
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photosynthetic production of the nitrogen-rich compound guanidine†
Bo
Wang
 a,
Tao
Dong
a,
Aldon
Myrlie
b,
Liping
Gu
b,
Huilan
Zhu
b,
Wei
Xiong
a,
PinChing
Maness
a,
Ruanbao
Zhou
b and
Jianping
Yu
*a
a,
Tao
Dong
a,
Aldon
Myrlie
b,
Liping
Gu
b,
Huilan
Zhu
b,
Wei
Xiong
a,
PinChing
Maness
a,
Ruanbao
Zhou
b and
Jianping
Yu
*a
aBiosciences Center, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, CO 80401, USA. E-mail: jianping.yu@nrel.gov
bDepartment of Biology & Microbiology, South Dakota State University, Brookings, SD 57007, USA
First published on 14th May 2019
Abstract
The development of an sustainable economy calls for improved energy utilization and storage technologies. Although battery- and carbon-based routes have gained tremendous attention, nitrogen-based routes have rarely been exploited so far. Guanidine (CH5N3) which contains 71.1% nitrogen by mass is an exemplary chemical to explore the nitrogen-based routes of energy utilization and storage. Guanidine has a variety of applications including its use as a slow-release fertilizer, a propellant, or as a precursor to pharmaceuticals and antimicrobial polymers. The conventional chemical synthesis of guanidine through the Frank–Caro process is energy-intensive, consumes fossil fuels, and is detrimental to the environment. Herein, a de novo guanidine biosynthesis (GUB) cycle is proposed with CO2 and nitrate/ammonium as the carbon and nitrogen sources, respectively. The ATP and NAD(P)H needed to drive the GUB cycle are generated via photosynthesis in an engineered cyanobacterium Synechocystis sp. PCC 6803 expressing an ethylene-forming enzyme (EFE). Up to 586.5 mg L−1 (9.9 mM) guanidine was produced after seven days of photoautotrophic cultivation, with an average productivity of 83.8 mg L−1 day−1. In addition, guanidine was directly biosynthesized from CO2, N2 and H2O in an engineered N2-fixing cyanobacterium Anabaena sp. PCC 7120 expressing the EFE. This work demonstrates the first biological conversion of renewable solar energy into chemical energy stored in the nitrogen-rich compound guanidine, which could shed light on harnessing the biological nitrogen metabolism for energy utilization and storage.
Introduction
Both carbon- and nitrogen-based compounds can serve as energy carriers to harness the energy flow in a sustainable economy. N2 (80%) is far more abundant than CO2 (0.04%) in the atmosphere and therefore nitrogen-based energy storage and utilization, i.e., nitrogen economy, could be an alternative approach in harnessing the energy flow.1 However, harnessing energy flow into nitrogen-rich compounds has seldom been reported in biotechnology. Guanidine (CN3H5) contains 71.1% nitrogen by mass and could be an exemplary nitrogen-based energy carrier. It serves as a prominent functional group in essential metabolites including arginine, creatine, guanine, as well as in a wide range of secondary metabolites.2 Due to its high pKa (13.6), it is a strong base and is positively charged under physiological conditions. Many of its applications are related to this unique property. Guanidine salts are used as slow-release fertilizers,3,4 components of propellants,5–7 flame retardants and protein denaturants.8 Guanidine also serves as a building block for the synthesis of pharmaceuticals, pesticides8 and antimicrobial polymers.9–12The industrial production of guanidine, in its current form, is energy-intensive, environmentally damaging, and unsustainable. The process requires fossil fuels exclusively as the raw material, emits CO2 into the atmosphere, and necessitates high-temperature and high-pressure conditions (Fig. S1†).13 The Frank–Caro process is a chemical nitrogen fixation method developed in the late 19th century, and its product cyanamide serves as a precursor to guanidine synthesis (Fig. S1†). However, this form of chemical nitrogen fixation consumes about eight times more energy per fixed nitrogen atom than the Haber–Bosch process,13,14 and the cyanamide synthesis module contributes to >90% of the energy cost in guanidine synthesis (Fig. S1†). Additionally, the process generates hazardous intermediates including cyanamide.8,15
