
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidation of a wood extractive betulin to biologically active oxo-derivatives using supported gold catalysts†
Ekaterina N.
Kolobova
 a,
Ekaterina G.
Pakrieva
a,
Sónia A. C.
Carabineiro
b,
Nina
Bogdanchikova
c,
Andrey N.
Kharlanov
d,
Sergey O.
Kazantsev
e,
Jarl
Hemming
f,
Päivi
Mäki-Arvela
f,
Alexey N.
Pestryakov
a and
Dmitry Yu.
Murzin
*f
a,
Ekaterina G.
Pakrieva
a,
Sónia A. C.
Carabineiro
b,
Nina
Bogdanchikova
c,
Andrey N.
Kharlanov
d,
Sergey O.
Kazantsev
e,
Jarl
Hemming
f,
Päivi
Mäki-Arvela
f,
Alexey N.
Pestryakov
a and
Dmitry Yu.
Murzin
*f
aResearch School of Chemistry & Applied Biomedical Sciences, Tomsk Polytechnic University, Lenin Avenue 30, 634050 Tomsk, Russia
bLaboratory of Catalysis and Materials (LCM), Associate Laboratory LSRE-LCM, Department of Chemical Engineering, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias s/n, 4200-465 Porto, Portugal
cCentro de Nanociencias y Nanotecnología, UNAM Post box 14, 22860 Ensenada, Mexico
dDepartment of Chemistry, M.V. Lomonosov Moscow State University, Leninskie Gory, 119991 Moscow, Russia
eInstitute of Strength Physics and Materials Science of Siberian Branch of Russian Academy of Sciences, (ISPMS SB RAS) pr. Akademicheskii 2/4, 634055 Tomsk, Russia
fJohan Gadolin Process Chemistry Centre, Abo Akademi University, FI-20500 Turku, Finland. E-mail: dmurzin@abo.fi; Fax: +358 2 215 4479; Tel: +358 2 215 4985
First published on 14th May 2019
Abstract
Betulin (90–94%) was extracted from birch with a non-polar solvent and recrystallized from 2-propanol. Liquid-phase oxidation of betulin aimed at obtaining its biologically active oxo-derivatives (betulone, betulonic and betulinic aldehydes), exhibiting e.g. antitumor, anti-inflammatory, antiparasitic, anticancer and anti-HIV properties, was demonstrated for the first time over gold-based catalysts. Gold was deposited on pristine TiO2 and the same support modified with ceria and lanthana, followed by pretreatment with a H2 or O2 atmosphere. The catalysts were characterized by XRD, BET, ICP, TEM, XPS, DRIFT CO, TPD of NH3 and CO2 methods. The nature of the support, type of modification and the pretreatment atmosphere through the metal–support interactions significantly influenced the average particle size of gold, its distribution and the electronic state of gold, as well as the acid–base properties and, thereby, the catalytic performance (activity and selectivity) in betulin oxidation. Au/La2O3/TiO2 pretreated in H2 displayed the highest catalytic activity in betulin oxidation among the studied catalysts with selectivities to betulone, betulonic and betulinic aldehydes of 42, 32 and 27%, respectively, at 69% conversion. Side reactions resulting in oligomerization/polymerization products occurred on the catalyst surface with the participation of strong acid sites, diminishing the yield of the desired compounds. The latter was improved by adding hydrotalcite with the basic properties to the reaction mixture containing the catalyst. Kinetic modelling through numerical data fitting was performed to quantify the impact of such side reactions and determine the values of rate constants.
Introduction
Utilisation of natural compounds for chemical transformations to obtain biologically active compounds has become one of the promising and actively developing areas of fine organic synthesis and pharmaceutical chemistry. Triterpenoids are a class of compounds that combine accessibility, i.e., availability in nature and easiness of isolation, with valuable biological activity. Betulin (lup-20 (29)-ene-3, 28-diol, C30H50O2, CAS: 473-98-3) – a pentacyclic triterpenoid of the lupane series, is found in almost two dozen plants belonging to different genera and families, wherein the main source of betulin is birch bark with the content varying from 10 to 35%. The methods of betulin extraction from birch bark are widely reported in the literature including extraction of the bark outer layer using various solvents, bark alkaline hydrolysis followed by ethanol extraction of betulin, “explosive” autohydrolysis, etc.1–6 Betulin and especially its oxo-derivatives (betulone, betulinic and betulonic aldehydes, and betulinic and betulonic acids) have valuable biologically active properties, and are of exceptional interest for the pharmaceutical, cosmetic and food industries.6–9 For example, betulinic acid and its derivatives exhibit anti-cancer, anti-HIV, antiviral, anti-inflammatory, antiseptic, antimicrobial, anti-malarial, anti-leishmaniasis, anthelmintic and fungicidal activities, while betulonic acid shows pronounced anti-inflammatory, antimelanoma and antiviral effects. Antiviral and anti-leukemic activities have also been reported for betulonic aldehyde, which is, moreover, active against diseases of the liver and the digestive tract and disorders of reproductive function. The 3-oxo derivative of betulin – betulone and its derivatives, exhibiting antitumor, anti-inflammatory, aniparasitic, and anti-HIV properties, also demonstrate in vitro cytotoxic activity against different cancer cell lines.10–22 Recent studies indicate a clear demand for betulone as a building block for creating effective anticancer agents with minimal side effects.20–22Currently, the main method of synthesis of betulin oxo-derivatives is its oxidation. The work of Csuk et al.23 described the formation of betulinic acid via betulinic aldehyde by oxidation of betulin with a mixture comprising TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl)–NaClO2–NaOCl at 35 °C with 92% yield. Synthesis of betulinic acid from betulin can be carried out in one stage using butyl acetate with 4-acetamido-TEMPO and Bu4NBr·H2O as oxidants in an aqueous solution of NaClO2 and NaOCl at 50 °C, regulating the pH with phosphate buffer. Methods of obtaining betulinic acid and betulinic aldehyde by betulin oxidation with chromium oxide(VI), chlorochromate or pyridinium dichromate fixed on a solid silica gel or alumina have also been reported. Betulinic acid was synthesized by the oxidation of betulinic aldehyde with potassium permanganate.24 A method for selective oxidation of betulin with pyridinium dichromate (PDC), pyridinium chlorochromate (PCC) or K2Cr2O7–9 M H2SO4 in the presence of tetrabutylammonium bromide (TBAB) to betulinic aldehyde, or its mixture with betulonic aldehyde and ketol was developed by Komissarov et al.25 The yield of oxidation products did not exceed 75%. The method of obtaining betulonic acid by oxidation of betulin, in the first step with the Jones reagent in acetone, should also be mentioned.26,27 Alternatives include application of the pyridine dichromate complex and acetic anhydride in dimethylformamide,28 and chromium(VI) oxide in acetic acid, followed by reduction to betulinic acid.29 Two-stage methods for the synthesis of betulinic acid have significant shortcomings. The low solubility of betulin in acetic acid, acetone and methylene chloride and of betulonic acid and its salts in alcohols, tetrahydrofuran and water, imposes several limitations, preventing oxidation and reduction and, thus, resulting in poor yields and purity. In the majority of the proposed oxidation methods, highly toxic Cr(VI) is used. Moreover, separation of the products containing toxic Cr(III) ions is very laborious and time consuming.
In addition to chemical modifications, attempts to transform betulin using microorganisms were carried out, mainly for the synthesis of betulone.10,30–33 However, such betulin biotransformation processes using conditionally pathogenic yeasts, fungi, etc. have significant drawbacks, requiring complex nutrient media, long duration, and low concentration levels of products as the biocatalyst might not tolerate higher concentrations.
Recently,34,35 some of the authors of this work demonstrated for the first time the possibility to selectively oxidize betulin to betulinic aldehyde using Ru/C as a catalyst mixed with the basic hydrotalcite and SiO2 as a dehydrating agent at 108 °C in toluene, with air as an oxidant. Under these conditions, the conversion of betulin after 24 hours was 41% with 67% selectivity to betulinic aldehyde, whereas without the addition of SiO2, the conversion and selectivity were 20% and 66%, respectively. A higher conversion was achieved when the reaction was carried out in an acidic medium, giving, however, allobetulin as the main product. It was found that the presence of a basic agent and elimination of water are crucial for selective oxidation of betulin to betulinic aldehyde on Ru catalysts. Selective oxidation of betulin to betulone has also been reported using silver supported on unmodified titania and modified with ceria under mild conditions, e.g. atmospheric pressure, relatively low temperature (140 °C), synthetic air as an oxidant, and mesitylene as the solvent. Conversion of betulin over a Ag/CeO2/TiO2 catalyst reached 27% after 6 hours, which was substantially larger than 11% obtained for Ag/TiO2. In all cases the main product was betulone with the selectivity exceeding 60%.
Based on the analysis of published studies on betulin oxidation, it is obvious that currently there are no economically and ecologically acceptable methods for producing oxo-betulin derivatives. Such methods based on heterogeneous catalysis should replace the existing stoichiometric processes which lead to the formation of large amounts of toxic waste, being able to provide preferably a quantitative yield of the desired product. In the present work, the possibility of heterogeneous catalytic oxidation of betulin with synthetic air, using catalysts based on gold nanoparticles supported on unmodified and modified titania, will be demonstrated for the first time. Some of these catalysts have been synthesized before and used for oxidation of octanol37 which has, however, chemical and physical properties different from those for betulin. No reports on betulin oxidation over gold catalysts are available in the literature; thus it was interesting to explore the possibility of their utilization for a much more complex case than oxidation of octanol.
The aim of the present study is thus to evaluate the applicability of gold-based catalysts in the liquid phase selective oxidation of betulin, to elucidate the influence of the support and nature of the additives, and the impact of redox pretreatment on catalytic properties.
Moreover, a comparative analysis of the catalytic properties of ruthenium, silver and gold catalysts in betulin oxidation was performed showing that the latter ones are more active and stable.
