
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improvements to the production of ZIF-94; a case study in MOF scale-up†
Timothy
Johnson
 *a,
Magdalena M.
Łozińska
b,
Angelica F.
Orsi
b,
Paul A.
Wright
b,
Sheena
Hindocha
a and
Stephen
Poulston
a
*a,
Magdalena M.
Łozińska
b,
Angelica F.
Orsi
b,
Paul A.
Wright
b,
Sheena
Hindocha
a and
Stephen
Poulston
a
aJohnson Matthey Technology Centre, Blount's Court, Sonning Common, RG4 9NH UK. E-mail: timothy.johnson@matthey.com
bEaStCHEM School of Chemistry, University of St Andrews, Purdie Building, North Haugh, St Andrews, Fife, KY16 9ST, UK
First published on 1st October 2019
Abstract
The ability to produce large scale quantities of MOF materials is essential for the commercialisation of these frameworks to continue. Herein we report how the production of ZIF-94 can be improved from a ∼1 g laboratory preparation to a scalable procedure allowing for large scale production of the desired framework. The synthesis of ZIF-94 was completed at room temperature, atmospheric pressure and without the use of DMF as a solvent. This method offers improvements over the current literature synthesis routes and affords a product at 18 wt% solids. To demonstrate the robustness of the derived methodology a 60 g, large scale, batch of this framework was produced which possessed a surface area of 468 m2 g−1. This large scale sample has superior CO2 uptake of 3.3 mmol g−1 at 1 bar, an improvement of 30% over literature reports.
Introduction
Metal organic frameworks (MOF) have long been promised as attractive materials for a wide range of applications including gas storage1–3 and separation,4–7 catalysis8–11 and chemical sensors.12–14 While many materials do, on the laboratory scale, show remarkable properties, the ability to translate that into real world, commercial applications, has long been a stumbling block. Recently MOFs have been commercialised in certain applications however there remain few reports of scale-up development or the synthetic considerations required for the scale-up of MOF materials.This paper presents a case study for the scale-up of MOFs. Here the synthetic methodology of a zeolitic imidazolate framework (ZIF), a subclass of the MOF family of materials, was studied and improved. This will show how small, low concentration, reactions can be built upon to produce materials in a scaleable and green fashion.
ZIF-94 is a framework composed of zinc tetrahedral metal ions connected by the organic linker 4-methyl-5-imidazolecarboxaldehyde in a sod topology, Fig. 1. ZIF-94, also known as SIM-1 (Substituted imidazolate material), is of interest due to its excellent chemical stability and high CO2 adsorbance at low pressure.15
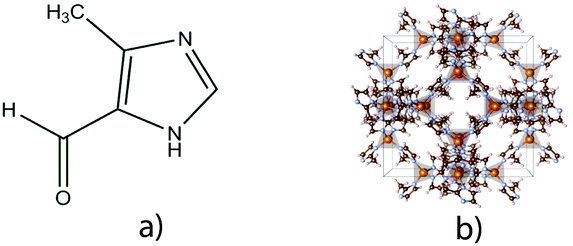 | ||
| Fig. 1 (a) ZIF-94 linker 4-methyl-5-imidazolecarboxaldehyde and (b) representation of ZIF with a sod toplogy. | ||
Previous reports regarding the synthesis of ZIF-94 consist of very dilute solutions, generally between 5–12 wt% solids in DMF, under solvothermal conditions.5,15–18 When dealing with the scale-up of this material, these factors present a logistical barrier due to the large solvent volumes and the need for large reactors. In addition, for applications in gas separation membranes, the particle size of the MOFs utilized is an important factor. For improved processability sub <100 nm particle sizes are optimal however reports of MMM using sub-micron particles have been reported.19
Recently we reported how nano particles of ZIF-94 could be produced in a green, scalable fashion. This route utilised methanol and tetrahydrofuran as solvents. While methanol is considered a “recommended” solvent, tetrahydrofuran is “problematic”.20 This should be viewed in the context that, prior to work conducted by the authors, the production method to produce this material required dimethylformamide, a solvent with a “hazardous” rating due to its health effects. This signifies a significant improvement especially considering this material may be produced industrially at large scale. Importantly previous reports produced material using very dilute reaction conditions of 1.4 wt% solids.21
4-Methyl-5-imidazolecarboxaldehyde and zinc acetate, the linker and metal source for ZIF-94, are currently commercially available. Importantly 4-methyl-5-imidazolecarboxaldehyde is a good candidate for production using mechanochemical routes,22 suggesting this can be produced with minimal environmental impact and zinc acetate, being a salt, can be precipitated from aqueous conditions – further reducing the use of organic solvents.