Guanidine is a natural metabolite of a variety of organisms. Guanidine metabolism in microbes in soil was studied three decades ago.16 However, genes responsible for guanidine metabolism or transportation in bacteria had not been discovered and characterized until most recently.17,18 On the other hand, trace amounts of guanidine have been found in human urine,19 and one recent study suggested that guanidine is naturally produced in E. coli and probably in a wide range of microorganisms.18 Nevertheless, the guanidine biosynthesis pathways are still elusive, and to our knowledge no living organisms have been engineered to produce guanidine despite emerging interest and growing demand.20
Guanidine can be formed by the ethylene-forming enzyme (EFE) which exists in a small cluster of microorganisms such as Pseudomonas syringae.21 This enzyme has gained significant attention in recent years owing to its role in the biological formation of ethylene,21–28 a ubiquitous plant signaling molecule and a platform compound in the chemical industry. The EFE reaction generates one molecule of ethylene from each molecule of α-ketoglutarate (AKG), and simultaneously converts arginine to guanidine and 1-pyrroline-5-carboxylic acid (P5C) (Fig. S2†).29 Despite the detection of guanidine in vitro from the EFE reaction,29 its fate in vivo in EFE-expressing organisms has not yet been reported. It has been assumed that the guanidine produced in vivo by EFE could be readily degraded non-enzymatically, followed by re-integration of the degradation products into metabolic pathways.30
In this study, we uncover the accumulation of significant levels of guanidine in a strain of the cyanobacterium Synechocystis sp. PCC 6803 (hereafter Synechocystis) engineered to overexpress EFE. We propose a guanidine biosynthesis (GUB) cycle, which consists of 16 enzymatic reactions and uses CO2 and ammonium as the carbon, nitrogen and hydrogen sources. In cyanobacteria, the GUB cycle is driven by ATP and NAD(P)H generated through photosynthesis. We further demonstrate that guanidine can be directly biosynthesized from CO2 and N2 under photosynthetic N2-fixing conditions in the N2-fixing cyanobacterium Anabaena sp. PCC 7120 engineered to express EFE. Such biological production of nitrogen-rich guanidine from exclusively renewable resources could contribute to the sustainable development of a nitrogen economy.1
Results and discussion
The ethylene-forming enzyme (EFE) closes a guanidine biosynthesis cycle
Preliminary findings showed guanidine accumulated in the culture medium of the engineered EFE-expressing cyanobacterium Synechocystis, but not in that of the wild-type (WT) strain. This indicated that guanidine was produced by EFE under physiological conditions and was not readily recycled into the metabolism in Synechocystis. Genome annotation31 and study of the metabolic network32–34 have led us to propose that a guanidine biosynthesis (GUB) cycle, which consists of 16 enzymatic reactions, operates in the engineered cyanobacterium (Fig. 1). All the genes (except step 9 catalyzed by the enzyme encoded by efe) associated with the GUB cycle are present in the Synechocystis WT (Table S1†), and the efe gene has been overexpressed in the engineered Synechocystis strain to close the cycle. This cycle may be harnessed for the biological production of guanidine in living cells.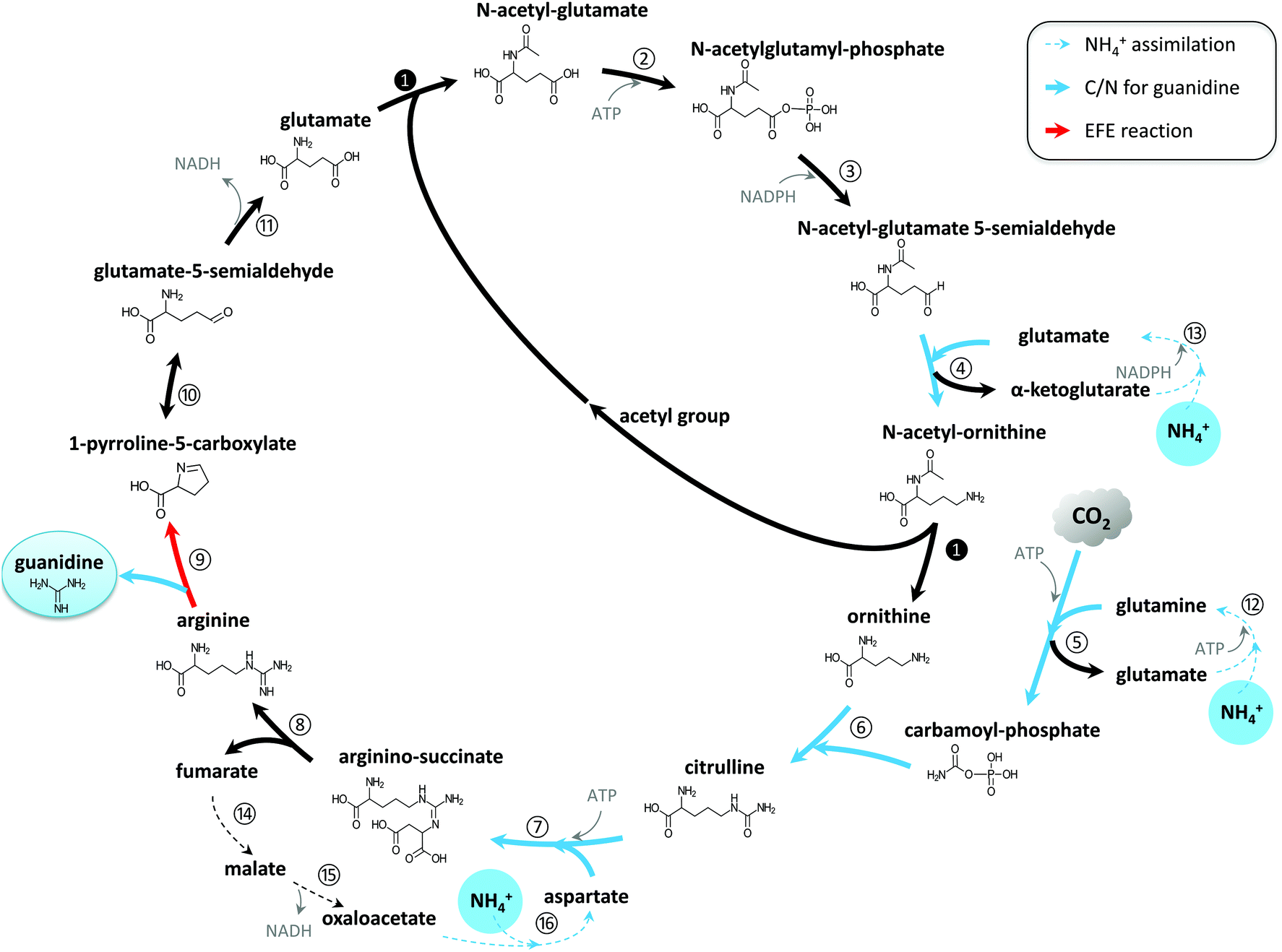 | ||
| Fig. 1 Guanidine biosynthesis (GUB) cycle for the de novo synthesis of guanidine directly from CO2 and ammonium. | ||
Through this biological route, one molecule of guanidine can be directly synthesized from three molecules of ammonium and one molecule of CO2. The GUB cycle consists of three segments: the biosynthesis of arginine from glutamate, the cleavage of arginine into guanidine and 1-pyrroline-5-carboxylic acid (P5C), and the recycling of P5C into glutamate. Starting from glutamate, the biosynthesis of arginine occurs through eight enzymatic reactions, along which three ammonium molecules and one carbon dioxide are assembled together (steps 4–7) forming the guanidine group in arginine. Next, EFE catalyzes the pivotal reaction (step 9) that cleaves arginine to release guanidine and P5C. P5C can then be spontaneously converted to glutamate-5-semialdehyde, which will be converted to glutamate to recycle this compound (reaction 11). In this study, we used the WT version of EFE from Pseudomonas syringae, which simultaneously produces guanidine and ethylene, to demonstrate the photosynthetic production of guanidine from renewable resources. However, it is noteworthy that a single amino acid substitution (e.g. A198 V) in the EFE could eliminate its capability of producing ethylene while retaining a significant portion of its activity in producing P5C (e.g., approximately 60% for variant A198 V).35 EFE variants with such altered properties could be explored to maximize the metabolic flux towards guanidine while minimizing AKG consumption in reaction 9.