Results and discussion
XRD was used to study the phase composition of the investigated catalysts (ESI Fig. S1†). The XRD patterns showed the absence of the reflections characteristic of gold and modifiers, indicating small sizes of gold and metal oxide particles (lower than the sensitivity XRD threshold of 3–4 nm) or their X-ray amorphous structure.Table 1 shows the specific surface area of supports and catalysts (SBET), the Au content and gold particle size data. The surface area of pristine TiO2 was diminished by 13% after modification (48 m2 g−1) with both modifiers. Further gold deposition did not significantly change the specific surface area of the supports, except Au/La2O3/TiO2, for which there was a noticeable decrease by 10% (Table 1). ICP analysis showed that Au contents were close to the nominal ones.
| Sample |
S
BET![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a, m2 g−1 a, m2 g−1 |
Au content wt.% by ICPa | Au average nanoparticle size, nm | Dispersion, % |
|---|---|---|---|---|
| a Data from ref. 37. | ||||
| TiO2 | 55 | — | — | |
| La2O3/TiO2 | 48 | — | — | |
| CeO2/TiO2 | 48 | — | — | |
| Hydrotalcite | 43 | — | — | |
| Au/TiO2_pH2 | 50 | 4.0 | 2.9 | 34 |
| Au/TiO2_pO2 | 50 | 4.0 | 3.3 | 30 |
| Au/CeO2/TiO2_pH2 | 46 | 4.1 | 2.8 | 36 |
| Au/CeO2/TiO2_pO2 | 46 | 4.1 | 2.4 | 42 |
| Au/La2O3/TiO2_pH2 | 43 | 3.3 | 2.6 | 38 |
| Au/La2O3/TiO2_pO2 | 43 | 3.3 | 2.7 | 37 |
The average size of gold nanoparticles is lower than 3 nm for most of the studied materials, except Au/TiO2_pO2 (Table 1). The largest size of gold nanoparticles and the broadest distribution were observed in the case of gold supported on unmodified titania (ESI Fig. S2†). In contrast, for La-modified materials, the size of Au particles and the range of their distribution are the smallest. These values for Ce-modified materials were in between those for other catalysts. In addition to the nature of the support, the pretreatment atmosphere (H2 or O2) also affects the uniformity of particles and their relative size. At the same time, the impact of pretreatment depends also on the support. For unmodified and lanthana-modified materials, smaller particles were obtained after pretreatment in H2 (300 °C), and for ceria-modified materials after pretreatment in O2 (300 °C). These effects can be attributed to the specificity of gold interactions with different supports during catalyst preparation, as previously confirmed,36 and to the different nature of the gold precursor decomposition under reducing and oxidizing pretreatments, previously revealed by TPR.37 It should also be taken into account that a certain fraction of gold is in the form of highly dispersed oxidized gold species, which are quite difficult to be detected by electron microscopy because of the lower contrast of oxidized species compared to that of the reduced ones. The presence of such oxidized species was previously validated by TPR.37 Moreover, gold in the ionic state (Au+ or Au3+) not detected by TEM could still be present, according to DRIFT CO and XPS (Fig. 1 and Table 2). The amount of these gold species (ionic and oxidized gold species) depends on the support and pretreatment.
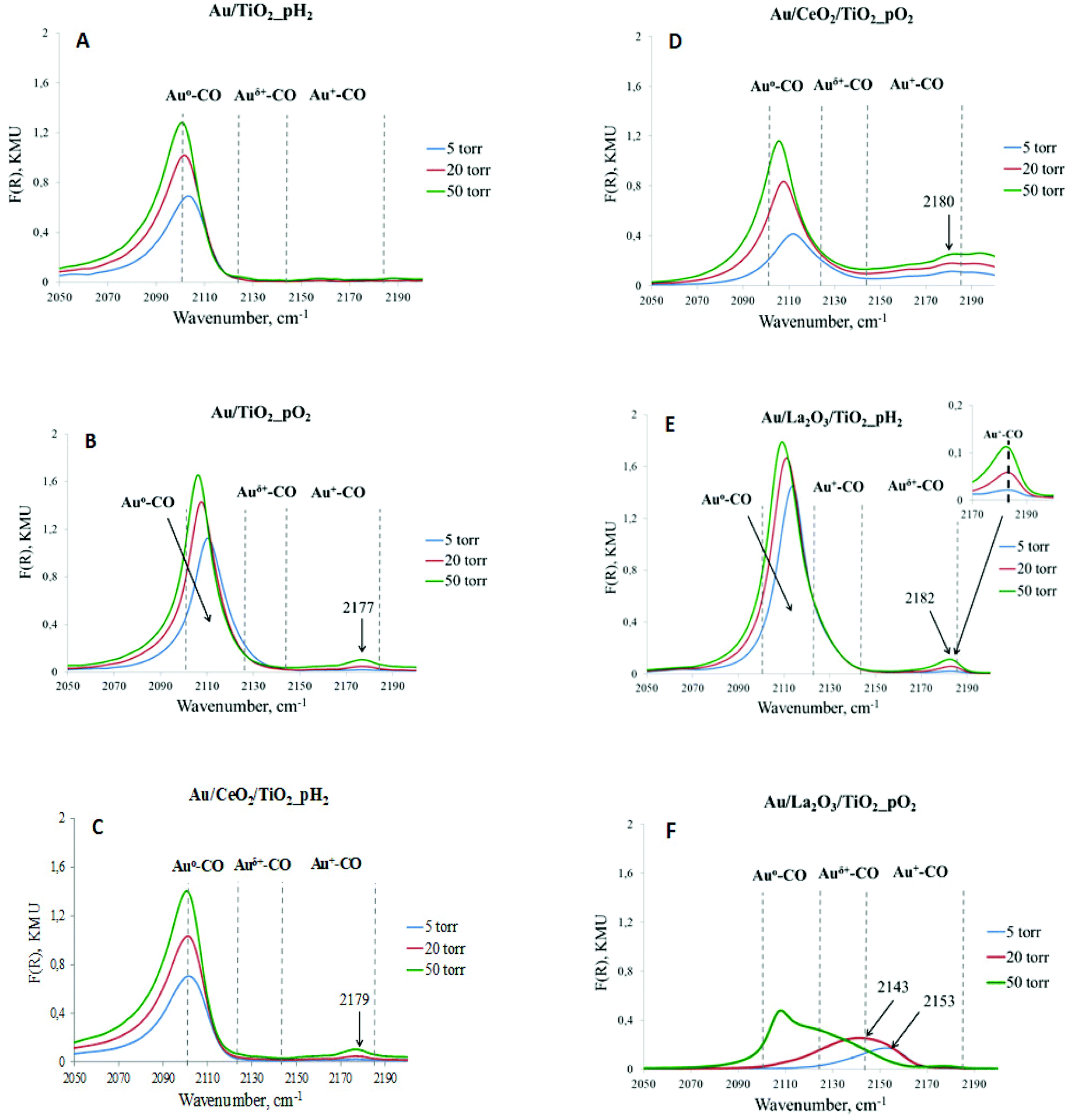 | ||
| Fig. 1 DRIFT spectra of CO adsorbed on catalysts pretreated at 300 °C for 1 h under a H2 or O2 atmosphere. The spectra of adsorbed CO were recorded at different CO pressures – 5, 20, and 50 Torr. | ||
Table 2 shows the relative atomic concentrations of various electronic states of gold, calculated according to XPS. As can be seen, the relative values of different gold states depend strongly on the support and pretreatment conditions. On the surface of all studied catalysts, most of the gold (68–89%) is in a metallic state with BE(Au 4f7/2) in the range of 84.2–84.3 eV, but also a part of gold (11–20%) is in the form of singly charged ions (Au+) with BE(Au 4f7/2) in the range of 85.2–85.5 eV. In the case of unmodified and Ce-modified samples pretreated in H2, another state related to three-charged gold (Au3+) with BE(Au 4f7/2) equal to 86.5 and 86.3 eV appears in the XPS spectrum (11 and 12%, respectively). These data confirm that the formation of the active surface on different supports, under the action of various pretreatments, occurs differently, and is in good agreement with TEM (Table 1).
For a more detailed study of the electronic state of gold in the investigated catalysts, and also as for the evaluation of the strength and stability of the adsorption centers, DRIFT spectroscopy of adsorbed CO was applied. CO adsorption was carried out at different pressures: 5, 20, and 50 Torr, making it possible to evaluate the strength of the centers. Pure supports did not exhibit bands of adsorbed CO in this region of the spectrum under the studied conditions. From Fig. 1, it can be concluded that for all catalysts, regardless of the pretreatment, one absorption band with the maximum in the range of 2100–2120 cm−1, attributed to the surface carbonyl groups of gold atoms Au0–CO,38 was observed. The intensity of this band increased with increasing CO pressure. Differences in the signal positions are caused by CO adsorption on the metal clusters of different sizes, and bands with a low-frequency are associated with larger nanoparticles. CO starts to adsorb on larger gold clusters as the pressure increases. Moreover, carbon monoxide is very weakly adsorbed on metallic gold because of some features of σ–π binding in M0–CO for Au, in comparison with other noble metals (Pt, Pd, Ru, Rh, and Cu).39 Only highly dispersed gold clusters or atoms can be sites for CO adsorption. This explains the different intensities of the absorption bands corresponding to Au0–CO at a CO pressure of 50 Torr. Subsequently, it can be assumed that there are larger particles in Au/TiO2_pH2, Au/CeO2/TiO2_pH2, Au/CeO2/TiO2_pO2, and especially Au/La2O3/TiO2_pO2 catalysts, which were not taken into account when analyzing TEM images because of their relatively low abundance. In our previous study,40 there was a similar discrepancy between the average particle size obtained by TEM and SR-XRD. The average particle size of gold obtained from SR-XRD was in good agreement with the catalytic results, and for some catalysts it was larger than the values determined by TEM. Another absorption band with the maximum in the range of 2140–2185 cm−1, related to the complexes of ions Au+–CO,41,42 was observed in almost all cases, except Au/TiO2_pH2. However, the intensity of this absorption band and its change with pressure variation are different. This absorption band is less intense than that attributed to Au0–CO, and also strongly depends on CO pressure. The intensity increases with the pressure increase. It is interesting to note that, for Au/La2O3/TiO2_pO2 (Fig. 1f), reduction of Au+ sites is observed under a CO atmosphere, which indicates their very low stability. The absence of this absorption band for Au/TiO2_pH2 can be due to the presence of only weak Au+ sites, and even a CO pressure of 50 Torr is not enough for their identification by DRIFT CO, while according to XPS (Table 2), Au+ is 15% of the total amount of gold. It should also be noted that the XPS method determines the ionic states of gold in the near-surface layer, some of which may not be accessible for adsorbed molecules. At the same time, the method of DRIFT adsorbed CO allows the identification of the active sites on the surface available for the reactions.
