
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow-based enzymatic synthesis of melatonin and other high value tryptamine derivatives: a five-minute intensified process†
Martina Letizia
Contente
 a,
Stefano
Farris
b,
Lucia
Tamborini
*c,
Francesco
Molinari
b and
Francesca
Paradisi
*ad
a,
Stefano
Farris
b,
Lucia
Tamborini
*c,
Francesco
Molinari
b and
Francesca
Paradisi
*ad
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
bDepartment of Food, Environmental and Nutritional Sciences (DeFENS), University of Milan, via Mangiagalli 25, 20133, Milan, Italy
cDepartment of Pharmaceutical Sciences (DISFARM), University of Milan, via Mangiagalli 25, 20133, Milan, Italy. E-mail: lucia.tamborini@unimi.it
dDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: francesca.paradisi@dcb.unibe.ch
First published on 31st May 2019
Abstract
To increase the uptake of biocatalytic processes by industry, it is essential to demonstrate the reliability of enzyme-based methodologies directly applied to the production of high value products. Here, a unique, efficient, and sustainable enzymatic platform for the multi-gram synthesis of melatonin, projected to generate around 1.5 billion U.S. dollars worldwide by 2021, and its analogues was developed. The system exploits the covalent immobilization of MsAcT (transferase from Mycobacterium smegmatis) onto agarose beads increasing the robustness and longevity of the immobilized biocatalyst. The fully-automated process deriving from the integration between biocatalysis and flow chemistry is designed to maximize the overall yields (58–92%) and reduce reaction times (5 min), overcoming the limitation often associated with bioprocesses and bridging the gap between lab scale and industrial production.
Melatonin (N-acetyl-5-methoxytryptamine) is a natural mammalian hormone that plays a crucial role in regulating the human body's circadian sleep-wake cycle.1 Clinically, it exhibits positive effects as a sleep aid2,3 and a powerful antioxidant as an over-the-counter dietary supplement.4 Recent studies on tryptamine derivative redox activity show their involvement to re-establish the equilibrium between pro- and antioxidants. Additionally, the fact that melatonin readily crosses the blood–brain-barrier suggests that it could be an adjuvant in treating oxidative stress, a key factor of neurodegenerative disorders.5 Thanks to its antiestrogenic properties as well as its ability to improve the efficacy and reduce the side effects of conventional antioestrogens, melatonin can be safely associated with breast cancer therapies currently in use.6 The first source of commercially available melatonin was the animal pineal glands.7 However, a major drawback encountered during the extraction process was contamination from animal tissue. Recently, a microbial cell factory approach for the preparation of recombinant melatonin in Saccharomyces cerevisiae was reported,8 but the engineered yeast strain produced melatonin only in very low concentration (15 mg L−1), following four enzymatic steps in a 76 h fermentation process. Consequently, melatonin and its analogues are almost exclusively chemically synthesized. Industrial procedures involve a final step where tryptamines are acetylated with acetic acid anhydride or acetyl chloride in anhydrous environment,9,10 at low temperature11 and using bases to carry out the reaction.9–11 Although these reagents are inexpensive, their environmental toxicity and hygroscopicity reduce their appeal in greener protocols; moreover, due to their high and non-specific reactivity, low selectivity is often observed in the acetylation of amines bearing other reacting functional groups.12 Alternatively, amide bonds can be formed by using heterogeneous and homogeneous metal catalysts, which are often expensive and need further purification steps.13 A green chemistry perspective on catalytic amide bond formation was recently assessed, pointing out that biocatalysis is among the techniques better suited to achieve sustainable processes for amide formation.14 Within this framework, the further development of biocatalysis in biodegradable, unconventional reaction media such as deep eutectic solvents (DESs) has been reported.15,16 Some hydrolases, for example, have proven to be effective in catalyzing amidation of esters in DESs.17 Following more traditional methodologies, amide synthesis was achieved in DESs with Pd/C without further purification steps.18 An acetyltransferase from Mycobacterium smegmatis (MsAcT) was proposed as biocatalyst for the synthesis of amides starting from primary amines and short-chain esters in water.19,20 MsAcT was initially discovered and fully characterized for its ability to transfer acetyl group to alcohols.21,22 A very interesting feature of MsAcT is its remarkable activity even at high substrate concentrations.23De facto, biocatalysis can be considered a reliable tool for organic and industrial chemists only if selectivity and mild operational conditions (typical of biotransformations) meet green and intensified processes, characterized by high product concentration and favourable atom economy.