Photosynthetic production of guanidine from ammonium and CO2
Biosynthesis of guanidine directly from CO2 and ammonium via the GUB cycle is driven by ATP and NAD(P)H (Fig. 1, Table S1†), which could be provided biologically by glycolysis via the degradation of organic compounds such as sugar feedstock (while releasing CO2). Alternatively, ATP and NAD(P)H can be generated through photosynthesis to drive the GUB cycle (Fig. 1). Herein, Synechocystis, a model cyanobacterium species capable of oxygenic photosynthesis, was utilized as the microbial platform to study guanidine production from ammonium and CO2. Synechocystis was engineered to harbor one copy (strains JU547 and PB646W)23 or two copies (strain PB646) of the efe gene on the chromosome, corresponding to increasing levels of EFE in cellular proteins (Fig. 2A). Both JU547 and PB646 strains as well as the WT were grown in modified BG11 medium (i.e., mBG11; see details in Materials and methods) with CO2 and light as the sole carbon and energy sources. The JU547 and PB646 strains exhibited slightly slower growth compared to the WT (Fig. 2B). However, guanidine was detected in the culture media of both engineered strains but not in that of the WT. The guanidine titers reached 0.21 mM and 0.49 mM for JU547 and PB646, respectively, within 6 days of photoautotrophic cultivation (Fig. 2C). It is noteworthy that at the beginning of the cultivation (day 0–day 2), the guanidine specific productivity was relatively low, at rates of 0.001 and 0.01 mmol L−1 day−1 OD730−1 for strains JU547 and PB646, respectively. The guanidine productivities from strains JU547 and PB646 then increased dramatically, reaching 0.01 and 0.024 mmol L−1 day−1 OD730−1, respectively, by day 6 (Fig. 2D). To investigate if guanidine can be biosynthesized directly from ammonium rather than using nitrate as the nitrogen source, the 17.6 mM NaNO3 in the mBG11 medium was replaced with 5 mM NH4Cl. Subsequently, free guanidine was detected in the culture supernatant of the PB646 strain within three days of photoautotrophic cultivation (Fig. S3†). Since guanidine biosynthesis requires a significant nitrogen input while moderate-high levels of ammonium in the culture medium may cause damage to the photosystems in cyanobacteria,36 nitrate rather than ammonium was used as the nitrogen source in all of the following experiments. Once nitrate is taken up by Synechocystis cells, it is reduced to ammonium by the endogenous nitrate reductase and nitrite reductase before being integrated into the downstream metabolism.37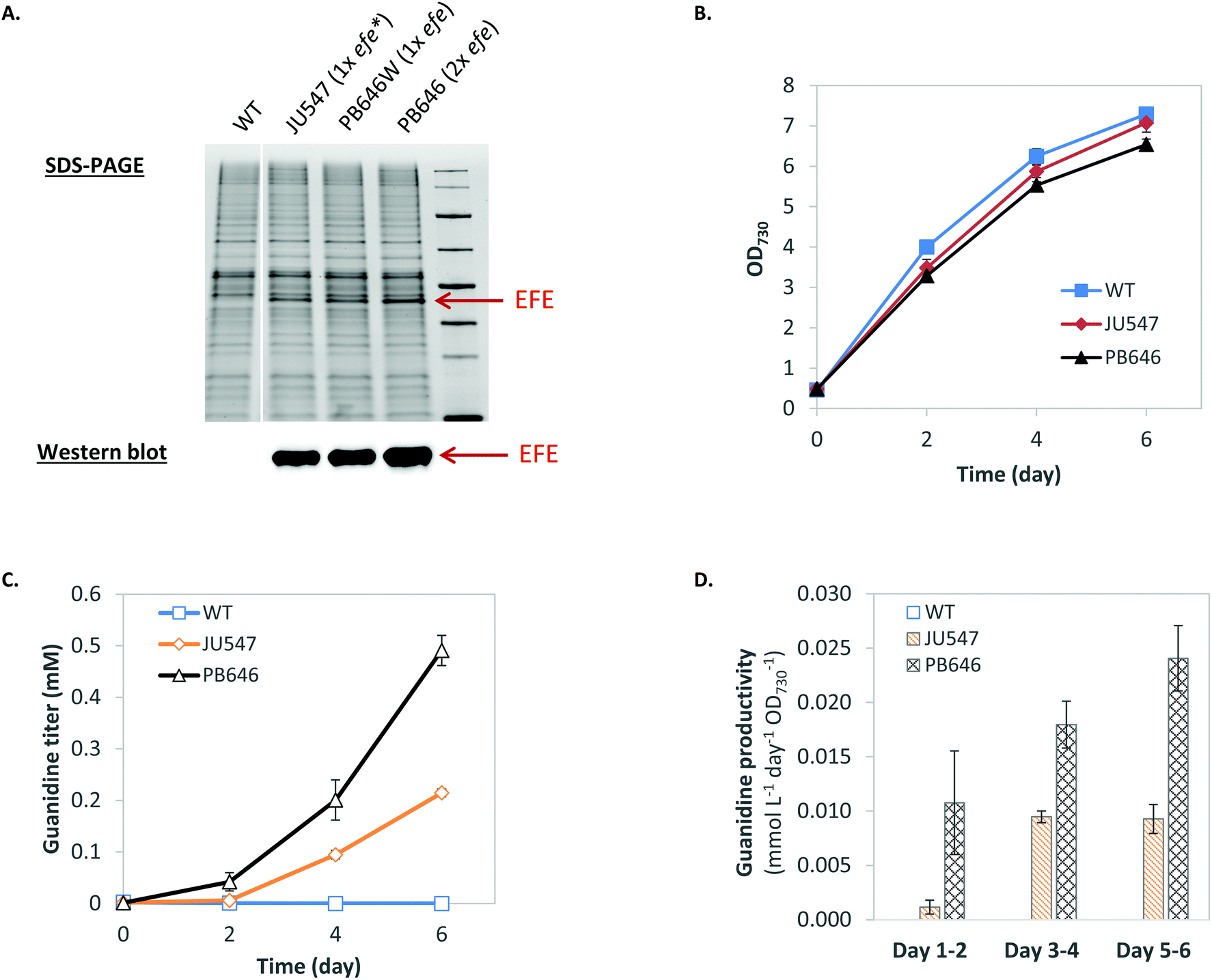 | ||
| Fig. 2 Production of guanidine from CO2 and ammonium in engineered Synechocystis. (A) Expression of EFE confirmed by protein gel and western blot. (B) Cell growth curves. (C) HPLC analysis of guanidine in the culture medium of the WT and engineered Synechocystis strains grown under a light intensity of 50 μE m−2 s−1. (D) Guanidine specific productivities. Data represent mean value and standard deviation of three independent biological replicates. | ||
We speculated that increased photosynthetic generation of ATP and NAD(P)H under stronger light conditions would accelerate the turnover rate of the GUB cycle. Therefore, we increased the light intensity from 50 to 500 μE m−2 s−1, which is about 25% of the maximal solar radiation in Denver, Colorado. In addition, the culture was initiated with a relatively high cell density to minimize possible high light stress. Synechocystis PB646 continued growing for seven days, along with the continuous accumulation of guanidine in the culture medium analyzed by HPLC with a multi-wavelength detector. The in-flask titer peaked at 6.86 mM by the end of day 6 (Fig. 3A). Taking the daily sampling and medium-replenishing into account, the accumulative guanidine titer reached 9.94 mM, i.e., 586.5 mg L−1 by the end of day 7, with an average productivity of 1.42 mmol L−1 day−1, i.e., 83.8 mg L−1 day−1. The sample collected at the end of day 7 was further analyzed on LC/MS, which verified that the product was indeed guanidine (Fig. 3B–D).
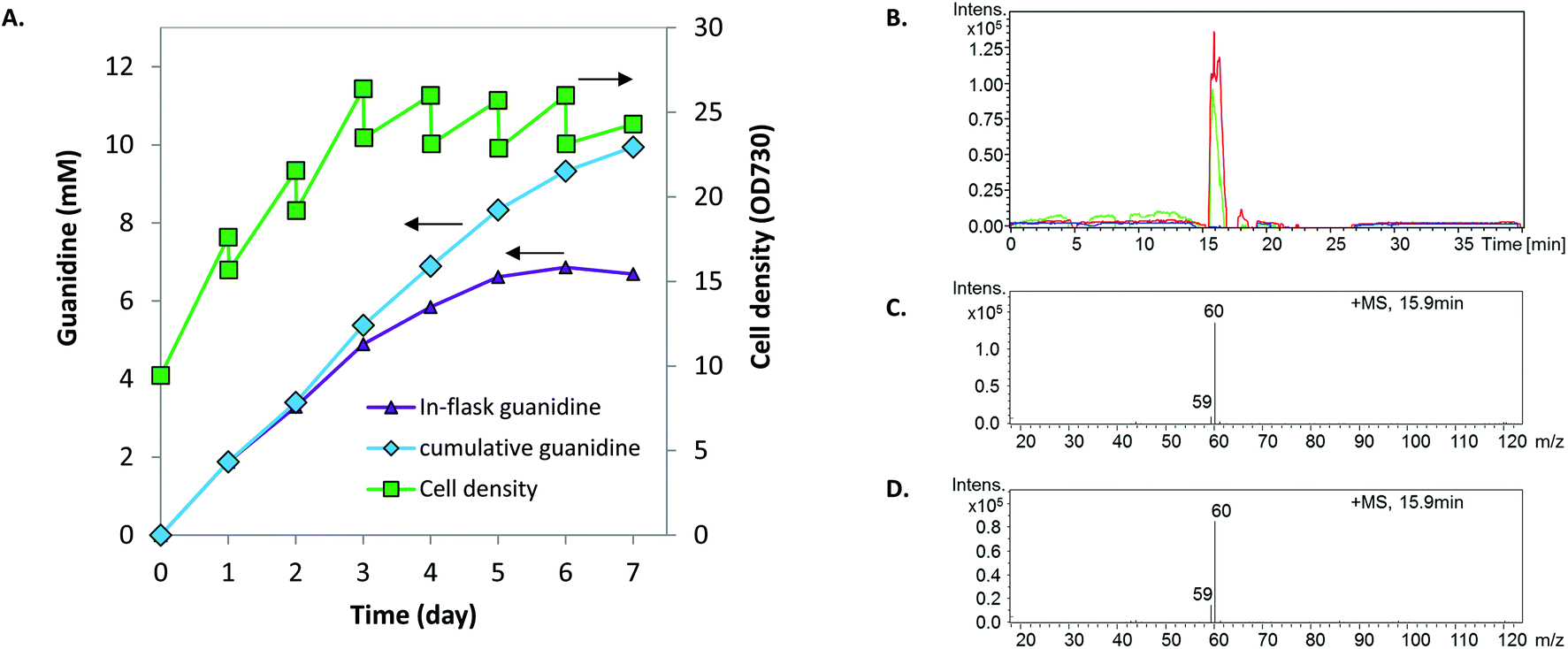 | ||
| Fig. 3 Photosynthetic production of guanidine under high light and high cell density conditions. (A) In-flask optical density of cells and accumulation of guanidine. (B) Guanidine elution from the LC/MS column. Green line shows the culture medium with de novo synthesized guanidine by engineered Synechocystis. Red line shows guanidine standard. (C) Mass spectrometry of guanidine standard eluted in (B). (D) Mass spectrometry of de novo synthesized guanidine by engineered Synechocystis eluted in (B). | ||