The TPD of NH3 was used to determine the acidity of supports and respective gold catalysts, namely the concentration and strength of acid sites (Table 3 and ESI Fig. S†). Physical adsorption can take place in the case of ammonia TPD, being, however, typical of low temperatures. Therefore, to avoid the contribution of physical adsorption, the analysis started from 100 °C.
| Sample | Concentration of acid sites, mol 10−4 m−2 | ||||
|---|---|---|---|---|---|
| Weak | Medium | Strong | Total | ||
| TiO2 | 378 | 144 | 55 | 577 | |
| CeO2/TiO2 | 304 | 90 | 115 | 509 | |
| La2O3/TiO2 | 302 | 90 | 8 | 400 | |
| Au/TiO2_pH2 | 528 | 162 | 16 | 706 | |
| Au/TiO2_pO2 | 520 | 132 | 18 | 670 | |
| Au/CeO2/TiO2_pH2 | 496 | 356 | 80 | 932 | |
| Au/CeO2/TiO2_pO2 | 421 | 187 | 69 | 677 | |
| Au/La2O3/TiO2_pH2 | 146 | 135 | 239 | 193 | 713 |
| Au/La2O3/TiO2_pO2 | 276 | 286 | 170 | 732 | |
Three types of acid sites are detected for the initial supports, but their concentration and strength are different (Table 3). The pristine titania showed the highest acidity among the used supports with the majority of acid sites being of weak strength, while the concentrations of the medium and strong acid sites are 2.6 and 6.9 fold lower than the previous one. Therewith, they are all Brønsted acid sites (acidic OH groups).43–45 However, it is possible that the strong acid sites are of aprotic nature and are Lewis acid sites (e.g. tetrahedral coordinated Ti4+).46
Modification of titania with ceria and lanthana led to a decrease in the concentration of weak and medium acid sites. This is most likely a consequence of surface dehydration after calcination at 550 °C during preparation. Alongside that, the amount of strong acid sites increased for Ce-modified titania, but decreased for La-modified titania. In the case of ceria-modified TiO2, this can be explained by the appearance of new Lewis sites, due to the presence of Ce4+/Ce3+, whose existence was indirectly confirmed by TPR.37 For the lanthana-modified material, no hydrogen consumption was observed in TPR profiles. Thus, it can be assumed that lanthana blocked the acid sites on the pristine titania surface leading to a decrease in acidity. After gold deposition, in all cases, there was a redistribution of acid sites. Regardless of the pretreatment for unmodified and Ce-modified materials, an increase in the concentration of weak acid sites and a significant decrease of strong acid sites were observed. The amount of medium sites in the case of Au/CeO2/TiO2 was increased, while for Au/TiO2 it remained almost unchanged. The distribution of acid sites is noticeably different for the La-modified catalyst, compared with the other materials. This is most clearly seen for strong acid sites, whose concentration significantly increased. Moreover, there was a decrease of weak acid sites in comparison with unmodified and Ce-modified materials. Such changes in acidity after metal deposition may originate from several reasons. One of the options can be associated with a change in the support properties during catalyst preparation resulting in the mutual influence of the support and the metal precursor, as previously discussed.36,47–49 Another possibility is blocking the acid sites, previously existing on the surface, by newly formed metal nanoparticles. It should be noted that, in the case of the La-modified catalyst, a part of the strong acid sites (90 × 10−4 mol m−2) may be associated with the Lewis acid sites, namely Au+. This is confirmed by comparing XPS (Table 2), DRIFT CO (Fig. 1) and NH3-TPD (Table 3) data. Considering that lanthana is a non-reducible oxide, it can be suggested that another part of the strong acid sites is associated with Brønsted acidity and belongs to the support or the modifier.
In order to assess the basic properties of the studied materials, the TPD of CO2 was used. Based on the literature,50–53 depending on the temperature range in which CO2 desorption occurs, the basic sites are divided into three types: weak, medium and strong, reflecting their nature. The weak basic sites (25–200 °C) are usually attributed to surface hydroxyl groups, medium ones (200–400 °C) to metal oxide pairs, and strong sites (400–600 °C) to low-coordinated oxygen anions. All types of basic sites mentioned above were observed for the supports studied in this work (Table 4 and ESI Fig. S4†). Pristine titania exhibited the average total basicity among the studied supports, with the dominance of the basic sites of medium strength and almost absent strong sites. A similar distribution of the basic sites was also observed for Ce-modified titania, while the amount of these sites was lower. After modification of titania by lanthana, there was an increase in the concentration of weak and strong basic sites, while the amount of medium sites remained almost unchanged. Table 4 also presents the results for hydrotalcite, the basicity of which is 2–3 fold higher than that of the used supports. The gold deposition on the support surface led to a redistribution of the basic sites similar to the acidic ones (Table 3). For almost all studied catalysts, there was an increase in the amount of basic sites while, in all cases, the strong basic sites increased. The reasons for the changes in basicity after gold deposition are apparently the same as for acidity in a sense that they originate from the exposure of the support to the metal precursor during preparation, mutual influence of the support and the precursor, and base site blocking.
| Sample | Concentration of basic sites, mol 10−4 m−2 | |||
|---|---|---|---|---|
| Weak | Medium | Strong | Total | |
| TiO2 | 53 | 93 | 9 | 155 |
| CeO2/TiO2 | 56 | 83 | 6 | 145 |
| La2O3/TiO2 | 117 | 96 | 35 | 248 |
| Hydrotalcite | 284 | 230 | 37 | 555 |
| Au/TiO2_pH2 | 30 | 85 | 34 | 149 |
| Au/TiO2_pO2 | 68 | 58 | 42 | 168 |
| Au/CeO2/TiO2_pH2 | 130 | 137 | 67 | 334 |
| Au/CeO2/TiO2_pO2 | 43 | 67 | 52 | 162 |
| Au/La2O3/TiO2_pH2 | 88 | 107 | 177 | 372 |
| Au/La2O3/TiO2_pO2 | 51 | 88 | 74 | 213 |
In addition, as shown in ref. 54, 55, CO2 is also capable of being adsorbed on small gold nanoparticles, with the abstraction of oxygen by Au0, giving CO and Au2+O2− species, which cannot be considered as the basic sites. When comparing CO2-TPD (Table 4) and TEM (Table 1), it can be concluded that the highest increase in the amount of basic sites was observed for samples with the smallest particle sizes. Subsequently, a part of the strong basic sites can be associated with CO2 adsorption on small gold nanoparticles.
The catalytic behavior of Au supported catalysts in the betulin liquid phase oxidation (Fig. 2) was studied at 140 °C and a synthetic air pressure of 1 bar in mesitylene. To assess the influence of the nature of the support and pretreatment atmosphere, gold was deposited on pristine titania and TiO2 modified with ceria and lanthana. The obtained materials were pretreated in H2 or O2.
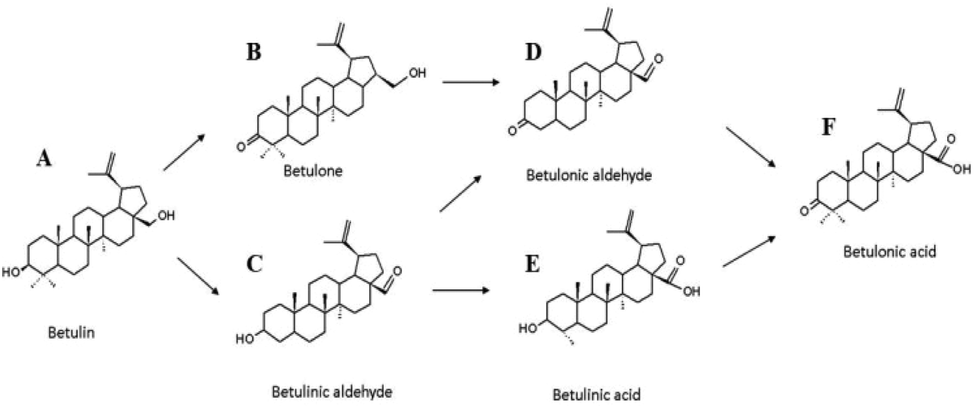 | ||
| Fig. 2 Reaction scheme for betulin oxidation. | ||
It was found that both the nature of the support and the pretreatment atmosphere have a significant influence on the catalytic behavior of the studied catalysts (Table 5). Among the studied gold containing materials, Au/TiO2 pretreated in H2 (Table 5, entry 1) showed the lowest activity. Betulin (A) conversion was 25%, with selectivity to betulone (B) and betulinic aldehyde (C) of 40 and 53%, respectively, at this conversion level. Herewith, the total yield of products was only 15%, which is 1.7 fold lower than the observed conversion (Table 5 – entry 1, Fig. 3a and d). This difference is due to incomplete mass balance (the sum of the masses of the reactants and products visible in GC and GCLPA). This is most likely caused by the strong adsorption of reactants or products on the catalyst surface. The mass balance closure was different for the various catalysts, being determined by the catalytic properties. In this particular case, the GCLPA was 90%. Betulin conversion and selectivity for the same material (Au/TiO2), but pretreated in O2, were almost the same (Table 5, entry 2). However, due to a better GCLPA – 96%, the total yield of products for this catalyst turned out to be 1.7 fold higher than that for the one pretreated in H2.