Integration of biocatalytic methods and continuous flow reactors (either micro- or meso-reactors) is designed to intensify the overall process and to overcome low productivity, which is one of the more common limitation of biocatalysis.24 In this work, we have developed a unique, rapid, and robust enzymatic method to prepare melatonin and analogous amides using immobilized MsAcT in a flow reactor to set-up highly productive biotransformations on a multi-gram scale.
Firstly, a highly tailored immobilization of the octameric MsAcT, key to achieve a robust and durable catalyst, was developed. To date, MsAct has been immobilized onto carbon nanotubes to fulfil its perhydrolase activity,25 and siliceous monolithic microreactors have been used for the acetylation of neopentylglycol.26 Although continuous-flow microchannel reactors can be efficient, the synthesis and functionalization of silica monoliths is anything but eco-friendly (e.g., requirement of HNO3, high temperature, inert atmosphere, long reaction times). In the present study, strategies forming covalent bonds between the matrix and the enzyme (see ESI†) were screened to achieve a durable catalyst to be used specifically at high flow rate for scalable productivity.27,28 Hydrophilic carriers (i.e., agarose, cellulose, 3-aminopropylic silica, epoxy resins) with different pore diameter (40–60/10–20 nm) and spacer size were chosen. Hydrophobic supports have been avoided as they can create non-specific interactions between the enzyme and non-functionalized hydrophobic regions of the carrier.25 Different concentration of MsAcT were tested (1, 2, and 5 mg gmatrix−1) for each carrier; the highest recovered activity (73%), was obtained at low enzyme concentration using activated glyoxyl agarose as support (see ESI†). This can be explained taking into consideration that MsAcT is a large molecule (72 × 72 × 60 Å) which needs tight association of dimer pairs to work. The supports showing the best retained activity (>30%) were examined before and after enzyme immobilization by scanning electron microscopy (SEM) to detect any surface change (see ESI†). In Fig. 1, the characterization of the agarose matrix is reported. Particles supported with 1 mgMsAcT gmatrix−1 did not present any aggregation phenomena, thus maximizing the contact area during the biotransformation. While epoxy resins seem to have the same behavior, SEM analysis of cellulose demonstrates that the fibers create aggregates with less surface available for the interaction with the substrates (see ESI†).
 | ||
| Fig. 1 SEM images of agarose matrix. Line a: Activated glyoxyl agarose before MsAcT immobilization 60×; 500×; 1000× magnification respectively. Line b: Activated glyoxyl agarose after MsAcT immobilization (1 mg gmatrix−1) 60×; 500×; 1000× magnification respectively. | ||
Additionally, spatial distribution of fluorophore-labelled enzyme was investigated (see ESI†). MsAcT is localized across the porous surface of the agarose beads favoring an intimate contact with the substrates (Fig. 2). The spatial enzyme organization remained stable after two-week storage demonstrating that the enzyme is covalently bound to the matrix and unable to migrate.
 | ||
| Fig. 2 Confocal microscopy of glyoxyl agarose particles immobilizing MsAcT (1 mg gmatrix−1) 20× magnification. | ||
Batch reactions to test the catalytic performance of agarose immobilized MsAcT were carried out. Three amines (2-phenylethylamine, p-ethylaniline, 3,4-dimethoxybenzylamine), already studied using the free MsAcT, were selected.20 The reaction was carried out in phosphate buffer (0.1 M pH 8.0) using non-activated acyl donors (ethyl formate, ethyl acetate, ethyl propionate) and activated esters (vinyl acetate, vinyl propionate). The substrate concentration was fixed at 0.25 M (>30 g L−1), while 10% v/v (∼1 M) of acyl donor was used, observing maximum conversions between 75 and 100% in times ranging between 30 min and 2 h (see ESI†). It is of note that the immobilized system afforded much higher activity and substrate tolerance than the free enzyme: in fact, the enzyme loading (0.2 mg mL−1) required for this level of reactivity was five times lower than for free MsAcT, even if a 5-fold higher substrate concentration was used.