In this study, guanidine was readily excreted into the culture medium of the EFE-expressing Synechocystis without any efforts to engineer Synechocystis for guanidine export across the cell membrane and cell wall. This implies that either there are endogenous transporters that export guanidine out of the cell, or that guanidine diffuses or leaks out of the cell due to a concentration gradient or cell death. We compared the intracellular and extracellular guanidine concentrations when the guanidine-producing Synechocystis strains JU547 and PB646 were in the late exponential growth phase (OD730 = 0.6–0.8). It was found that the guanidine concentrations inside cells reached 2.2 and 3.2 mM in strain JU547 and PB646, respectively, whereas the extracellular guanidine concentration (in the culture supernatant) was below the detection limit which is roughly two orders of magnitude lower than the intracellular guanidine concentration (Fig. S4†). This suggests that the export of guanidine from Synechocystis is inefficient. Researchers have identified guanidine transporters from various microorganisms17,18 that could be overexpressed in the engineered Synechocystis to increase the guanidine productivity by reducing product inhibition or intracellular toxicity effects, or to facilitate guanidine harvest without cell lysis.
Photosynthetic production of guanidine from N2 and CO2
Microbial nitrogen-fixation contributes to more than half of the fixed nitrogen on Earth.13 It has the potential to substitute the energy-intensive, CO2-emitting Haber–Bosch process for ammonium production, as well as to directly produce many other nitrogen-containing compounds. Despite decades of studies on microbial nitrogen fixation and the bioproduction of ammonium,38,39 few have focused on the production of other nitrogen-containing compounds through engineering nitrogen-fixing microorganisms. We hypothesized that guanidine can be photosynthetically produced by nitrogen-fixing cyanobacteria directly from N2, CO2 and H2O, which are among the most abundant resources on Earth. To test this hypothesis, the efe gene was expressed under the dual Pnir and PpsbA1 promoter on the replicative plasmid pZR1429 in the nitrogen-fixing cyanobacterium Anabaena sp. PCC 7120 (hereafter Anabaena 7120) (Fig. 4A and B). Transformation of Anabaena 7120 was confirmed via colony PCR (Fig. S5†). Expression of EFE in the resulting engineered Anabaena strain A1429 was shown by SDS-PAGE and western blot (Fig. 4C). To test if Anabaena A1429 was able to produce guanidine directly from N2, BG11 medium was modified to completely remove all sources of combined nitrogen, resulting in medium BG1100 as detailed in the Methods section.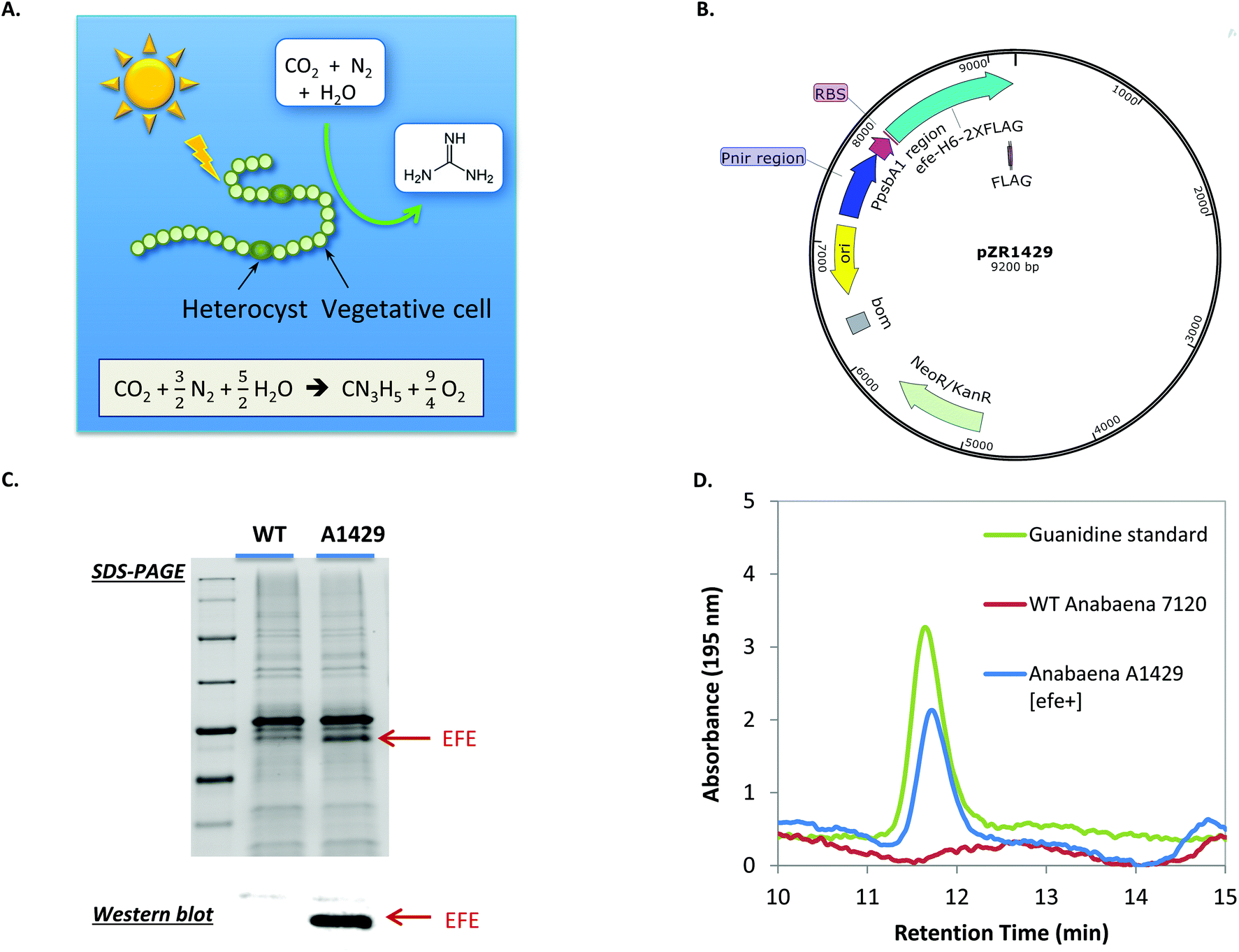 | ||
| Fig. 4 Photosynthetic biosynthesis of guanidine from CO2, H2O and N2 in the engineered N2-fixing cyanobacterium Anabaena 7120. (A) The concept of guanidine production from N2-fixing cyanobacteria. (B) Recombinant plasmid used to transform Anabaena WT. (C) Protein gel and western blot for confirming heterologous expression of EFE in the engineered Anabaena strain A1429. (D) HPLC results showing that guanidine was produced from Anabaena strain A1429. | ||
Anabaena A1429 was found to excrete up to 0.43 mg L−1 (7.3 μM) guanidine into the culture medium after 7 days of photoautotrophic cultivation under N2-fixing conditions (Fig. 4D). Ethylene production was also detected from the Anabaena A1429 culture (Fig. S6†). In contrast, neither guanidine nor ethylene was detected from the culture of Anabaena 7120 WT under the experimental conditions. This represents the first time that guanidine, which contains 71.1% of nitrogen by mass, has been synthesized from CO2 and atmospheric N2, using light as the sole energy source. Current guanidine production from the engineered Anabaena has much room for optimization. For instance, the light intensity was only 15 μE m−2 s−1 when 0.43 mg L−1 guanidine was produced, because Anabaena 7120 derived strains are sensitive to higher light under the tested N2-fixing conditions in our lab. We expect that higher guanidine productivities could be achieved when EFE is expressed in other N2-fixing cyanobacterial strains that exhibit more robust growth.40 This work thereby provides the first proof-of-concept toward realizing a renewable nitrogen economy based on atmospheric nitrogen.