ZIF-94 also offers excellent scope for post synthetic modification (PSM) due to the abundance of aldehyde groups within the pores of the material. The use of post-synthetic modification is of commercial interest since targeted functionality can be introduced into materials.
For ZIF-94, the addition of a long, hydrocarbon chain primary amine has demonstrated improvements in N2/CO2 separation under humid conditions5 and in Knoevenagel condensations23 where the MOF was used as the catalyst.
Herein we report the scale-up and production of a 60 g batch of ZIF-94. The procedures were specifically chosen to ensure scalability which includes the greening of solvents, room temperature synthesis and improved reaction concentration. This results in reduced demand for solvents and energy, while ensuring a pure nano-ZIF is produced.
Experimental
Materials
Zinc acetate dihydrate was purchased from Acros Chemicals (98% purity) and 4-methyl-5-imidazolecarboxaldehyde was obtained from Active Scientific UK (≥97% purity). Methanol (99.8%) and anhydrous tetrahydrofuran (≥99.9%) were supplied by Sigma-Aldrich. All chemicals were used without further purification.Synthesis of ZIF-94
Table 1 gives a summary of the reaction conditions used for the synthesis of ZIF-94. Below is a representative experimental method for entry 11 in Table 1.| Entry | Solid content (wt%) | Reaction time (min) | Zinc acetatea (g) | MeOH (mL) | THF (mL) | Mass of product (g) | Est. yieldc (%) |
|---|---|---|---|---|---|---|---|
| a The amount of linker (g) is equal to the amount of zinc acetate (g). b Mass not reported due to unintentional loss during transfer. For an explanation of how solid content was calculated, please see ESI.† c Estimated yield obtained from TGA data. | |||||||
| 1 | 0.3 | 15 | 1.07 | 228 | 571 | — | — |
| 2 | 0.3 | 60 | 1.07 | 228 | 571 | — | — |
| 3 | 0.3 | 360 | 1.07 | 228 | 571 | — | — |
| 4 | 4 | 30 | 2.64 | 40 | 100 | 3.18 | 86.9 |
| 5 | 4 | 60 | 2.64 | 40 | 100 | 3.12 | 85.7 |
| 6 | 4 | 180 | 2.64 | 40 | 100 | 3.4 | 93.9 |
| 7 | 4 | 960 | 2.64 | 40 | 100 | 3.43 | 95.2 |
| 8 | 4 | 960 | 1.32 | 20 | 50 | —b | — |
| 9 | 9 | 960 | 2.64 | 20 | 50 | 3.74 | — |
| 10 | 18 | 960 | 5.28 | 20 | 50 | 7.49 | — |
| 11 | 18 | 960 | 52.8 | 200 | 500 | 74.51 | 84.7 |
The zinc acetate dihydrate (52.8 g, 0.24 mol) was dissolved in 200 mL methanol. A separate solution of 4-methyl-5-imidazolecarboxaldehyde (52.8 g, 0.48 mol) was dissolved in 400 mL tetrahydrofuran. The methanol solution was added to the tetrahydrofuran solution under vigorous stirring. The mixture was continuously stirred at room temperature for 960 minutes. The product was collected by centrifugation (10 minutes, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) and washed with methanol three times. The resulting sample was dried at room temperature in air for 48 hours.
000 rpm) and washed with methanol three times. The resulting sample was dried at room temperature in air for 48 hours.