To demonstrate the robustness and longevity of the immobilized biocatalyst, stability of the immobilized enzyme towards different esters (acting as acyl donors) and temperature was investigated. Agarose-immobilized MsAcT showed a remarkable increase of the tolerance to the assayed esters (more than 50% of retained activity after 24 h) and slightly higher thermal stability when incubated up to 70 °C (see ESI†).
Despite amide formation being dramatically favored over hydrolysis due to the characteristic hydrophobicity of the active site, product cleavage was observed over prolonged times; accordingly, continuous product removal can guarantee high yield of the desired amide. With this in mind and taking into consideration that flow processing has also the potential to accelerate biotransformations (due to enhanced mass transfer and better control of the reaction parameters), a continuous flow reactor using immobilized MsAcT was set-up and optimized for the preparation of N-(2-phenylethyl)acetamide using EtOAc as acetylating agent. A segmented liquid–liquid phase composed of buffer/EtOAC 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 was flowed into the packed-bed column containing immobilized MsAcT. Keeping constant the residence time (5 min), concentration of the amine in the feeding flow was varied. Complete conversion was observed using an amine feed between 0.25–0.50 M (30–60 g L−1). When 2-phenylethylamine concentration was increased up to 1.0 M (120 g L−1), the biphasic flow was slightly modified to ensure a constant excess of acyl donor (85:15 aqueous phase/EtOAc), allowing for N-acetylation with 90% molar conversion (Scheme 1).
:10 was flowed into the packed-bed column containing immobilized MsAcT. Keeping constant the residence time (5 min), concentration of the amine in the feeding flow was varied. Complete conversion was observed using an amine feed between 0.25–0.50 M (30–60 g L−1). When 2-phenylethylamine concentration was increased up to 1.0 M (120 g L−1), the biphasic flow was slightly modified to ensure a constant excess of acyl donor (85:15 aqueous phase/EtOAc), allowing for N-acetylation with 90% molar conversion (Scheme 1).
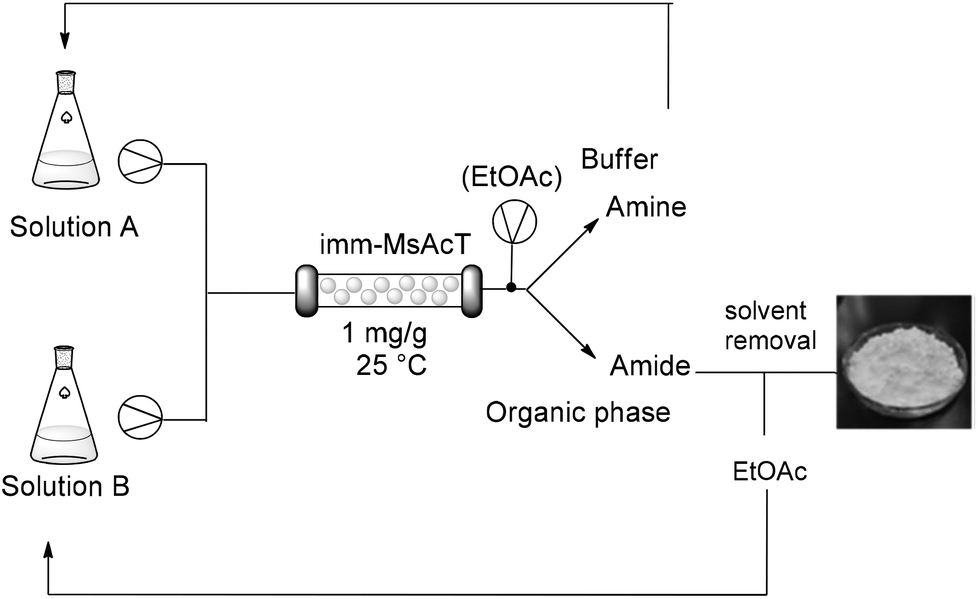 | ||
| Scheme 1 Flow synthesis of amides. Solution A: 0.5 M or 1.0 M amine solution in phosphate buffer (0.1 M, pH 8.0) containing 5–10% DMSO. Solution B: EtOAc or vinylOAc. T = 25 °C, P = atmospheric pressure. | ||