Conclusions
A biological route for guanidine production has been demonstrated by rewiring cyanobacterial photosynthesis and nitrogen metabolism. Compared to the current industrial production of guanidine, this route, via the proposed guanidine biosynthesis (GUB) cycle, represents a sustainable alternative to pathways that involve fossil fuels and toxic intermediates. This is also an example of re-purposing a well-known enzymatic reaction for a new biotechnology product. Future research to optimize biological guanidine production should explore enzymology, transporters, and robust host strains. Further development of this technology could contribute to development of the nitrogen economy, where nitrogen-rich compounds are used to address energy utilization and storage.Materials and methods
Bacterial strains and growth conditions
E. coli NEB5α (New England BioLabs, MA, USA) was routinely grown in LB medium and was used as the microbial host for cloning and maintaining all recombinant plasmids. Synechocystis sp. PCC 6803 and the recombinant strains were typically grown in modified BG11 medium (mBG11) containing the following components in 1 liter: 1.5 g NaNO3, 75 mg MgSO4·7H2O, 36 mg CaCl2·2H2O, 6.6 mg citric acid·H2O, 11.2 mg EDTA·Na2, 20 mg Na2CO3, 30.5 mg K2HPO4, 6 mg ferric ammonium citrate, 2.86 mg H3BO3, 1.81 mg MnCl2·4H2O, 0.22 mg ZnSO4·7H2O, 0.39 mg Na2MoO4·2H2O, 0.08 mg CuSO4·5H2O, and 0.04 mg CoCl2·6H2O. 4.6 g N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (20 mM TES) and 8.4 g (100 mM) NaHCO3 were added into the mBG11 for buffering purposes unless otherwise noted. Synechocystis cultures were placed on a rotary shaker at 150 rpm at 30 °C under a constant light intensity of about 50 μE m−2 s−1 in a Percival chamber (Percival Scientific, Inc., IA, USA) aerated with 5% CO2 unless otherwise specified. When Synechocystis was grown on solid medium, TES was adjusted to 10 mM, 3 g L−1 thiosulfate was added, and NaHCO3 was omitted. The medium pH was adjusted to 8.2 before 1.5% (w/v) agar was added followed by autoclaving at 121 °C for 30 min. When appropriate, antibiotics were added to the solid medium to final concentrations of 50 mg L−1 and 10 mg L−1 for spectinomycin and kanamycin, respectively. Anabaena sp. PCC 7120 and the engineered strain were grown in mBG11 medium for general maintenance purpose. When growing Anabaena under N2-fixing conditions, the medium was modified to completely remove all sources of combined nitrogen, resulting in medium BG1100: 1.5 g L−1 NaNO3 was replaced by 1.5 g L−1 NaCl, 6 μg mL−1 ferric ammonium citrate was replaced by 5.1 μg mL−1 FeCl3·6H2O, and EDTA was removed from the medium. In addition, bicarbonate and TES were not added into the BG1100 medium. All strains and plasmids used in this study are listed in Table 1.| Strains | Genotype or features | Sources |
|---|---|---|
| E. coli HB101 [pRL623 + pRL443] | Encodes methylases AvaiM, Eco47iiM, and Ecot22iM that target sites subject to restriction in Anabaena sp. strain PCC 7120; pRL443 is a shuttle vector suitable for mobilizing plasmids to cyanobacterial strains. | 44 |
| NEB10β | Δ(ara-leu) 7697 araD139 fhuA ΔlacX74 galK16 galE15 e14- ϕ80dlacZΔM15 recA1 relA1 endA1 nupG rpsL (StrR) rph spoT1 Δ(mrr-hsdRMS-mcrBC) | NEB |
| NEB5α | fhuA2 Δ(argF-lacZ)U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | NEB |
| Synechocystis sp. PCC 6803 | Wild-type | ATCC |
| Synechocystis JU547 | 1× efe; PpsbA-RBSv4-efe-TT7-SmR inserted at the slr0168 neutral site | 23 |
| Synechocystis PB646W | 1× efe; PpsbA-RBSv4-efe-TT7-TrrnBT1-KanR inserted between slr1362 and sll1274 of the Synechocystis chromosome | This study |
| Synechocystis PB646 | 2× efe; PpsbA-RBSv4-efe-TT7-SmR inserted at the slr0168 neutral site; PpsbA-RBSv4-efe-TT7-TrrnBT1-KanR inserted between slr1362 and sll1274 of the Synechocystis chromosome | This study |
| Anabaena sp. PCC 7120 | Wild-type | 43 |
| Anabaena A1429 | Harboring plasmid pZR1429 | This study |
| Plasmids | ||
| pJU158 | slr0168-PpsbA-RBSv4-efe-TT7-SmR-slr0168, pUC ori | 22 |
| pBS-S2 | Contains homologous arms targeting slr1362 and sll1274 of the Synechocystis chromosome, derived from pBlueScript II SK(+) | 42 |
| pSrT1PK | rrnB T1-KanR inserted between MluI and SalI sites of pBS-S2 | 41 |
| pPB146 | AmpR, slr1362-PpsbA-RBSv4-efe-TT7-TrrnBT1-KanR-sll1274 | This study |
| pCR2.1-TOPO | Invitrogen | |
| pZR1188 | KmR, NmR; Pnir/PpsbA1; MCS; F2 | 43 |
| pZR1428 | Gene efe cloned into pCR2.1-TOPO | This study |
| pZR1429 | Gene efe inserted between the BglII and SalI sites on plasmid pZR1188 | This study |
Construction of recombinant plasmids
The high fidelity Q5 DNA polymerase (New England BioLabs, MA, USA) was used to amplify DNA fragments in PCR for cloning purposes. Plasmid pPB146 was constructed by PCR amplification of the ethylene forming enzyme (EFE) expression cassette from plasmid pJU15823 using primers PetE-F1 and T7-R2, and the purified PCR product was inserted between the BamHI and MluI restriction sites on plasmid pSrT1PK. Gibson Assembly Kit (New England BioLabs, MA, USA) was used for the DNA recombination. pSrT1PK was previously constructed for the expression of the tesB gene in the Synechocystis strain TTrK.41 Briefly, the kanamycin resistance cassette containing its promoter was PCR amplified using primers Kan9 and Kan2. The PCR product was purified and further amplified with primers rT1KP and Kan2 using LongAmp Taq DNA polymerase (New England BioLabs, Ipswich, MA) before being digested with the MluI and SalI-HF restriction enzymes and inserted between the MluI and SalI restriction sites on plasmid pBS-S2.42 Plasmid pZR1429 was constructed by first cloning the efe gene using primers ZR791 and ZR792 into plasmid pCR2.1-TOPO (Invitrogen) to produce the recombinant plasmid pZR1428, restriction digesting pZR1428 with BglII and SalI, and inserting the resultant efe-containing fragment between the BglII and SalI sites on plasmid pZR1188.43 The resulting pZR1429 plasmid was verified by colony PCR with primers ZR45 and ZR792 (data not shown). All DNA fragments of interest were validated via DNA sequencing. All primers used in this study are listed in Table S2.†Genetic engineering of cyanobacteria