SEM
SEM micrographs were collected and analysed using a Zeiss ultra 55 Field emission electron microscope equipped with in-lens secondary electron and backscattered detectors. High-resolution low-accelerating voltage imaging. Accelerating voltage: 1.6 kV Aperture used: 20–30 μm Working distance (WD): 2–3 mm detectors: In-lens secondary electron and In-lens backscattered electron detectors. The samples were carbon coated prior to analysis to provide a conductive layer for charge dissipation. The sample powders were dusted directly onto SEM stubs.PXRD
Powder X-ray diffraction (PXRD) data were collected in reflection geometry using a Bruker AXS D8 diffractometer using Cu Kα radiation (λ = 1.5406 + 1.54439 Å) over the 5 < 2θ < 50° range in 0.02° steps. Le Bail refinement was performed using Topas24 with reflection profiles modelled using a fundamental parameters approach25 with reference data collected from NIST660 LaB6.TGA
Thermal gravimetric analysis (TGA) was conducted between room temperature and 1000 °C at a heating rate of 3 °C min−1 under an Ar:O2 (80:20) atmosphere on a Netzsch TG 209 F1 Libra instrument.
Physisorption
Samples were subjected to analysis by N2 and CO2 physisorption measurements. Isotherms were collected on a Quantachrome Autosorb iQ instrument with experiments being conducted at −196 °C for N2 and 0 °C for CO2. All samples were degassed to 200 °C for 12 hours prior to analysis. N2 Isotherms were subject to BET analysis for surface area calculation. The Rouquerol method was used to determine the correct P/P0 range for BET calculations.26Results and discussion
Low concentration experiments
Initial experimentation on a low solid weight percent reaction was conducted with relevant experiment details provided in entries 1–3 (Table 1). Fig. 2 shows the XRD patterns collected from samples produced over a range of times at 0.3 wt% solids.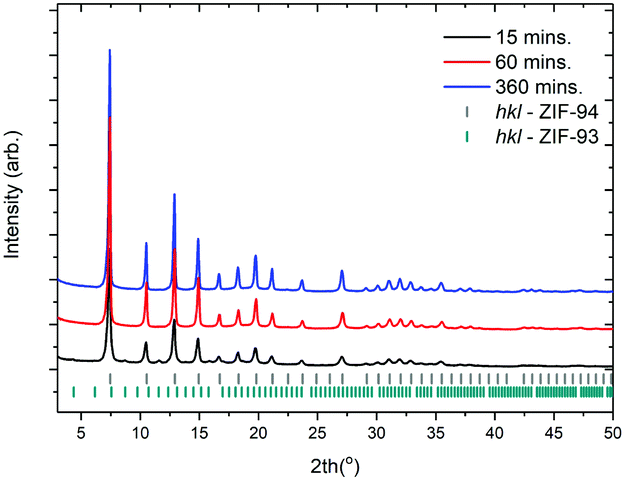 | ||
| Fig. 2 XRD patterns collected on samples synthesised at 0.3 wt% solid. Entries 1–3 (Table 1). | ||
Importantly it can be seen that at short reaction times, 15 minutes or less, an impure sample is produced. The impurity observed can be indexed to ZIF-93 – a framework produced from the same linker and metal as ZIF-94 but possessing the rho topology. Simulated PXRD patterns for both ZIF-94 and ZIF-93 can be seen in S1. Fig. 3 shows SEM micrographs which demonstrate that, with a reaction time of 15 minutes, poorly defined nano particles are produced. It is only when reaction times reach in excess of 30 minutes that nano spheres start to form. It is also of note that with a reaction time of 360 minutes nano particles continue to be observed.