Continuous product removal completely eliminated hydrolysis of the newly formed amide, improving the overall process yield. The process was further implemented with the addition of an in-line extraction. The biphasic system exiting from the flow reactor was continuously separated using a Zaiput liquid/liquid separator and the desired amide was recovered in the organic phase, significantly simplifying and accelerating the work-up procedure, as no further purification was required. The designed process can be considered an ultra-efficient, closed-loop strategy,29 as all the materials involved in the biosynthesis can be reused. The recovered waste water containing the unreacted amine can be easily re-circulated in the system, while evaporation of the organic phase allows for the recovery of ethyl acetate, used as acyl donor. Further optimization of the reaction conditions (such as addressing the buffer formulation and shifting towards more volatile mixtures) is ongoing. The operational stability of the immobilized MsAcT under continuous flow was also assayed by running the reactor over prolonged time. Samples (4 mL) were collected and analyzed by HPLC. The consistency of the results obtained over time was verified in terms of N-(2-phenylethyl)acetamide produced, showing excellent stability under the chosen working conditions. A total volume of more than 400 mL was collected without any loss of enzymatic activity. This allowed for an extensive utilization of the pre-packed column with an unprecedented amide production of 56 per day (using 1.6 mg of enzyme), thus demonstrating the intensification of the process with excellent atom efficiency and automation.
The established methodology was then applied to the synthesis of the target pharmaceuticals; N-acetyl amines derived from commercially available tryptamine, 5-methoxy triptamine and serotonin (Fig. 3). Melatonin analogues, as well as playing an important biological role as agonist of melatonin receptors, are promising neuroprotectant drugs for the treatment of neurodegenerative disorders.30,31
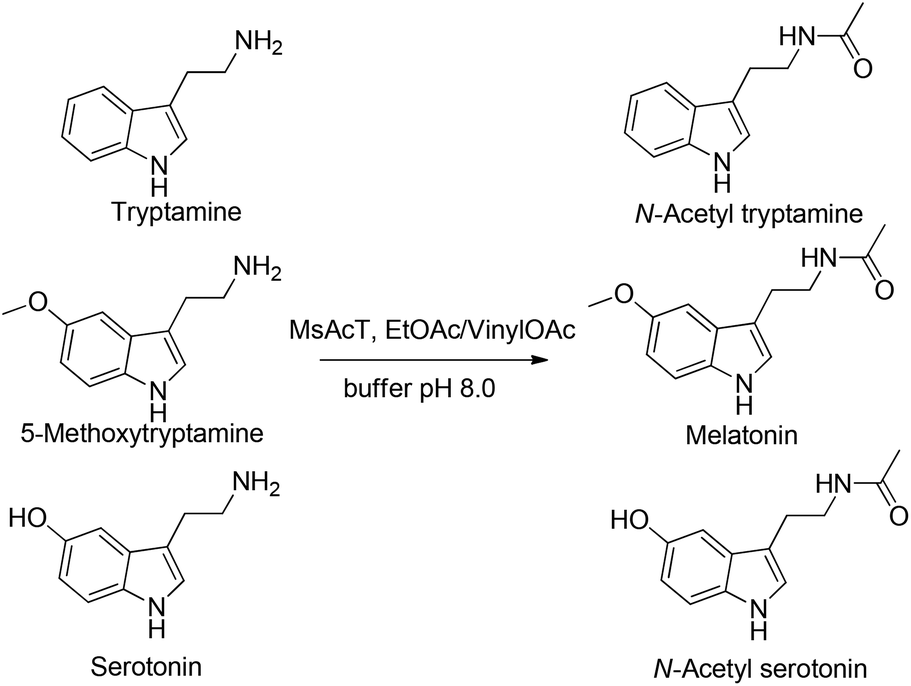 | ||
| Fig. 3 Flow production of melatonin analogues. | ||
Using EtOAc as acetyl donor (10% v/v) and 0.5 M amine solutions, melatonin analogues were prepared with very good molar conversions (70%, 62% and 60% respectively) with 5-minute residence time (further increasing the reaction time did not give any significantly better result). Activated vinylOAc was also employed for the N-acetylation of 0.5 M tryptamines: the desired amides were rapidly produced (5 min of residence time) with excellent conversion (95%, 96% and 89%, respectively). Downstream liquid extraction afforded products with high chemical purity (>95%) and 90%, 92% and 85% isolated yields, respectively. Notably, N-acetylation of tryptamines catalyzed by MsAcT occurred with complete chemoselectivity, since no reaction involving the phenolic OH was observed, demonstrating that our process can be considered a clean, mild and virtually zero-waste procedure for amide production.