Transformation of Synechocystis was accomplished via natural transformation.22 Briefly, strain JU54723 was inoculated into mBG11 medium with an initial OD730 of 0.1 and was grown until the OD730 reached approximately 0.4. Then, 2.5 mL of culture was condensed to about 0.2 mL via centrifugation and resuspended with the same culture medium. Samples were transferred into a 1.5 mL Eppendorf tube, and about 2 μg pPB146 plasmid DNA was mixed into the resuspended cells. The sample was incubated under low light for about 5 hours, and mixed once in the middle of the incubation. Cells were then spread onto BG11 plates supplemented with 10 mM TES-NaOH and 10 mg L−1 kanamycin. Strain PB646 W was constructed by transforming the Synechocystis WT with the integration plasmid pPB146. The complete segregation of Synechocystis genomes was verified via colony PCR, and DNA sequencing of the purified PCR product validated the correct DNA sequence.The Anabaena A1429 strain was constructed through the conjugal transfer of plasmid pZR1429 into Anabaena 7120. Tri-parental mating was initiated by mixing E. coli HB101 [pRL623 + pRL443] with E. coli NEB10β containing the cargo plasmid pZR1429. The E. coli strains were combined in a single 1.5 mL tube and incubated at room temperature for 30 min to allow the strains to mate. Anabaena 7120 culture was then added to the E. coli mating mixture and allowed to mate for another 1 hour. Then, the culture solution was plated on a nitrocellulose membrane (HATF 08550) onto BG11 agar supplemented with 5% LB and incubated at 30 °C under continuous light (50 μE m−2 s−1) for 24 hours. Next, the membrane was transferred to a BG11 plate containing 100 μg mL−1 neomycin (Nm100) to select for transformed Anabaena 7120. Plates were incubated at 30 °C under light until single colonies formed. On a weekly basis, membranes were transferred to new BG11 plates supplemented with Nm100. More details are described in the literature.44 The Anabaena colonies containing pZR1429 were verified by colony PCR with primers EFE1 and EEF4, resulting in a 0.7 kb PCR product (Fig. S5†).
Production of guanidine under high light and high cell density conditions
Synechocystis strain PB646 was grown in 50 mL mBG11 in a baffled 250 mL flask under a constant light intensity of about 50 μE m−2 s−1 in a Percival chamber (Percival Scientific, Inc., IA, USA) aerated with 5% CO2. Once the OD730 reached about 1.0, the culture was centrifuged, and the cell pellet was resuspended with 50 mL 5× concentrated mBG11 supplemented with 20 mM TES and 100 mM NaHCO3 in a 250 mL baffled flask. The flask was placed on a rotary shaker (at 150 rpm) in the AlgaeTron AG230 incubator (Photon Systems Instruments, Czech Republic) aerated with humidified 5% CO2 at a rate of 50 mL min−1. The culture was initially illuminated with about 50 μE m−2 s−1, and then the light intensity was increased stepwise to 500 μE m−2 s−1 within one day. Subsequently, the light intensity was held at 500 μE m−2 s−1 for another day to allow the culture to grow to a higher cell density. When the OD730 reached about 8, cells were harvested by centrifugation, and then resuspended with 38 mL of 5× mBG11 supplemented with 20 mM TES and 100 mM NaHCO3 in a 250 mL baffled flask. 3 mL was sampled out immediately (as day 0), and by the end of day 1, 2 mL of culture was sampled out followed by the addition of 4 mL of 10× mBG11 right after sampling on day 1. Starting from day 2 until day 7, 4 mL of culture was sampled for analysis of the cell density (OD730) and guanidine and 4 mL of 10× mBG11 medium was added back into the flask at the end of each day. Upon determining the OD730, samples were first diluted with the medium to a range between 0.05 and 0.5 and then the absorbance at the wavelength of 730 nm was taken using a Beckman Coulter DU-800 UV/Vis spectrophotometer; culture OD730 was finally back calculated. The culture volume was maintained at about 35 mL throughout the seven days of cultivation.Production of guanidine using engineered Anabaena
The Anabaena 7120 WT and the engineered Anabaena strain A1429 were inoculated at a low cell density and were grown in the BG1100 medium under a continuous light of 15 μE m−2 s−1 at 30 °C in a Percival chamber (Percival Scientific, Inc., IA, USA) aerated with 5% CO2. Neomycin was added to a final concentration of 50 μg mL−1. After seven days, cultures were centrifuged and supernatants were filtered first through filters with a pore size of 0.2 μm and then through a 5 kDa membrane before being analyzed by HPLC.SDS-PAGE and western blotting
Protocols were adapted from our previous work.22 Briefly, when the cyanobacterial culture was grown to an OD730 of 0.5–1.0, approximately 5 OD730 mL (i.e., 5 mL culture sampled if the OD730 of the culture equals 1) of cells were collected after centrifugation at 3220g, 24 °C for 5 min. The supernatants were discarded, and the cell pellets were stored at −80 °C for later use. Upon running SDS-PAGE, cells were resuspended with 0.5 mL of cold 0.1 M potassium phosphate buffer (pH7.0) supplemented with DTT (0.2 mM) and Halt Protein Inhibitor Cocktail (Thermo Fisher Scientific, MA, USA), and subsequently lysed by sonication in an ice-water bath using a Q500 Sonicator (Qsonica L.L.C, CT, USA) programed for 100 cycles of 3-sec-on-3-sec-off at an amplitude of 20%. The cell lysate was then centrifuged at 4 °C, 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min, and then the cell extract (supernatant) was taken into a new Eppendorf tube placed on ice. The protein concentrations of the cell extract were quantified using Bradford assay (Thermo Fisher Scientific, MA, USA). Approximately 2.5 μg proteins of each cell extract were mixed with same volume of the 2× SDS-PAGE sample buffer (950 μl BioRad 2× Laemmli Sample Buffer + 50 μl BME) in PCR tubes and incubated at 99 °C for 5 min using a thermocycler. Samples were then loaded onto Mini-PROTEAN® TGX Stain-Free™ precast gels (Bio-Rad Laboratories, CA, USA), and the electrophoresis was conducted at 150 V for about 45 min. Gels were imaged using UV excitation in a FluorChem Q imager (ProteinSimple, CA, USA).