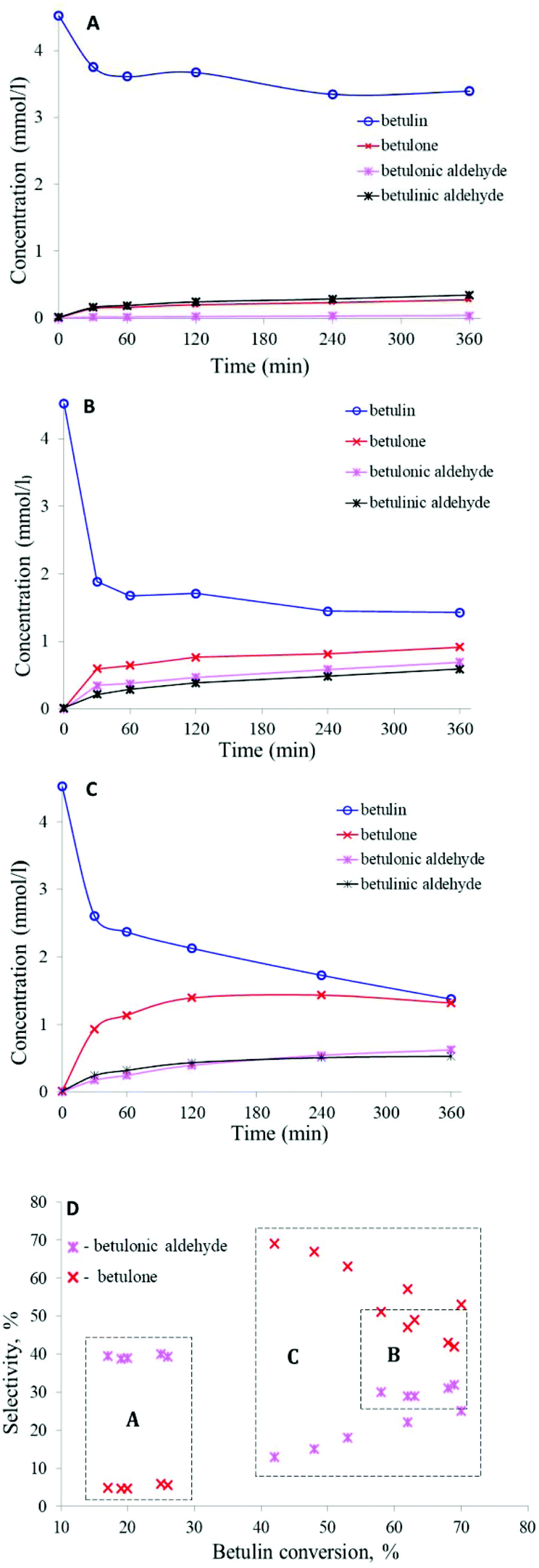 | ||
| Fig. 3 Kinetics of betulin oxidation on (a) Au/TiO2_pH2, (b) Au/La2O3/TiO2_pH2 and (c) Au/La2O3/TiO2_pH2 with 0.2 g of hydrotalcite; data are presented taking into account the mass balance; and (d) selectivity towards betulone and betulonic aldehyde as a function of betulin conversion. Conditions: 140 °C, synthetic air (50 ml min−1) as the oxidant, initial betulin concentration – 4.5 mmol l−1, solvent – mesitylene, 0.2 g catalyst. | ||
| Entry | Catalyst | X (%) | GCLPA (%) | Selectivity, % | ∑Yproduct (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| S B | S D | S F | S C | S E | S allobetulin | |||||
| Conditions: 140 °C, synthetic air (50 ml min−1) as the oxidant, solvent – mesitylene, entries 1–12, 18 and 19; X – conversion of betulin after 6 h (%); GCLPA – the sum of the reactant and product masses in GC analysis (%); SB – selectivity to betulone (%); SD – selectivity to betulonic aldehyde (%); SF – selectivity to betulonic acid (%); SC – selectivity to betulinic aldehyde (%); SE – selectivity to betulinic acid (%); Sallobetulin – selectivity to allobetulin (%); ∑Yproduct – total product yield after 6 h (%); tr. – traces.a With 0.2 g of hydrotalcite.b With 0.2 g of hydrotalcite and 0.2 g SiO2.c Second run.d Conditions: 108 °C, synthetic air (50 ml min−1) as the oxidant, solvent – toluene, entries 13–17.e With 0.5 g of hydrotalcite.f Conditions are the same as for entries 1–12.g With 0.5 g of hydrotalcite + 0.5 g SiO2.h Conversion of betulin after 5 h (%).i Conversion of betulin after 24 h (%); data of 14–17 entries from ref. 34; data of 18 and 19 entries from ref. 35. | ||||||||||
| 1 | TiO2 | 13 | 93 | 65 | 3 | 5 | 15 | 2 | 0 | 6 |
| 2 | CeO2/TiO2 | 17 | 91 | 69 | 9 | 0 | 0 | 22 | 0 | 8 |
| 3 | La2O3/TiO2 | 4 | 97 | 53 | 47 | 0 | 0 | 0 | 0 | 3 |
| 4 | Au/TiO2_pH2 | 25 | 90 | 40 | 6 | tr. | 53 | 1 | 0 | 15 |
| 5 | Au/TiO2_pO2 | 27 | 96 | 38 | 10 | 1 | 51 | tr. | 0 | 25 |
| 6 | Au/CeO2/TiO2_pH2 | 45 | 77 | 48 | 11 | tr. | 40 | 1 | 0 | 22 |
| 7 | Au/CeO2/TiO2_pO2 | 33 | 95 | 34 | 11 | 1 | 54 | 0 | 0 | 27 |
| 8 | Au/La2O3/TiO2_pH2 | 69 | 80 | 42 | 32 | tr. | 27 | 0 | 0 | 48 |
| 9 | Au/La2O3/TiO2_pO2 | 25 | 97 | 44 | 7 | 1 | 48 | 0 | 0 | 22 |
| 10 | Au/La2O3/TiO2_pH2a | 70 | 85 | 54 | 25 | 0 | 21 | 0 | 0 | 55 |
| 11 | Au/La2O3/TiO2_pH2b |
71 | 87 | 52 | 27 | 0 | 21 | 0 | 0 | 58 |
| 12 | Au/La2O3/TiO2_pH2c |
48 | 87 | 57 | 24 | 0 | 19 | 0 | 0 | 35 |
| 13 | Au/La2O3/TiO2_pH2d |
58 | 88 | 64 | 19 | tr. | 17 | tr. | 0 | 46 |
| 14 | Ru/C, Degussad | 54h | — | 0 | 2 | 1 | 16 | 2 | 72 | — |
| 15 | Ru/C, Degussad,e | 16h | — | 0 | 9 | 5 | 77 | 0 | 2 | — |
| 16 | Ru/C, Degussaf | 12h | — | 0 | 13 | 49 | 37 | 0 | 1 | — |
| 17 | Ru/C, Degussad,g | 41i | — | 0 | 8 | 4 | 67 | 4 | 4 | — |
| 18 | Ag/TiO2 | 11 | — | 59 | 8 | 3 | 21 | 3 | 0 | — |
| 19 | Ag/CeO2/TiO2 | 27 | — | 60 | 4 | 1 | 6 | 7 | 0 | — |
In contrast to the unmodified material, betulin conversion for the Ce-modified catalyst was higher after the pretreatment in H2 (betulin conversion – 45%, Table 5, entry 3). However, the mass balance closure in this case was the worst – 77%, ultimately giving only 22% total yield of the main products. For the same catalyst, but pretreated in oxygen, a lower conversion – 33% was achieved (Table 5, entry 4). The best 95% GCLPA was, however, reached for this case, with the total yield of products being 1.2 fold higher than that after pretreatment in H2. The pretreatment atmosphere even affected the selectivity for the primary products, such as betulone (B) and betulinic aldehyde (C), which should be less sensitive to conversion levels. For Au/CeO2/TiO2_pO2 in particular, the main product was betulinic aldehyde (C), while for Au/CeO2/TiO2_pH2 betulone (B) was mainly obtained.
Au/La2O3/TiO2 pretreated in hydrogen showed the highest activity among the studied catalysts. Betulin conversion was 69% and the main products were betulone (B), betulonic (D) and betulinic (C) aldehydes, with selectivities of 42, 32 and 27%, respectively (Table 5 – entry 5, Fig. 3b and d). It should be noted that due to a GCLPA of 80%, the total yield of the main products turned out to be 1.4 fold lower than the observed conversion, being 48%. Betulin conversion for the same material (Au/La2O3/TiO2), but pretreated in O2, was 2.8 fold lower than that after treatment in H2 (Table 5, entry 6).
For this catalyst, the product distribution was also different. It can also be related to the conversion as betulinic aldehyde (C) can be transformed to betulonic aldehyde (D). In this case, betulonic aldehyde (D) was practically not formed and the main products were betulone (B) and betulinic aldehyde (C), with a higher GCLPA (97%).
Table 5 also presents the results of the previous studies on betulin oxidation over Ru and Ag catalysts.34,35 As can be seen from the data (Table 5, entries 4–9, 15, 17 and 18), under the same experimental conditions, the activity of supported gold catalysts significantly exceeds the activity of Ru and Ag counterparts. Selectivity of Ru is significantly different from that for Au and Ag catalysts. Over the majority of Au(Ag)/(modifier)/TiO2 catalysts the main reaction products were betulone (B) and betulinic aldehyde (C), while betulonic acid (F) and betulinic aldehyde (C) were obtained for Ru/C. It is also worth noting that allobetulin was not observed in the reaction products for Au(Ag)/(modifier)/TiO2 in comparison with Ru. Moreover in the previous work the mass balance closure was not explicitly accounted for, making a direct comparison of the yields difficult.
In the work,34 it was also shown that the catalytic behavior (activity and selectivity) of Ru catalysts depends strongly on the reaction conditions. In toluene as a solvent at 108 °C, betulin conversion over Ru/C (entry 14) was 54% after 5 hours with allobetulin (77%), a structural isomer of betulin, as the main product. To evaluate how the reaction conditions affect the catalytic behavior of gold materials, a similar experiment was carried out using Au/La2O3/TiO2_pH2 in toluene at 108 °C (entry 13). Compared with entry 8, betulin conversion decreased 1.2 fold; however, the main reaction products were still betulone (B), betulonic (D) and betulinic (C) aldehydes. It is worth noting that in this case, the mass balance closure was higher (88%) compared with entry 8 (80%); therefore, the difference in ∑Yproduct between entries 8 and 13 was only 2%.