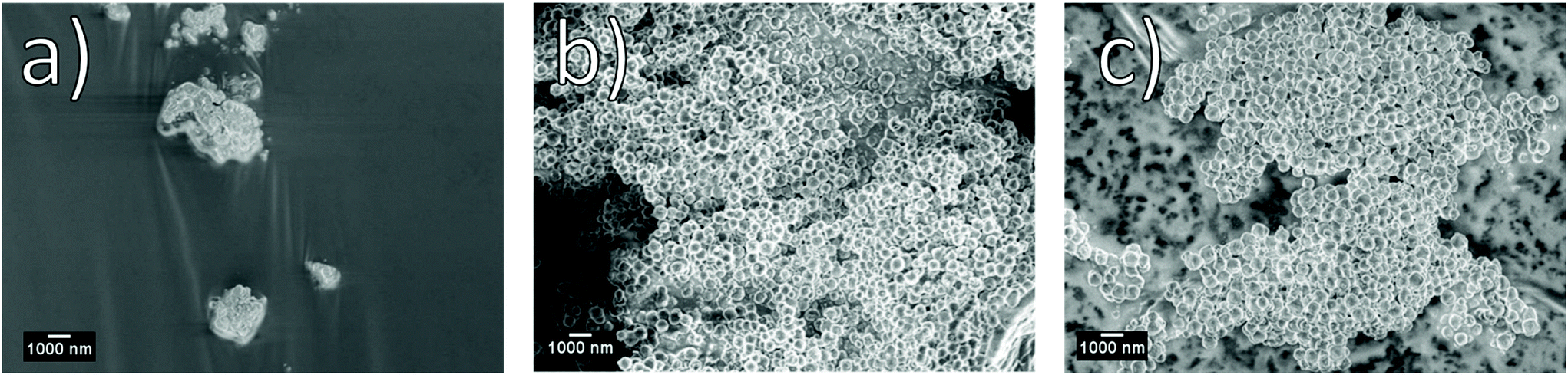 | ||
| Fig. 3 SEM micrographs of samples produced at 0.3 wt% solids at (a) 15, (b) 60 and (c) 360 min respectively, Entries 1–3 (Table 1). | ||
The effect of reaction time on purity and morphology
Fig. 4 shows the XRD patterns collected for samples synthesised at 4 wt% solids, experimental details can be found in entries 4–7 (Table 1). Only after 16 h is a phase pure material produced with samples produced at times less than 16 h showing Bragg diffraction from the ZIF-93 phase (2th = 4.3°). To confirm the nano morphology of the materials a series of SEM micrographs were taken (Fig. 5). Fig. 5a shows that short reaction times below 30 min do not produce a nano material, this morphology is consistent with samples produced at lower concentrations. Indeed, it is shown that nano spheres are only observed at reaction times of 60 min or more. Even after 16 h nano spheres are still obtained – as shown in Fig. 5d. TGA analysis confirms all samples possess the same mass loss profiles, S2. All samples show an initial mass loss of ∼5%, this is assigned to solvent trapped within the framework. Thermal decomposition occurs at 225 °C demonstrating the stability of ZIF-94 at high temperatures. All samples also show decomposition to ∼29% original mass. This is in line with the theoretical value of 27%, based on full linker decomposition and ZnO formation.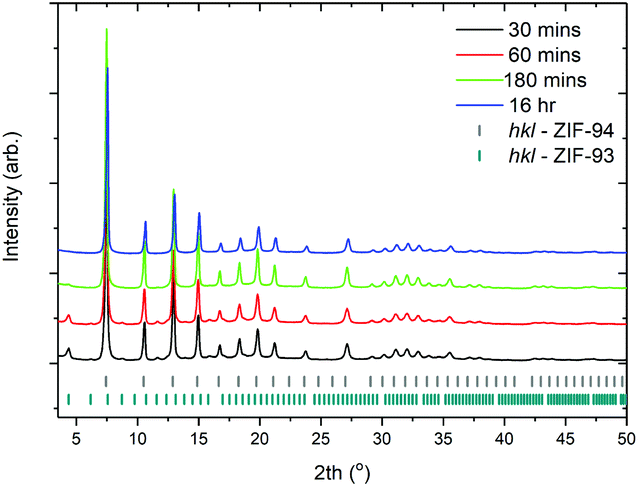 | ||
| Fig. 4 XRD patterns collected on samples synthesised at 4 wt% solid (entries 4–7, Table 1). | ||
 | ||
| Fig. 5 SEM micrographs of samples produced at 4 wt% solids at (a) 30, (b) 60, (c) 180 and (d) 960 min respectively. Entries 4–7 (Table 1). | ||
N2 isotherms collected on samples obtained from entries 4 and 8 (Table 1), S3 and 4, show BET surface areas of 432 and 571 m2 g−1, respectively. These values are comparable to literature values of 415 m2 g−1.18 This indicates that reaction time does not adversely affect the surface area of the framework produced and shows that this method is comparable to the use of low concentration reactions in DMF under solvothermal conditions.
