, Cellulose, 2018, 25, 179–194 CrossRef.
- M. A. Hubbe, P. Tayeb, M. Joyce, P. Tyagi, M. Kehoe, K. Dimic-Misic and L. Pal, BioResources, 2017, 12, 9556–9661 CAS.
- D. Yang, X.-W. Peng, L.-X. Zhong, X.-F. Cao, W. Chen and R.-C. Sun, Cellulose, 2013, 20, 2427–2437 CrossRef CAS.
- C. Du, H. Li, B. Li, M. Liu and H. Zhan, BioResources, 2016, 11, 5276–5284 CAS.
- S. Matsuoka, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2014, 106, 138–146 CrossRef CAS.
- M. B. Agustin, F. Nakatsubo and H. Yano, Cellulose, 2016, 23, 451–464 CrossRef CAS.
- N. Subair, T. V. Jinitha, V. Shaniba, M. P. Sreejith, K. B. Aparna and E. Purushothaman, Carbohydr. Polym., 2018, 180, 13–20 CrossRef PubMed.
- M. A. C. Mhd Haniffa, Y. C. Ching, C. H. Chuah, K. Yong Ching, N. Nazri, L. C. Abdullah and L. Nai-Shang, Carbohydr. Polym., 2017, 173, 91–99 CrossRef CAS PubMed.
- M.-J. Chen, X.-Q. Zhang, A. Matharu, E. Melo, R.-M. Li, C.-F. Liu and Q.-S. Shi, ACS Sustainable Chem. Eng., 2017, 5, 7278–7283 CrossRef CAS.
- J. Xu, H. Hou, J. Hu and B. Liu, Appl. Energy, 2018, 229, 745–755 CrossRef CAS.
- J.-H. Chen, J.-K. Xu, P.-L. Huang and R.-C. Sun, Cellulose, 2016, 23, 2727–2739 CrossRef CAS.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass, Laboratory analytical procedure of National Renewable Energy Laboratory Golden, 2008.
| This journal is © The Royal Society of Chemistry 2019 |