000g for 10 min, and then the cell extract (supernatant) was taken into a new Eppendorf tube placed on ice. The protein concentrations of the cell extract were quantified using Bradford assay (Thermo Fisher Scientific, MA, USA). Approximately 2.5 μg proteins of each cell extract were mixed with same volume of the 2× SDS-PAGE sample buffer (950 μl BioRad 2× Laemmli Sample Buffer + 50 μl BME) in PCR tubes and incubated at 99 °C for 5 min using a thermocycler. Samples were then loaded onto Mini-PROTEAN® TGX Stain-Free™ precast gels (Bio-Rad Laboratories, CA, USA), and the electrophoresis was conducted at 150 V for about 45 min. Gels were imaged using UV excitation in a FluorChem Q imager (ProteinSimple, CA, USA).
Western blotting was conducted using a Pierce™ G2 Fast Blotter (Thermo Fisher Scientific, MA, USA). Mouse monoclonal anti-FLAG antibody (Rockland Immunochemicals Inc., PA, USA) was used as the primary antibody (at 1:1000 dilution) to probe the FLAG-Tag fused to the C-terminus of EFE. Clean-Blot™ IP Detection Reagent (HRP; Thermo Fisher Scientific, MA, USA) was used as the secondary antibody (at 1:1000 dilution). The chemiluminescent blots were imaged using a FluorChem Q imager (ProteinSimple, CA, USA).
Quantification of guanidine
Guanidine was quantified using HPLC equipped with a cation exchange column.45 Guanidine hydrochloride (Cat.#G4505, Sigma-Aldrich, USA) was used to prepare standard solutions, and standards and filtrated culture samples (via pass through 0.2 μm diameter filters) were analyzed using an Agilent 1200 Series HPLC (Agilent, USA) equipped with a multi-wavelength detector and a set of Dionex IonPac™ CS14 cation-exchange guard (4 mm × 50 mm) and analytical columns (4 mm × 250 mm; Thermo Fisher Scientific, MA, USA). The column temperature was kept at 30 °C. The mobile phase was 3.75 mM methanesulfonic acid. The flow rate was kept at 1.0 mL min−1 for 30 min for each run. The sample injection volume was 100 μL. The guanidine concentrations were routinely quantified via the multi-wavelength detector at an absorbance of 195 nm (Fig. S7†).Measurement of Synechocystis intracellular guanidine
Cultures of Synechocystis WT, JU547 and PB646 were inoculated into 30 mL mBG11 with an initial OD730 of approximately 0.08. Cultures were grown under a light intensity of 40 μE m−2 s−1 for 2 days (OD730 reaching 0.6–0.8). Then 27 mL of each culture was quickly taken into a pre-chilled 50 mL centrifuge tube and immediately placed into a −50 °C bath (50% methanol) for 20 seconds to cool down the sample. Samples were then centrifuged at 5000g, 4 °C for 5 min. The supernatants were aspirated, 300 μL −50 °C 80% methanol was added to the cell pellet, and the pellet was transferred into a clean pre-chilled 1.5 mL Eppendorf tube. The samples were then kept on dry ice for 15 min, vortexed at 4 °C for 3 min, and centrifuged at 18000g, 4 °C for 5 min. After centrifugation the extracts (supernatants) were transferred into a clean pre-chilled 1.5 mL Eppendorf tube and were kept on ice. The extraction of metabolites was repeated on the same sample once with 300 μL −50 °C 80% methanol, and the supernatant was pooled together with the previous one. Next, an additional 400 μL −50 °C 80% methanol was added, and the cell pellet was sonicated with 3-sec-on-3-sec-off for 30 cycles in an ice/water bath. The sonicated samples were centrifuged at 18000g, 4 °C for 5 min and the supernatant was pooled with the previous one for each strain. Extracts were then dried using a speed-vacuum, and were then resuspended with 80 μL 80% methanol by vortexing for 30 s before being transferred to glass vials for LC-MS.Detection of guanidine via LC-MS
Culture supernatants and metabolite extracts were analyzed by using an Agilent 1100 Series liquid chromatograph (LC) coupled with an electrospray ionization (ESI) mass spectrometer. A Luna NH2 (Phenomenex) 250 mm × 2 mm × 5 μm column (Phenomenex, Aschaffenburg, Germany) was utilized for chromatographic separation. The mobile phase was comprised of solvent A: 20 mM ammonia acetate + 20 mM ammonia hydroxide in 95:5 water:acetonitrile, pH 9.25; solvent B: acetonitrile. The gradients are as follows: t = 0, 85% B; t = 15 min, 0% B; t = 28 min, 0% B; t = 30 min, 85% B; t = 40 min, 85% B. The injection volume was 10 μl. The flow rate was 0.15 ml min−1. Metabolites were detected using an Agilent Ion Trap mass spectrometer (LC/MSD Trap SL) operated in the positive-ion mode. The instrument settings were: source, ESI; capillary,+3500 V; end plate offset, −500 V; scan range: 20–400 m/z; nebulizer, 20 psi; dry gas flow, 8.0 L min−1; dry temperature, 325 °C.
Measurement of ethylene produced from the engineered Anabaena
2 mL Anabaena cultures were transferred into a 13 mL Hungate tube, supplemented with 50 mM sterile bicarbonate, and was sealed immediately and incubated under the same growth conditions with shaking. After overnight incubation, 500 μL gas was sampled from the headspace using a sample-lock syringe and injected into an Agilent 7980A gas chromatograph (GC). The GC was equipped with a TCD detector and an 80/100 Porapak N column (SUPELCO Stock #13141-U; 6 FT × 1/8 IN × 2.1 MM SS), and was operated under the following conditions: carrier gas, helium; inlet temperature, 115 °C; inlet total flow, 30 mL min−1; inlet septum purge flow, 3 mL min−1; oven temperature, 60 °C; detector temperature, 190 °C; detector reference flow 30 mL min−1; detector makeup flow (He): 10 mL min−1.Author contributions
B. W., P. M., R. Z. and J. Y. conceived the work. B. W. designed and performed most of the experiments and drafted the manuscript. T. D. and W. X. analyzed guanidine by LC-MS. A. M., L. G., H. Z. and R. Z. constructed the recombinant Anabaena strain. All authors read, revised and approved the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was authored in part by Alliance for Sustainable Energy, LLC, the manager and operator of the National Renewable Energy Laboratory for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding was provided by the DOE Office of Energy Efficiency and Renewable Energy BioEnergy Technologies Office (B. W., T. D., W. X., P. C. M. and J. Y.). This study was supported in part by the South Dakota Agricultural Experiment Station (L. G., R. Z.) and by USDA-NIFA GRANT11665597: N2-Fixing Cyanobacteria Harnessed for Biosolar Production of Nitrofertilizer (to R. Z). Sunnyjoy Dupuis helped with language editing. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- O. Elishav, D. R. Lewin, G. E. Shter and G. S. Grader, Appl. Energy, 2017, 185, 183–188 CrossRef CAS.
- R. G. S. Berlinck, A. F. Bertonha, M. Takaki and J. P. G. Rodriguez, Nat. Prod. Rep., 2017, 34, 1264–1301 RSC.
- Z. Zhang, M. Nyborg, M. Worsley, K. M. Worsley and D. A. Gower, Commun. Soil Sci. Plant Anal., 1992, 23, 431–439 CrossRef CAS.