Due to excess solvent within the pores of the material estimated yields have been calculated from the TGA data S2. It can be seen how estimated yields for these materials range from 85–95%. While these data show the yield increases with reaction time, most of the product is formed very quickly. This suggests that the formation of ZIF-94 is under thermodynamic control, with the kinetic product, ZIF-93, being formed initially and very quickly. These data indicate that with extended reaction time, equilibrium can be reached and the favoured thermodynamic product formed.
Improvements to solid content
The observation that longer reaction times does not adversely affect the morphology of samples produced suggested that higher solid concentrations could be employed and, with sufficient reaction time, phase pure, nano ZIF-94 could be produced. Experiments to investigate this were conducted as detailed in entries 8–10 (Table 1). XRD patterns from the obtained samples are shown in Fig. 6. It can be seen that across the range of solid weight percents phase pure ZIF-94 is produced with no observable diffraction from an impurity phase. Importantly SEM micrographs, shown in S5, indicate that despite longer reaction times nano spheres are still produced. This parameter is key if ZIF-94 is to be used in the production of mixed matrix membranes.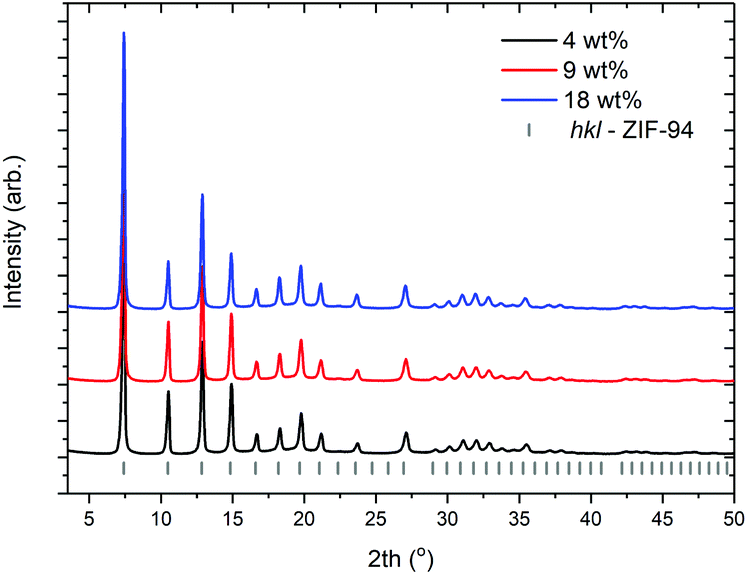 | ||
| Fig. 6 XRD patterns of samples produced at (a) 4 wt% (b) 9 wt% (c) 18 wt% respectively produced with reaction times of 960 min. Entries 8–10 (Table 1). | ||
Large scale reaction
A large scale reaction was conducted using the optimised reaction conditions discussed previously, details of this can be found in Table 1, entry 11. The XRD pattern of this sample can be seen in Fig. 7 and shows that a phase pure material is obtained from this large scale reaction. Using an integral breadth approach, subsequent Le Bail fitting of the pattern collected shows that this sample has a crystallite size of 39 nm – suggesting the particles are not single crystals. In addition, SEM micrographs, Fig. 8, show that, despite a high concentration and a long reaction time, nanoparticle morphology was still obtained.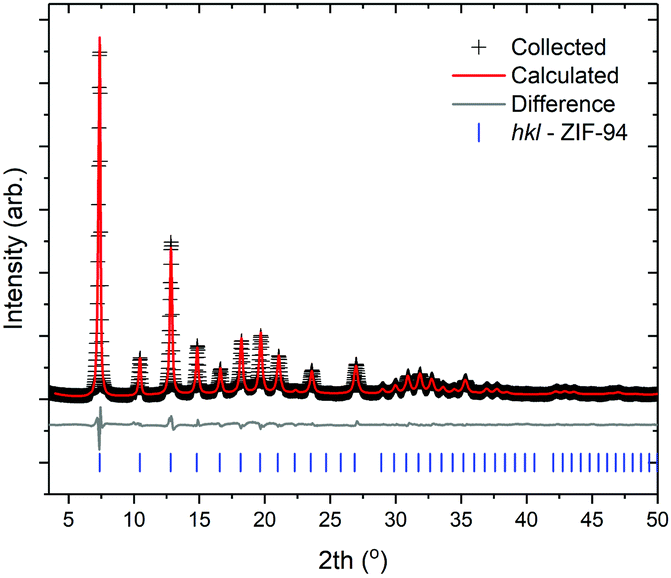 | ||
| Fig. 7 XRD pattern and subsequent Le Bail fit for large scale ZIF-94, produced at 18% solids with a reaction time of 960 min, entry 11 (Table 1). | ||
 | ||
| Fig. 8 SEM micrograph of ZIF-94 produced at 18% solids with a reaction time of 960 min, entry 11 (Table 1). | ||
TGA, S6, shows that the large scale batch of ZIF-94 contains a proportionately larger amount of trapped solvent than the smaller scale batches. A mass loss of 10% was observed before thermal decomposition of the framework occurred at 230 °C. This observation indicates that the drying procedure, which was adequate at small scales, did not transfer to larger scale experiments. A more rigorous drying procedure will be required in future, for example heating the framework below the thermal decomposition temperature in a vacuum oven.