Selectivity to a more desired product (betulinic aldehyde as opposed to allobetulin) was increased34 by adding basic hydrotalcite to the reaction mixture, even if there was a negative influence on activity decreasing the betulin conversion from 54% to 16% (Table 5, entry 15). The betulin conversion reached 41% with a selectivity to betulinic aldehyde of 67% (Table 5, entry 17) with an increase in the reaction time up to 24 hours, adding hydrotalcite and silica as dehydrating agents. In the present work, a similar experiment was carried out, and hydrotalcite was added to the reaction mixture containing Au/La2O3/TiO2_pH2 (Table 5, entry 10, Fig. 3c and d). However, there were no significant changes in betulin conversion or in the product distribution. Betulin conversion increased by only 1% after adding hydrotalcite. Despite a slight increase in betulin conversion, the TOF increased 1.4 fold and was 0.010 s−1, compared to the experiment without hydrotalcite, for which it was 0.007 s−1. Moreover, due to the better GCLPA – 85%, the total yield of the main products increased to 55% compared with entry 5, being 48% (Table 5). Addition of silica to the reaction mixture, along with Au/La2O3/TiO2_pH2 and hydrotalcite, additionally increased the betulin conversion by 1% and the product yield by 3%, as well as the GCPLA to 87% (Table 5, entry 11). Thus, it can be assumed that the addition of hydrotalcite, leading to apparently local changes in concentrations of a proton and hydroxyl groups in the vicinity of the catalyst surface, can thereby affect the properties of the catalyst surface. In turn, silica prevents the inhibitory action of water. However, in the case of betulin oxidation over gold catalysts, inhibition by water is much less pronounced than that for Ru catalysts.34 In general, gold materials were less sensitive to changes in the reaction conditions than ruthenium ones. When Au/La2O3/TiO2_pH2 was recycled (Table 5, entry 12), a 30% drop in activity compared to entry 8 was observed, indicating some catalyst deactivation. However, the gold catalyst was still more stable than ruthenium, for which the activity decreased by 44% in the second run (from 41% to 23%).34 As mentioned above in Introduction betulone is equally important as betulinic aldehyde or betulinic acid. Selectivity to betulone can be increased by adding to the reaction mixture besides the catalyst also hydrotalcite and silica or by replacing the solvent and lowering the reaction temperature.
When comparing the GCLPA for the same catalyst, but pretreated under different atmospheres (e.g. Au/La2O3/TiO2_pH2 and Au/La2O3/TiO2_pO2, Table 5, entries 4–8) with acid–base properties (Tables 3 and 4), it can be assumed that the GCLPA is determined by the acidity of the materials, namely the concentration of medium and strong acid sites. The lower this concentration, the higher the mass balance closure (Fig. 4), which can be explained by side reactions, promoted on stronger sites leading to a lower GCLPA. This is also confirmed by comparing the catalytic and ammonia TPD data for the supports applied in this work (Tables 3 and 5 – entries 1–3). For Ce-modified TiO2, the concentration of medium and strong acid sites was the highest among supports, and the GCPLA was the lowest. For La-modified TiO2, the opposite situation was observed. From this point of view, hydrotalcite indirectly affected the catalytic properties, in particular the strong acid sites, preventing the side reactions and increasing the product yield.
 | ||
| Fig. 4 Dependence of the GCLPA on the concentration of medium and strong acid sites. | ||
Along with the acidity of the materials, their basicity also plays an important role (Table 4). Betulin conversion was higher for catalysts with more pronounced basic properties. Herewith, H2-pretreated materials were more basic, but at the same time, they also demonstrated higher acidity. The exception was Au/TiO2, for which conversion or the product distribution was almost independent of the pretreatment atmosphere. This can be explained based on the fact that the acid–base properties of this material vary only slightly upon different pretreatments.
It should be noted that while a certain correlation between the acid–base properties and catalytic performance was seen, the role of gold in betulin oxidation is decisive. Moreover, correlations between the TPD of ammonia and CO2 made in the gas-phase with catalytic properties should be taken with caution when the catalysts are employed in the liquid-phase processes. Nevertheless, TPD methods provide general information on the nature of solid surfaces and the types of sites and are often applied for characterizing the acid–base properties of solid catalysts even for the liquid phase reactions.
In order to find out the reason for a decrease in the GCPLA, namely, what was adsorbed on the catalyst surface, size exclusion chromatography (SEC) was used.
After carrying out the extraction and SEC analysis, it was found that polymers and oligomers with a molecular weight of 5000 and 1000 Da, respectively, were formed on the catalyst surface (ESI Fig. S5†). The weight of oligomers/polymers on the catalyst was not quantified, being, however, related to the mass imbalance between the theoretical GCLPA (100%) and the corresponding values of GCLPA reported in Table 5.
Thus, it can be concluded that a decrease in the GCPLA is associated with the side reactions of the oligomerization/polymerization of betulin or its derivatives on the catalyst surface, with the participation of strong acid sites. Formation of oligomers/polymers on the catalyst surface is also likely to cause partial deactivation of the recycled catalyst (Table 5, entry 12). Despite washing the catalyst after the first run in hot acetone, a part of the oligomers/polymers could remain on the surface thereby blocking partially active sites. This is also confirmed by an increase in the GCPLA after the second run (Table 5, entry 12).
In order to quantify the kinetic significance of various steps comprising the reaction network and a contribution of side reactions leading to oligomers/polymers, kinetic modelling was performed for betulin oxidation in the presence of Au/La2O3/TiO2_pH2 and hydrotalcite (Fig. 3c). The reaction scheme given in Fig. 2 was somewhat modified (Fig. 5) to incorporate formation of oligomers (O) and finally polymers (P) and account for a clear lack of mass balance closure in Fig. 3c. The reaction scheme was simplified as the concentration of acids was negligible for Au/La2O3/TiO2_pH2. In general, oligomers can originate not only from the reactant as in Fig. 5 but also from the products, as mentioned above. However, a lack of mass balance closure in some cases was already seen at the beginning of experiments justifying that the main contribution for the formation of oligomers comes from the reactants. To keep a more general character of the model, formation of oligomers was considered to be reversible, while generation of polymers as terminal species was supposed to be irreversible.
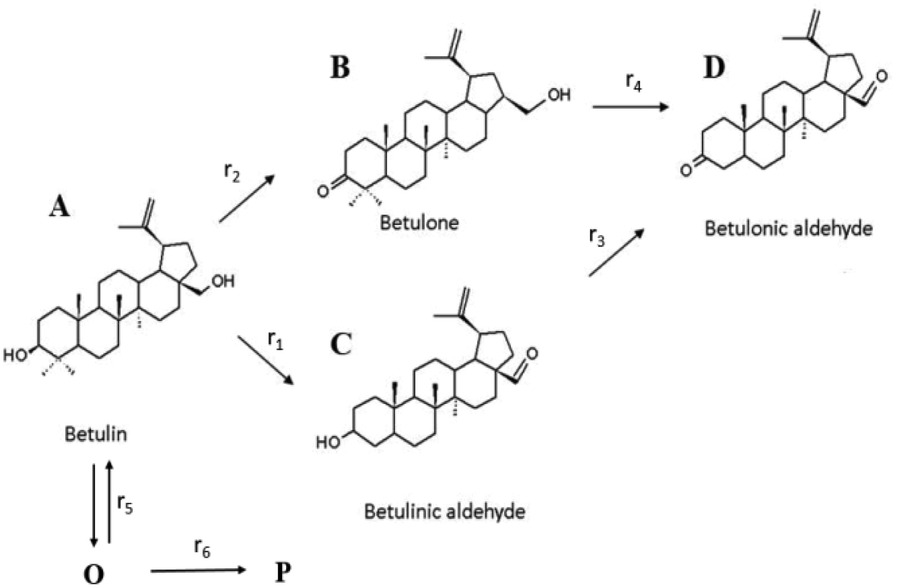 | ||
| Fig. 5 Modified reaction scheme for betulin oxidation, which includes formation of oligomers (O) and polymers (P). | ||
The equations for the reaction rates presented in Fig. 5 can be easily written:
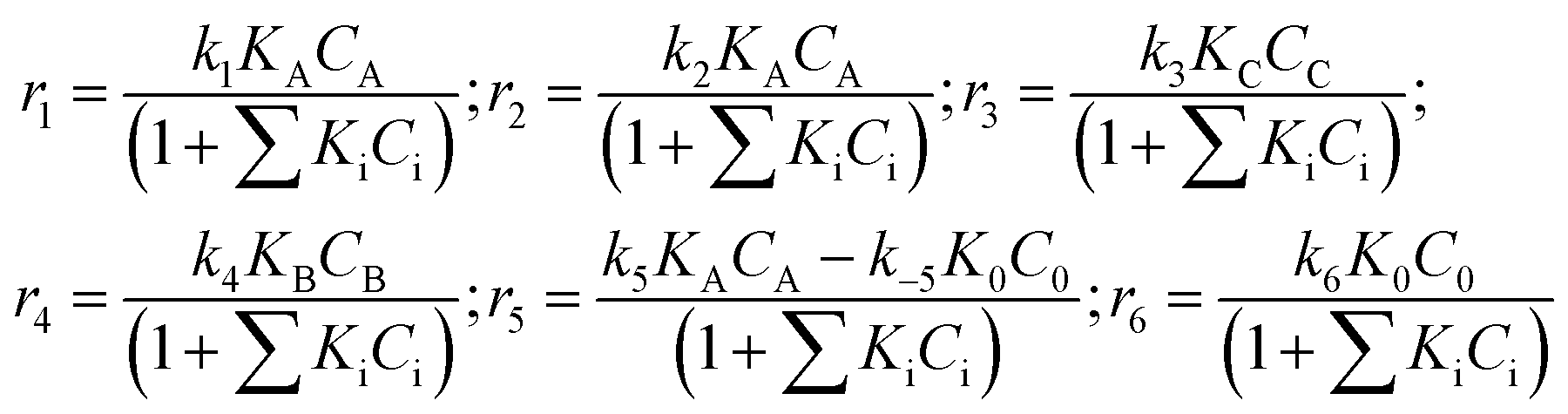 | (1) |
These equations correspond to the adsorption of all organic compounds and subsequent oxidation with noncompetitively adsorbed oxygen. Dependence of the oxygen concentration is thus implicitly incorporated in the rate constants ki.
In the preliminary development of the kinetic model (eqn (1)), adsorption of all reactants was considered. However, the initial parameter estimation showed that the calculated terms in the denominator involving adsorption coefficients, for all substances and their concentrations apart from betulonic aldehyde, are very low. This allows assuming that the coverage of these species is rather low. The constants in eqn (1) are lumped ones comprising implicitly also dependence on oxygen pressure.