Finally, continuous flow-based preparation of melatonin was carried out for 24 hours (Table 1), showing that multi-gram synthesis of melatonin was achievable using less than 2 mg of enzyme in a 1.2 mL reactor: 23.2 or 36.9 g of melatonin were isolated after automated work up described above.
Conclusions
The high substrate concentrations (and relative yields), together with the remarkable operational stability of the immobilized system described in this work, help to overcome the perception about the inefficiency of biocatalysis when compared with conventional chemical strategies. The method described is remarkably intensified and can compete with conventional (bio)processes for the production of melatonin and melatonin derivatives (and more in general amides) at industrial level.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 792804 AROMAs-FLOW (M. L. C.). The authors wish to thank Resindion S.R.L. for donating the EP403/S, HFA403/S, EC-HFA/S, EC-EP/S resins and UNITECH NO LIMITS for the SEM and confocal microscopy experiments.Notes and references
- R. Hardeland, Cell. Mol. Life Sci., 2008, 65, 2001–2018 CrossRef CAS PubMed.
- R. C. Leonardo-Mendonça, A. Martinez-Nicolas, C. de Teresa Galván, J. Ocaña-Wilhelmi, I. Rusanova, E. Guerra-Hernández, G. Escames and D. Acuña-Castroviejo, Chronobiol. Int., 2015, 32, 1–10 CrossRef PubMed.
- J. Xu, L. L. Wang, E. B. Dammer, C. B. Li, G. Xu, S. D. Chen and G. Wang, Am. J. Alzheimers Dis. Other Demen., 2015, 30, 439–447 CrossRef PubMed.
- D. X. Tan, C. L. C. Manchester, E. Esteban-Zubero, Z. Zhou and R. J. Reiter, Molecules, 2015, 20, 18886–18906 CrossRef CAS PubMed.
- E. Miller, A. Morel, L. Saso and J. Saluk, Curr. Top. Med. Chem., 2015, 15, 163–169 CrossRef CAS PubMed.
- A. González-González, M. D. Mediavilla and E. J. Sánchez-Barceló, Molecules, 2018, 23, 336–357 CrossRef PubMed.
- A. B. Lerner, J. D. Case and Y. Takahashi, J. Am. Chem. Soc., 1958, 80, 2587–2587 CrossRef CAS.
- S. M. Germann, S. A. Baallal Jacobsen, K. Schneider, S. J. Harrison, N. B. Jensen, X. Chen, S. G. Stahlhut, I. Borodina, H. Luo, J. Zhu, J. Maury and J. Forster, Biotechnol. J., 2016, 11, 717–724 CrossRef CAS PubMed.
- C. Prabhakar, N. V. Kumar, M. R. Reddy, M. R. Sarma and G. Om Reddy, Org. Process Res. Dev., 1999, 3, 155–160 CrossRef CAS.