Thermal decomposition progressed to 28% original mass, this is in line with theoretical value of 27% decomposition based on formation of ZnO and full linker destruction. The estimated yield observed for this reaction is also high at 84.7%. This is lower than that of past samples however this is likely due to the increased number of centrifuge pots needed during the separation – ultimately increasing transfer error. This yield is however still significant and, with improvements to separations, well within acceptable limits for large scale reaction procedures.
N2 isotherms, S7, were collected on the scaled up ZIF-94 sample. Subsequent BET analysis revealed the material possesses a surface area of 415 m2 g−1 which agrees with literature values as well as with samples produced at lower solid weight percent.18
The method of production reported herein utilises a centrifuge to separate the product from the mother liquor. It should be noted that this technique is a viable separation technique and used industrially to produce material at scale.27
CO2 adsorption data, Fig. 9, was also collected for this sample with a maximum uptake of 3.5 mmol g−1 observed. Importantly this large scale material outperforms previous literature reports of ZIF-94 which show CO2 uptakes of 2.5 mmol g−1 at 1 bar for samples produced hydrothermally in DMF, compared to 3.3 mmol g−1 at 1 bar for this work.15 This equates to a 30% performance improvement – increasing the validity of this material, and this synthetic method, for carbon capture applications.
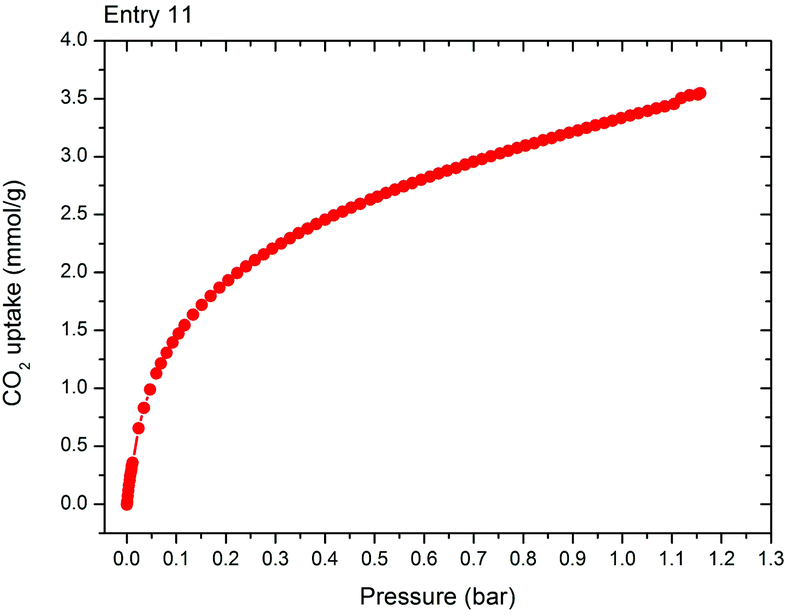 | ||
| Fig. 9 CO2 adsorption isotherm collected at 0 °C for scaled up ZIF-94 sample, entry 11 (Table 1). | ||
Conclusions
In conclusion it can be seen that improvements have been made to the synthesis of the ZIF-94 framework. The weight percent of solids within the reaction is higher than any previously reported synthetic route and allows for phase pure, nano, ZIF-94 to be obtained. The work presented lays the ground work for larger scale batch production. With the material produced at room temperature, at high solid to solvent ratios and using centrifugation, transfer to a larger production volume can now be conducted. This route has shown how ZIF-94 can be produced on a viable scale following green chemistry principles which could be extended to other ZIFs. The move away from DMF to lower volumes and more environmentally benign solvents, along with the room temperature synthesis shows how greening of MOF syntheses can be achieved while maintaining crystallinity and performance. This work could be applied to other, structurally analogous frameworks, such as ZIF-8, where nano sized, phase pure products are of interest. The ability to produce this material on a large scale, and the wealth of possible post synthetic modifications that can be conducted on the ZIF-94 system, means that this work is highly relevant for further study of the use of targeted functional MOF materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013), under grant agreement no. 608490, M4CO2 project. The authors are grateful to the JMTC analytical team for various data collected for this work including Adam Phillips and Emily Brooke for collection and presentation of the SEM micrographs.Notes and references
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2007, 129, 5314–5315 CrossRef CAS PubMed.