The reactor mass balances for each component in the reaction system are as follows:
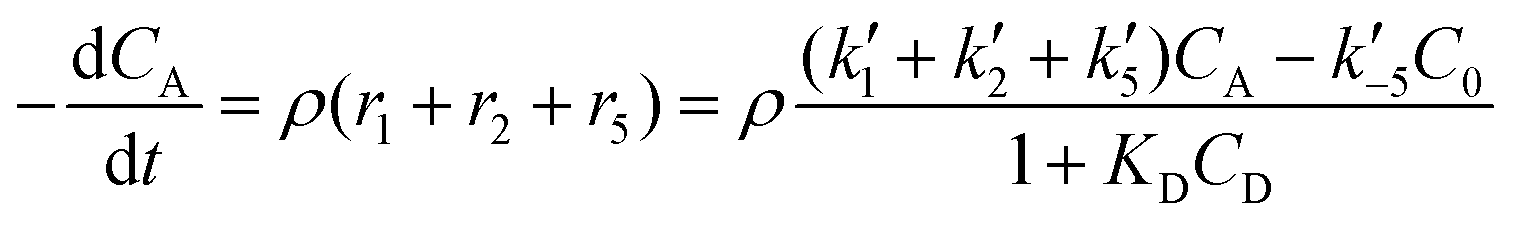 | (2) |
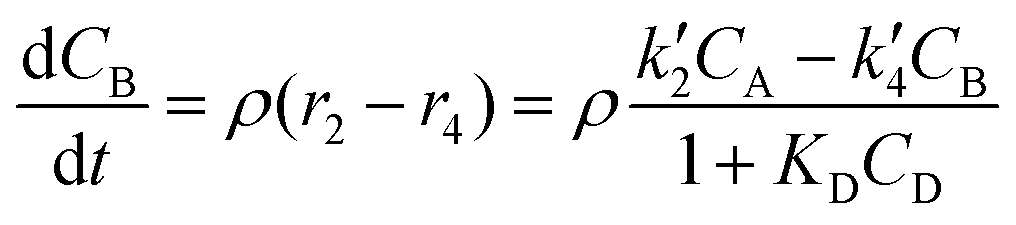 | (3) |
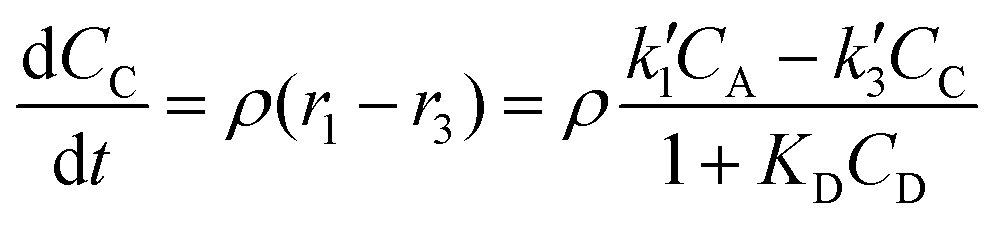 | (4) |
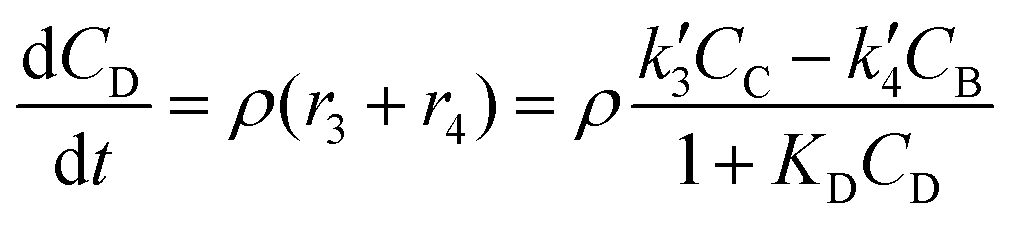 | (5) |
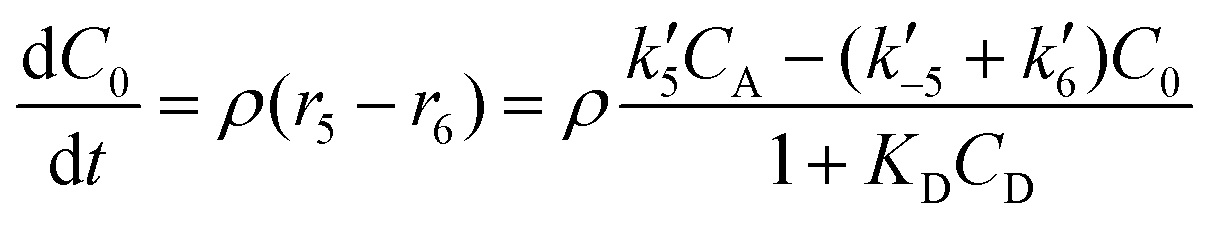 | (6) |
In eqn (1)–(6)Ci denotes the concentration of respective compounds, mol L−1, and ρ is the catalyst bulk density given in g L−1. Modified constants, etc. contain also the respective adsorption coefficients.
Differential eqn (2)–(6) were solved using the backward difference method and the parameter estimation was performed with the simplex and Levenberg–Marquardt methods. The numerical tools are inbuilt in the optimization software ModEst,56 in which the objective function Q is defined through experimental yi and calculated ŷi concentrations of the components in the reacting system:
| Q = ∑(yi − ŷi)2 | (7) |
The results (Fig. 6) show that this model can describe the experimental data rather well.
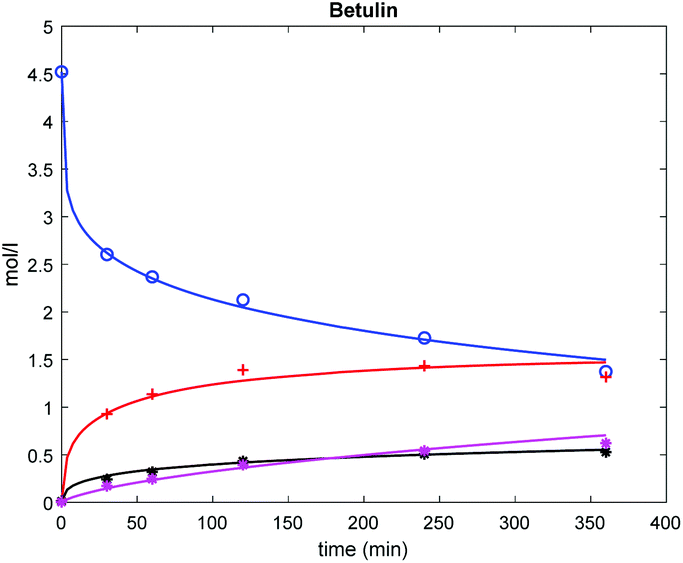 | ||
| Fig. 6 Comparison between experimental (points) and calculated (lines) concentration profiles in betulin oxidation on Au/La2O3/TiO2_H2. Conditions and notation of components are given in Fig. 3. | ||
For 24 data there were initially 8 adjustable parameters, namely 7 rate constants (k′1 to k′6 and k′−5) and one adsorption constant (KD). During the parameter estimation it turned out that some of these constants, namely k′3 and k′6, are negligible. Thus the final model comprised 6 parameters. Their values are given in Table S1.† Even if the number of data points is much larger than the number of parameters they were somewhat correlated with each other, preventing a detailed analysis of their physicochemical significance. Apparently, a separate kinetic study accounting for catalyst deactivation and more rigorous chemical analysis of oligomers/polymers is required, being, however, outside of the scope of the current work.
The degree of explanation R2:
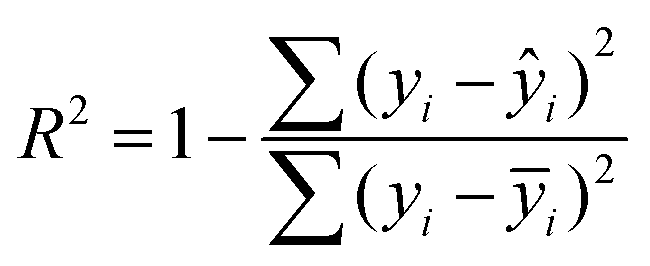 | (8) |
Conclusions
The current work is the first study dealing with the liquid-phase oxidation of betulin over gold-based catalysts. As a support, titania per se, or modified with CeO2 or La2O3, was used. The nature of the support and pretreatment atmosphere (H2 or O2) significantly affected the uniformity of gold particle distribution and their mean size, the electronic state of gold and acid–base properties and, as a consequence, the catalytic behavior (activity and selectivity) of the studied materials in betulin oxidation. The smallest gold nanoparticles with their narrow distribution and the strongest and most stable adsorption sites (Au0 and Au+) were formed on the La-modified TiO2 surface after H2 pretreatment. Additionally, this material exhibited the highest basicity and the highest concentration of medium and strong acid sites among the studied catalysts, and as a consequence the best catalytic results. Betulin conversion was 69% for 6 h at 140 °C, and the main products were betulone, betulonic and betulinic aldehydes, with selectivities of 42, 32 and 27%, respectively. However, the total yield of products was 1.4 fold lower than the observed conversion, which was due to an incomplete mass balance and was caused by the side reactions of oligomerization/polymerization on the catalyst surface, promoted on stronger acid sites. The product yield was increased by adding basic hydrotalcite to the reaction medium along with the catalyst. Such results can be explained by an indirect influence of hydrotalcite on the surface properties of the catalyst, in particular strong acid sites which, in turn prevents the side reactions and increases the product yield. Kinetic modelling was performed to quantify the significance of such side reactions.Experimental
Catalyst preparation
TiO2 P25 (nonporous, 70% anatase and 30% rutile, particle size: 21 nm, purity: 99.5%, Evonik Degussa GmbH) was used as the starting support. For comparative studies, titania was modified with ceria and lanthana by impregnation with solutions of the corresponding nitrates (molar ratio Ti/M = 40, where M = Ce or La). After impregnation, the samples were dried at room temperature for 48 h, then at 110 °C for 4 h, followed by calcination at 550 °C for 4 h.Gold catalysts (Au/TiO2, Au/CeO2/TiO2 and Au/La2O3/TiO2) were prepared by deposition–precipitation with urea, according to the procedure previously described.36,57–59 The nominal gold content in all catalysts was 4 wt%. The gold precursor (HAuCl4·3H2O, Merck) and urea (Merck) were dissolved in distilled water, and thereafter the support was added to the solution. The resulting mixture was heated to 80 °C and kept at constant temperature for 16 h, with stirring. Thereafter, the catalysts were pretreated at 300 °C for 1 hour under a H2 or O2 atmosphere.
The catalysts are denoted hereinafter as Au/(MxOy)/TiO2_P, where MxOy is CeO2 or La2O3 and P indicates the pretreatment atmosphere (O2 or H2).