- A. Siwicka, Z. Moleda, K. Wojtasiewicz, A. Zawadzka, J. K. Maurin, M. Panasiewicz, T. Pacuszka and Z. Czarnocki, J. Pineal Res., 2008, 45, 40–49 CrossRef CAS PubMed.
- D. C. Schuck, A. K. Jordão, M. Nakabashi, A. C. Cunha, V. F. Ferreira and C. R. S. Garcia, Eur. J. Med. Chem., 2014, 78, 375–382 CrossRef CAS PubMed.
- U. P. Saikia, F. L. Hussain, M. Suri and P. Pahari, Tetrahedron Lett., 2016, 57, 1158–1160 CrossRef CAS.
- S. Bartolucci, M. Mari, A. Bedini, G. Piersanti and G. Spadoni, J. Org. Chem., 2015, 80, 3217–3222 CrossRef CAS PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaksh and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- R. A. Sheldon, Chem. – Eur. J., 2016, 22, 12984–12999 CrossRef CAS PubMed.
- N. Guajardo, C. R. Müller, R. Schrebler, C. Carlesi and P. Domínguez de María, ChemCatChem, 2016, 8, 1020–1027 CrossRef CAS.
- J. T. Gorke, F. Srienc and R. J. Kazlauskas, Chem. Commun., 2008, 1235–1237 RSC.
- F. Messa, S. Perrone, M. Capua, F. Tolomeo, L. Troisi, V. Capriati and A. Salomone, Chem. Commun., 2018, 54, 8100–8103 RSC.
- H. Land, P. Hendil-Forssell, M. Martinelle and P. Berglund, Catal. Sci. Technol., 2016, 6, 2897–2900 RSC.
- M. L. Contente, A. Pinto, F. Molinari and F. Paradisi, Adv. Synth. Catal., 2018, 360, 4814–4819 CrossRef CAS.
- I. Mathews, M. Soltis, M. Saldajeno, G. Ganshaw, R. Sala, W. Weyler, M. A. Cervin, G. Whited and R. Bott, Biochemistry, 2007, 46, 8969–8979 CrossRef CAS PubMed.
- (a) N. de Leeuw, G. Torrelo, C. Bisterfeld, V. Resch, L. Mestrom, E. Straulino, L. van der Weel and U. Hanefeld, Adv. Synth. Catal., 2018, 360, 242–249 CrossRef CAS; (b) M. Kazemi, X. Sheng, W. Kroutil and F. Himo, ACS Catal., 2018, 8, 10698–10706 CrossRef CAS.
- I. Chiarelli Perdomo, S. Gianolio, A. Pinto, D. Romano, M. L. Contente, F. Paradisi and F. Molinari, J. Agric. Food Chem., 2019 DOI:10.1021/acs.jafc.9b01790.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS PubMed.
- C. Z. Dinu, G. Zhu, S. S. Bale, G. Anand, P. J. Reeder, K. Sanford, G. Whited, R. S. Kane and J. S. Dordick, Adv. Funct. Mater., 2010, 20, 392–398 CrossRef CAS.
- K. Szymańska, K. Odrozek, A. Zniszczol, G. Torrelo, V. Resch, U. Hanefeld and A. B. Jarzebski, Catal. Sci. Technol., 2016, 6, 4882–4888 RSC.
- M. Planchestainer, M. L. Contente, J. Cassidy, L. Tamborini, F. Molinari and F. Paradisi, Green Chem., 2017, 19, 372–375 RSC.
- M. L. Contente, F. Dall'Oglio, L. Tamborini, F. Molinari and F. Paradisi, ChemCatChem, 2017, 9, 3843–3848 CrossRef CAS.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef.
- R. Nonno, M. Pannacci, V. Lucini, D. Angeloni, F. Fraschini and B. M. Stankov, Br. J. Pharmacol., 1999, 127, 1288–1294 CrossRef CAS PubMed.
- G. Tosini, K. Ye and P. M. Iuvone, Neuroscientist, 2012, 18, 645–653 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc01374a |
| This journal is © The Royal Society of Chemistry 2019 |