- A. Noguera-Díaz, N. Bimbo, L. T. Holyfield, I. Y. Ahmet, V. P. Ting and T. J. Mays, Colloids Surf., A, 2016, 496, 77–85 CrossRef.
- R. Babarao and J. Jiang, Langmuir, 2008, 24, 6270–6278 CrossRef CAS PubMed.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- S. Aguado, J. Canivet and D. Farrusseng, J. Mater. Chem., 2011, 21, 7582 RSC.
- M. S. Denny, J. C. Moreton, L. Benz and S. M. Cohen, Nat. Rev. Mater., 2016, 1, 16078 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- D. S. Kim, W. J. Park and C. H. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed.
- W.-Y. Gao, H. Wu, K. Leng, Y. Sun and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 5472–5476 CrossRef CAS PubMed.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- M. Zhang, G. Feng, Z. Song, Y.-P. Zhou, H.-Y. Chao, D. Yuan, T. T. Y. Tan, Z. Guo, Z. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed.
- W. Morris, N. He, K. G. Ray, P. Klonowski, H. Furukawa, I. N. Daniels, Y. A. Houndonougbo, M. Asta, O. M. Yaghi and B. B. Laird, J. Phys. Chem. C, 2012, 116, 24084–24090 CrossRef CAS.
- S. Aguado, J. Canivet and D. Farrusseng, Chem. Commun., 2010, 46, 7999–8001 RSC.
- S. Aguado, C.-H. Nicolas, V. Moizan-Baslé, C. Nieto, H. Amrouche, N. Bats, N. Audebrand and D. Farrusseng, New J. Chem., 2011, 35, 41–44 RSC.
- A. M. Marti, M. Van and K. J. Balkus, J. Porous Mater., 2014, 21, 889–902 CrossRef CAS.
- R. Lin, B. V. Hernandez, L. Ge and Z. Zhu, J. Mater. Chem. A, 2018, 6, 293–312 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- M. Etxeberria-Benavides, O. David, T. Johnson, M. M. Łozińska, A. Orsi, P. A. Wright, S. Mastel, R. Hillenbrand, F. Kapteijn and J. Gascon, J. Membr. Sci., 2018, 550, 198–207 CrossRef CAS.
- M. Leonardi, M. Villacampa and J. C. Menéndez, Chem. Sci., 2018, 9, 2042–2064 RSC.
- J. Canivet, S. Aguado, C. Daniel and D. Farrusseng, ChemCatChem, 2011, 3, 675–678 CrossRef CAS.
- Topas v5.0: General Profile and Structure Analysis Software for Powder Diffraction Data, Bruker AXS, Karlsruhe, Germany, 2003–2015 Search PubMed.
- R. W. Cheary and A. Coelho, J. Appl. Crystallogr., 1992, 25, 109–121 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 CrossRef CAS.
- T. Beveridge, in Encyclopaedia of Separation Science, ed. I. D. Wilson, Academic Press, 1st edn, 2000, ch. 2, pp. 320–329 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc00783k |
| This journal is © The Royal Society of Chemistry 2019 |