Catalyst characterization
The specific surface area (SBET) of supports and catalysts was measured by nitrogen adsorption with a “TriStar 3000” analyzer (Micromeritics, USA). Prior to measurements, the samples were subjected to thermal vacuum treatment at 300 °C for 5 hours. To calculate the SBET, a multipoint BET method with linearization of the adsorption isotherm for the relative pressure between 0.005 to 0.25 was used.The phase composition of supports and catalysts was studied by the step-scanning procedure (step size: 0.02°; 0.5 s) with a Philips XPert PRO diffractometer, using CuKα radiation (λ = 0.15406 nm) and a Ni-filter. The measured diffractograms were analyzed with the ICDD-2013 powder diffraction database.
The morphology of catalysts and the size of gold particles were investigated by transmission electron microscopy (TEM) and STEM-HAADF (scanning transmission electron microscopy-High Angle Annular Dark Field) using a JEOL JEM-2100F. The samples were ground to a fine powder and sonicated in hexane at room temperature. Then a part of the suspension was placed on a lacey carbon-coated Cu grid. In order to obtain micrographs that most fully reflect the real structure of the samples, a thorough examination of the samples was carried out, after which the selected area was scanned at various resolutions. For each sample, at least 150 particles were registered.
The metal loading of the catalysts was determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using a PerkinElmer ICP-OES Optima 3300 DV spectrometer. The solids were dissolved by acid dissolution, digested in a microwave oven, diluted to 100 mL and analyzed in the spectrometer.
The catalysts were characterized by X-ray photoelectron spectroscopy (XPS) with a SPECS GmbH custom made system using a PHOIBOS 150 WAL hemispherical analyzer and a non-monochromated X-ray source. All the data were acquired using Al Kα X-rays (1486.6 eV, 200 W). A pass-energy of 50 eV, a step size of 0.1 eV per step and a high-intensity lens mode were selected. The diameter of the analysed area was 3 mm. Charging shifts were referenced against the Ti 2p3/2 peak of TiO2 at 458.8 eV. The pressure in the analysis chamber was kept lower than 1 × 10−8 mbar. The accuracy of the binding energy (BE) values was about ±0.1 eV. Peak areas were estimated by calculating the integral of each peak after subtracting a Shirley type background, fitting the experimental peak to a combination of Lorentzian/Gaussian lines with a 30/70 proportion and keeping the same width on all lines. Deconvolution of spectra was performed with the program CasaXPS.
Diffuse Reflectance Fourier Transform Infrared (DRIFT) spectra of CO adsorbed on the catalysts were recorded by using a Bruker EQUINOX 55/S FTIR spectrometer with a homemade accessory at 4 cm−1 resolution at room temperature. The powdery fraction of an oxide was placed in a quartz ampoule with a window of CaF2. The samples were preliminarily calcined at 100 °C under vacuum not less than 10−4 Torr for 1 h. For each catalyst three samples were investigated: as-prepared, and after pretreatments either in H2 or in O2 (100 Torr) at 300 °C for 1 h and then cooled down to room temperature. Then, H2 or O2 was evacuated and CO adsorption (>99%) was carried out. The spectra of adsorbed CO were recorded at several pressures – 5, 20, 50 Torr, at room temperature, with the pressure measurement accuracy of 5%. The obtained spectra were recalculated into Kubelka–Munk units (KMU). The background spectrum was subtracted from the spectrum of the sample with adsorbed CO and the baseline was corrected. All calculations were performed using the OPUS 6.0 software (Bruker).
Acidic and basic properties of the catalysts and corresponding supports were studied by the temperature-programmable desorption (TPD) of ammonia (“Chemosorb” chemical adsorption instrument) and CO2 (Autochem 2900 apparatus), respectively. The procedures in both cases were almost the same apart from the starting desorption temperature, which was 100 °C for ammonia TPD and 25 °C in the case of CO2 and the carrier gas, in the former case, was helium, and the latter argon. Prior to the analysis, the samples were treated at 300 °C under an inert atmosphere (helium or argon) for 1 h to remove the impurities adsorbed on the surface. Thereafter, the temperature was decreased to 100 °C (25 °C) followed by saturation with NH3 (CO2) for 60 min and flushing with He (Ar) for 1 h to remove physisorbed NH3 (CO2). The temperature was increased to 600 °C with a 10 °C min−1 ramp under a helium (argon) atmosphere.
For comparative analysis, NH3 and CO2 desorption profiles of the supports and corresponding catalysts are demarcated into temperature ranges: 100–200 °C (for TPD CO2 the starting temperature is 25 °C), 200–400 °C and 400–600 °C and are designated as weak, medium and strong acid or basic sites, respectively.
Catalytic testing
Betulin (90–94%) was extracted from birch with a non-polar solvent and recrystallized from 2-propanol in Åbo Akademi University.2 Betulin oxidation was performed over supported Au catalysts under atmospheric pressure with synthetic air (AGA, 20% oxygen, 80% nitrogen) as an oxidant in mesitylene at 140 °C or in toluene at 108 °C (Sigma Aldrich, >99%). Synthetic air was bubbled through the liquid with an inlet for the gas (flow rate: 50 ml min−1) located at the bottom of the reactor to enhance the gas–liquid mass transfer. Moreover, a metallic sinter was applied to diminish the size of air bubbles. Typically oxidation of betulin was carried out using 200 mg of the reagent in 100 ml of the solvent (the initial betulin concentration was 4.5 mmol l−1) using 200 mg of the catalyst. The reaction started when the desired temperature was reached, via turning on the stirring (450 rpm). Small catalyst particles (<63 μm) and a high stirring rate of 450 rpm were used to suppress the internal and external mass transfer limitations. In some experiments, hydrotalcite (Merck) was used together with the catalyst as a base-additive. Hydrotalcite was calcined for 3 h at 500 °C prior to its use.The samples for analysis were withdrawn from the reactor at regular intervals. Prior to GC-analyses, the samples (150 μL) were silylated by adding 150 μL of a mixture of pyridine (VWR International, Fontenay-sous-Bois, France), N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA, Supelco Analytical, Bellefonte, PA, USA), and trimethylsilyl chloride (TMCS, Merck KGaA, Darmstadt, Germany) in a 1:4:1 volume ratio, and the mixture was heated in an oven at 70 °C for 45 min. GC analysis was performed on a PerkinElmer AutosystemXL gas chromatograph using an Agilent HP-1 capillary column, 25 m (L) × 0.2 mm (ID), film thickness: 0.11 mm. Hydrogen was used as a carrier gas, with a flow rate of 0.8 ml min−1. Betulinic aldehyde and betulinic acid (90% purity), used as standards, were purchased from MedChem Express and Merck, respectively. The products were confirmed by GC-MS. The conditions of betulin oxidation and the analytical procedure were previously published.34,35
Size exclusion chromatography was performed to investigate oligomer and polymer formation on the spent catalyst surface.60 20 mg of the spent catalysts was added to a round flask together with 20 ml of the solvent heptane and a condenser. The flask was placed in an oil bath and heated to 98 °C. Thereafter, extraction occurred for four hours with a stirring rate of 400 rpm. The flow rate of the inert gas, consisting of 5% Ar in 95% N2, was set to 100 ml min−1. The solution obtained after the 4 h extraction was then kept at 40 °C, until complete evaporation of heptane. The resulting residue was then dissolved in 10 ml of tetrahydrofuran, and thereafter filtered for analysis. The resulting concentration of the residue was 2 mg ml−1. The analysis was carried out using a SEC-HPLC system equipped with two columns, a Guard column with the dimensions of 50 mm × 7.8 mm and a Jordi Gel DVB 500A column with the dimensions of 300 mm × 7.8 mm.
The TOF values were calculated as the number of converted moles of betulin per mole of exposed catalytic site per unit time, during the first 15 min, taking into account the metal dispersion:
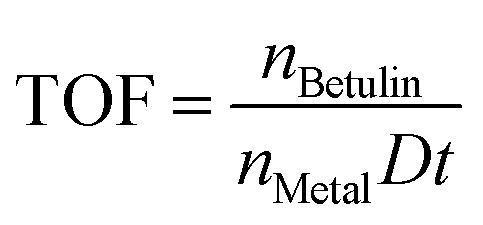 | (9) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research is funded from the Russian Science Foundation project No. 18-73-00019 and Tomsk Polytechnic University Competitiveness Enhancement Program, project VIU-ISHBMT-65/2019 (Russia). SACC acknowledges Fundação para a Ciência e a Tecnologia (FCT) for the Investigador FCT program (IF/01381/2013/CP1160/CT0007). This work was also financially supported by Associate Laboratory LSRE-LCM – UID/EQU/50020/2019 – funded by national (Portuguese) funds through FCT/MCTES (PIDDAC).References
- E. W. H. Hayek, U. Jordis, W. Moche and F. Sauter, Phytochemistry, 1989, 28, 2229 CrossRef CAS.
- C. Eckerman and R. Ekman, Pap. Puu, 1985, 67, 100 CAS.
- H. Pakdel, M. Murwanashyuka and C. Roy, J. Wood Chem. Technol., 2002, 22, 147 CrossRef CAS.
- S. Ohara, Y. Hayashi and M. Yatagai, Baiomasu Henkan Keikaku Kenkyu Hokoku, 1990, 24, 12 CAS . [C. A. 120 (1994) 301339f].
- B. N. Kuznetsov, V. A. Levdansky and N. I. Polezhaeva, Chem. Plant Raw Mater., 2004, 2, 21–24 Search PubMed.
- G. A. Tolstikov, O. B. Flekhter, E. E. Shultz, L. A. Baltina and A. G. Tolstikov, Chem. Sustainable Dev., 2005, 13, 1–29 CrossRef CAS.
- S. Alakurtti, T. Mäkelä and S. Koskimies, Eur. J. Pharm. Sci., 2006, 29, 1–13 CrossRef CAS PubMed.
- R. C. Santos, J. A. R. Salvador and S. Marín, Bioorg. Med. Chem., 2009, 17, 6241–6250 CrossRef CAS PubMed.
- S. C. Jonnalagadda, P. Suman, D. C. Morgan and J. N. Seay, Stud. Nat. Prod. Chem., 2004, 53, 45–84 Search PubMed.
- V. V. Grishko, E. V. Tarasova and I. B. Ivshina, Proc. Biochem., 2013, 48, 164–1644 CrossRef.