- M. Nyborg, M. Worsley and K. Worsley, WO Pat., 1992/04302, 1992 Search PubMed.
- A. Ulas, G. A. Risha and K. K. Kuo, Fuel, 2006, 85, 1979–1986 CrossRef CAS.
- G. K. Lund and R. Bradford, US Pat., 8 815029B2, 2014 Search PubMed.
- R. D. Taylor and I. V. Mendenhall, US Pat., 6103030, 2000 Search PubMed.
- T. Güthner, B. Mertschenk and B. Schulz, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006, DOI:10.1002/14356007.a12_545.pub2.
- S. Heydarifard, Y. Pan, H. Xiao, M. M. Nazhad and O. Shipin, Carbohydr. Polym., 2017, 163, 146–152 CrossRef CAS PubMed.
- S. Ghamrawi, J. P. Bouchara, O. Tarasyuk, S. Rogalsky, L. Lyoshina, O. Bulko and J. F. Bardeau, Mater. Sci. Eng., C, 2017, 75, 969–979 CrossRef CAS.
- Y. Mei, C. Yao and X. Li, Biofouling, 2014, 30, 313–322 CrossRef CAS.
- K. Liu, Y. Xu, X. Lin, L. Chen, L. Huang, S. Cao and J. Li, Carbohydr. Polym., 2014, 110, 382–387 CrossRef CAS.
- B. S. Patil, V. Hessel, L. C. Seefeldt, D. R. Dean, B. M. Hoffman, B. J. Cook and L. J. Murray, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, DOI:10.1002/14356007.a17_471.pub2.
- Technology Roadmap: Energy and GHG Reductions in the Chemical Industry via Catalytic Processes, International Energy Agency-IEA, 2013 Search PubMed.
- T. Güthner and B. Mertschenk, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006, DOI:10.1002/14356007.a08_139.pub2.
- W. R. Mitchell, Bull. Environ. Contam. Toxicol., 1987, 39, 974–981 CrossRef CAS.
- A. A. Kermani, C. B. Macdonald, R. Gundepudi and R. B. Stockbridge, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3060–3065 CrossRef CAS PubMed.
- J. W. Nelson, R. M. Atilho, M. E. Sherlock, R. B. Stockbridge and R. R. Breaker, Mol. Cell, 2017, 65, 220–230 CrossRef CAS PubMed.
- S. Natelson and J. E. Sherwin, Clin. Chem., 1979, 25, 1343–1344 CAS.
- R. R. Breaker, R. M. Atilho, S. N. Malkowski, J. W. Nelson and M. E. Sherlock, Biochemistry, 2017, 56, 345–347 CrossRef CAS.
- C. Eckert, W. Xu, W. Xiong, S. Lynch, J. Ungerer, L. Tao, R. Gill, P. C. Maness and J. Yu, Biotechnol. Biofuels, 2014, 7, 33 CrossRef PubMed.
- B. Wang, C. Eckert, P. C. Maness and J. Yu, ACS Synth. Biol., 2018, 7, 276–286 CrossRef CAS PubMed.
- W. Xiong, J. A. Morgan, J. Ungerer, B. Wang, P.-C. Maness and J. Yu, Nat. Plants, 2015, 1, 15053 CrossRef CAS.
- V. P. Veetil, S. A. Angermayr and K. J. Hellingwerf, Microb. Cell Fact., 2017, 16, 34 CrossRef PubMed.
- H. Mo, X. Xie, T. Zhu and X. Lu, Biotechnol. Biofuels, 2017, 10, 145 CrossRef.
- J. Ungerer, L. Tao, M. Davis, M. Ghirardi, P. C. Maness and J. P. Yu, Energy Environ. Sci., 2012, 5, 8998–9006 RSC.
- Z. Zhang, T. J. Smart, H. Choi, F. Hardy, C. T. Lohans, M. I. Abboud, M. S. W. Richardson, R. S. Paton, M. A. McDonough and C. J. Schofield, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4667–4672 CrossRef CAS PubMed.
- F. Guerrero, V. Carbonell, M. Cossu, D. Correddu and P. R. Jones, PLoS One, 2012, 7, e50470 CrossRef CAS PubMed.
- H. Fukuda, T. Ogawa, M. Tazaki, K. Nagahama, T. Fujii, S. Tanase and Y. Morino, Biochem. Biophys. Res. Commun., 1992, 188, 483–489 CrossRef CAS PubMed.
- T. Zavrel, H. Knoop, R. Steuer, P. R. Jones, J. Cerveny and M. Trtilek, Bioresour. Technol., 2016, 202, 142–151 CrossRef CAS PubMed.
- T. Kaneko, S. Sato, H. Kotani, A. Tanaka, E. Asamizu, Y. Nakamura, N. Miyajima, M. Hirosawa, M. Sugiura, S. Sasamoto, T. Kimura, T. Hosouchi, A. Matsuno, A. Muraki, N. Nakazaki, K. Naruo, S. Okumura, S. Shimpo, C. Takeuchi, T. Wada, A. Watanabe, M. Yamada, M. Yasuda and S. Tabata, DNA Res., 1996, 3, 109–136 CrossRef CAS PubMed.
- M. I. Muro-Pastor, J. C. Reyes and F. J. Florencio, Photosynth. Res., 2005, 83, 135–150 CrossRef CAS PubMed.
- J. H. Shin and S. Y. Lee, Microb. Cell Fact., 2014, 13, 166 CrossRef PubMed.
- H. Zhang, Y. Liu, X. Nie, L. Liu, Q. Hua, G. P. Zhao and C. Yang, Nat. Chem. Biol., 2018, 14, 575–581 CrossRef CAS PubMed.
- S. Martinez, M. Fellner, C. Q. Herr, A. Ritchie, J. Hu and R. P. Hausinger, J. Am. Chem. Soc., 2017, 139, 11980–11988 CrossRef CAS.
- M. Drath, N. Kloft, A. Batschauer, K. Marin, J. Novak and K. Forchhammer, Plant Physiol., 2008, 147, 206–215 CrossRef CAS PubMed.
- E. Flores, J. E. Frias, L. M. Rubio and A. Herrero, Photosynth. Res., 2005, 83, 117–133 CrossRef CAS.
- L. F. Razon, Bioresour. Technol., 2012, 107, 339–346 CrossRef CAS PubMed.
- M. Brouers and D. O. Hall, J. Biotechnol., 1986, 3, 307–321 CrossRef CAS.
- J. Moreno, M. A. Vargas, H. Rodriguez, J. Rivas and M. G. Guerrero, Biomol. Eng., 2003, 20, 191–197 CrossRef CAS.
- B. Wang, W. Xiong, J. P. Yu, P. C. Maness and D. R. Meldrum, Green Chem., 2018, 20, 3772–3782 RSC.
- B. Wang, S. Pugh, D. R. Nielsen, W. Zhang and D. R. Meldrum, Metab. Eng., 2013, 16, 68–77 CrossRef CAS PubMed.
- C. Halfmann, L. P. Gu and R. B. Zhou, Green Chem., 2014, 16, 3175–3185 RSC.
- K. Chen, H. Zhu, L. Gu, S. Tian and R. Zhou, Bio-Protoc., 2016, 6, e1890 Search PubMed.
- J. Qiu, H. Lee and C. Zhou, J. Chromatogr. A, 2005, 1073, 263–267 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc01003c |
| This journal is © The Royal Society of Chemistry 2019 |