- S. Alakurtti, P. Bergström, N. Sacerdoti-Sierra, C. L. Jaffe and J. Yli-Kauhaluoma, J. Antibiot., 2010, 63, 123–126 CrossRef CAS PubMed.
- M. S. Gachet, O. Kunert, M. Kaiser, R. Brun, M. Zehl, W. Keller, R. A. Munoz, R. Bauer and W. Schuehly, J. Nat. Prod., 2011, 74, 559–566 CrossRef CAS PubMed.
- C. P. Reyes, M. J. Núnez, I. A. Jiménez, J. Busserolles, M. J. Alcaraz and L. BazzocchiI, Bioorg. Med. Chem., 2006, 14, 1573–1579 CrossRef CAS PubMed.
- K. Hata, K. Hori and S. Takahashi, J. Nat. Prod., 2002, 65, 645–648 CrossRef CAS.
- A. Koohang, N. D. Majewski, E. L. Szotek, A. A. Mar, D. A. Eiznhamer, M. T. Flavin and Z. Q. Xu, Bioorg. Med. Chem. Lett., 2009, 19, 2168–2171 CrossRef CAS PubMed.
- M. Liu, S. Yang, L. Jin, D. Hu, Z. Wu and S. Yang, Molecules, 2012, 17, 6156–6169 CrossRef CAS PubMed.
- A. A. Mar, A. Koohang, N. D. Majewski, E. L. Szotek, D. A. Eiznhamer, M. T. Flavin and Z. Q. Xu, Chin. Chem. Lett., 2009, 20, 1141–1141 CrossRef CAS.
- I. C. Sun, J. K. Shen, H. K. Wang, L. M. Cosentino and K. H. Lee, Bioorg. Med. Chem. Lett., 1998, 8, 1267–1272 CrossRef CAS.
- I. C. Sun, H. K. Wang, Y. Kashiwada, J. K. Shen, L. M. Cosentino, C. H. Chen, L. M. Yang and K. H. Lee, J. Med. Chem., 1998, 41, 4648–4657 CrossRef CAS PubMed.
- E. Bębenek, M. Kadela-Tomanek, E. Chrobak, J. Wietrzyk, J. Sadowska and S. Boryczka, Med. Chem. Res., 2016, 26, 1–8 Search PubMed.
- E. Bębenek, M. Kadela-Tomanek, E. Chrobak, M. Latocha and S. Boryczka, Med. Chem. Res., 2018, 27, 2051–2061 CrossRef PubMed.
- V. V. Grishko, I. A. Tolmacheva, V. O. Nebogatikov, N. V. Galaiko, A. V. Nazarov, M. V. Dmitriev and I. B. Ivshina, Eur. J. Med. Chem., 2017, 125, 629–639 CrossRef CAS PubMed.
- R. Csuk, K. Schmuck and R. Schafer, Tetrahedron Lett., 2006, 47, 8769–8770 CrossRef CAS.
- A. Pichette, H. Liu, C. Roy, S. Tanguay, F. Simard and S. Lavoie, Synth. Commun., 2004, 34, 3925–3937 CrossRef CAS.
- N. G. Komissarova, N. G. Belenkova, O. V. Shitikova, L. V. Spirikhin and M. S. Yunusov, Chem. Nat. Compd., 2006, 338(1), 58–61 CrossRef.
- S. D. Kim, Z. Chen, T. V. Nguyen, J. M. Pezzuto, S. Qui and Z. Z. Lu, Synth. Commun., 1997, 27, 1607–1612 CrossRef.
- J. M. Pezzuto and S. H. L. Darrick Kim, Pat US5804575, 1998 Search PubMed.
- V. I. Roshchin, N. Yu. Shabanova and D. N. Vedernikov, Pat RU2190622, 2002, MGG C07J053/00, C07J063/00 Search PubMed.
- V. A. Levdansky, N. I. Polezhaeva and B. N. Kuznetsov, Pat RU2269541 2004, IPC C07J53/00, C07J63/00 Search PubMed.
- K. Muffler, D. Leipold, M. Schellera, C. Haas, J. Steingroewer, T. Bley, H. E. Neuhaus, M. A. Mirata, J. Schrader and R. Ulber, Proc. Biochem., 2011, 46, 1–15 CrossRef CAS.
- D. B. Mao, Y. Q. Feng, Y. H. Bai and C. P. Xu, J. Taiwan Inst. Chem. Eng., 2012, 43, 825–829 CrossRef CAS.
- H. Liu, X. L. Lei, N. Li and M. H. Zong, J. Mol. Catal. B: Enzym., 2013, 88, 32–35 CrossRef CAS.
- E. V. Tarasova, V. V. Grishko and I. B. Ivshina, Proc. Biochem., 2017, 52, 1–9 CrossRef CAS.
- P. Mäki-Arvela, M. Barsukova, I. Winberg, A. Smeds, J. Hemming, K. Eränen, A. Torozova, A. Aho, K. Volcho and D. Yu. Murzin, ChemistrySelect, 2016, 1, 3866–73869 CrossRef.
- E. Kolobova, Y. Kotolevich, E. Pakrieva, G. Mamontov, M. H. Farias, V. Cortés Corberán, N. Bogdanchikova, J. Hemming, A. Smeds, P. Mäki-Arvela, D. Yu. Murzin and A. Pestryakov, Fuel, 2018, 234, 110–119 CrossRef CAS.
- R. Zanella, L. Delannoy and C. Louis, Appl. Catal., A, 2005, 29, 62–72 CrossRef.
- E. Pakrieva, E. Kolobova, G. Mamontov, N. Bogdanchikova, M. H. Farias, L. Pascual, V. Cortés Corberán, S. Martinez Gonzalez, S. A. C. Carabineiro and A. Pestryakov, ChemCatChem, 2019, 11, 1–11 CrossRef.
- A. Penkova, K. Chakarova, O. H. Laguna, K. Hadjiivanov, F. Romero Saria, M. A. Centeno and J. A. Odriozola, Catal. Commun., 2009, 10, 1196–1202 CrossRef CAS.
- A. N. Pestryakov and A. A. Davydov, Appl. Catal., A, 1994, 120, 7–15 CrossRef CAS.
- Y. Kotolevich, E. Kolobova, E. Khramov, M. H. Farías, Ya. Zubavichus, H. Tiznado, S. Martínez-González, V. Cortés Corberán, J. D. Mota-Moralez, A. Pestryakov and N. Bogdanchikova, J. Mol. Catal. A: Chem., 2017, 427, 1–10 CrossRef CAS.
- A. Simakov, I. Tuzovskaya, A. Pestryakov, N. Bogdanchikova, V. Gurin, M. Avalos and M. H. Farıas, Appl. Catal., A, 2007, 331, 121–128 CrossRef CAS.
- N. Bogdanchikova, A. Pestryakov, I. Tuzovskaya, T. A. Zepeda, M. H. Farias, H. Tiznado and O. Martynyuk, Fuel, 2013, 110, 40–47 CrossRef CAS.
- S. V. Nayak and R. V. Chodhary, J. Catal., 1983, 81(1), 26–45 CrossRef.
- H. D. Olson, T. G. Kokotailo, L. S. Lawton and M. W. Meier, J. Phys. Chem., 1981, 85(15), 2238–2243 CrossRef.
- L. Zhu, Y. Zeng, Sh. Zhang, J. Deng and Q. Zhong, J. Environ. Sci., 2017, 54, 277–287 CrossRef PubMed.
- P. N. Amaniampong, K. Li, X. Jia, B. Wang, A. Borgna and Y. Yang, ChemCatChem, 2014, 6, 2105–2114 CrossRef CAS.
- D. Kubicka, N. Kumar, P. Mäki-Arvela, M. Tiitta, V. Niemi, H. Karhu, T. Salmi and D. Yu. Murzin, J. Catal., 2004, 227, 313–327 CrossRef CAS.
- D. Kubicka, N. Kumar, T. Venäläinen, H. Karhu, I. Kubickova, H. Österholm and D. Yu. Murzin, J. Phys. Chem. B, 2006, 110, 4937–4946 CrossRef CAS PubMed.
- J. I. Villegas, D. Kubicka, H. Karhu, H. Österholm, N. Kumar, T. Salmi and D. Yu. Murzin, J. Mol. Catal. A: Chem., 2007, 264, 192–201 CrossRef CAS.
- D. N. Thanh, O. Kikhtyanin, R. Ramos, M. Kothari, P. Ulbrich, T. Munshi and D. Kubicka, Catal. Today, 2016, 277, 97–107 CrossRef.
- J. I. Di Cosimo, V. K. Dıez, M. Xu, E. Iglesia and C. R. Apesteguıa, J. Catal., 1998, 178, 499–510 CrossRef CAS.
- M. Di Serio, M. Ledda, M. Cozzolino, G. Minutillo, R. Tesser and E. Santacesaria, Ind. Eng. Chem. Res., 2006, 45, 3009–3014 CrossRef CAS.
- C. O. Veloso, C. N. Pérez, B. M. de Souza, E. C. Lima, A. G. Dias, J. L. F. Monteiro and C. A. Henriques, Microporous Mesoporous Mater., 2008, 107, 23–30 CrossRef CAS.
- M. Mihaylov, E. Ivanova, Y. Hao, K. Hadjiivanov, B. C. Gates and H. Knozinger, Chem. Commun., 2008, 175–177 RSC.
- M. Mihaylov, E. Ivanova, Y. Hao, K. Hadjiivanov, H. Knozinger and B. C. Gates, J. Phys. Chem. C, 2008, 112, 18973–18189 CrossRef CAS.
- H. Haario, ModEst, Modelling and optimization software, Helsinki (Finland), 2011 Search PubMed.
- R. Zanella, S. Giorgio, C. R. Henry and C. Louis, J. Phys. Chem. B, 2002, 106, 7634–7642 CrossRef CAS.
- M. Hinojosa-Reyes, R. Camposeco-Solis, R. Zanella, V. Rodríguez-González and F. Ruiz, Catal. Lett., 2018, 148(1), 383–396 CrossRef CAS.
- R. Zanella and C. Louis, Catal. Today, 2005, 107–108, 768–777 CrossRef CAS.
- A. H. Abdullah, A. Hauser, F. A. Ali and A. Al-Adwani, Energy Fuels, 2006, 20(1), 320–323 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc00949c |
| This journal is © The Royal Society of Chemistry 2019 |