
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 conversion by high-dose rate electron beam irradiation: one-step, metal-free and simultaneous production of H2, CO, CH4, C2H6 and organic acids from an acid-decomposed CaCO3/additive EtOH mixture†
Yoichi
Hosokawa
 *a,
Shuji
Kajiya
a,
Ayako
Ohshima
a,
Nobuhiro
Ishida
*a,
Masakazu
Washio
b and
Arimitsu
Usuki‡
a
*a,
Shuji
Kajiya
a,
Ayako
Ohshima
a,
Nobuhiro
Ishida
*a,
Masakazu
Washio
b and
Arimitsu
Usuki‡
a
aToyota Central R&D Labs., Inc., 41-1 Nagakute, Aichi 480-1192, Japan. E-mail: e1305@mosk.tytlabs.co.jp; n-ishida@mosk.tytlabs.co.jp
bWaseda Research Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku, Tokyo 169-8555, Japan
First published on 15th May 2019
Abstract
The reduction in CO2 emissions is an important issue across many industries. Inspired by extraterrestrial organic matter formation, we herein report a CO2 conversion approach based on high-dose rate electron beam (EB) irradiation of an acid-decomposed CaCO3/additive EtOH mixture. With 13C-CaCO3, 12C-EtOH and 100 kGy s−1 EB, H2, CO, CH4, C2H6 and organic acids are simultaneously produced within a few seconds, except for 2,3-butanediol formation from excess EtOH. According to the organic analysis results, CO and organic acids contain 13C carbon derived from 13C-CaCO3. The high-dose rate EB gives increased CO2 conversion products compared to the low-dose rate EB. The CO2 conversion yield/energy efficiency (product energy/input electrical energy) at 300 kGy is 1.51/0.50% in total (CO: 0.03/0.01%, formic acid: 1.31/0.29%, acetic acid: 0.05/0.04% and propionic acid: 0.12/0.16%), and the total radiation energy efficiency (REE, product energy/net radiation energy) of CO2 at 300 kGy is 51.5% (CO: 0.90%, formic acid: 30.3%, acetic acid: 3.71% and propionic acid: 16.6%). The CO2 conversion yield is ∼15 times larger than that of the only known CO2 gas radiolysis (0.1%, CO only). Furthermore, the REE at 100 kGy is also ∼15 times higher than that obtained in the absence of EtOH. The energy input for the 100% conversion yield is estimated to be 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 GJ per t-CO2. The combination of the high-dose rate EB with organic additives facilitated CO2 capture by radicals to afford improved CO2 conversion efficiency/yield.
000 GJ per t-CO2. The combination of the high-dose rate EB with organic additives facilitated CO2 capture by radicals to afford improved CO2 conversion efficiency/yield.
Introduction
The reduction of CO2 emissions to suppress global warming entails the reduction of fossil fuel consumption, which is particularly an important issue for the automotive industry that heavily relies on the use of hydrocarbons and plastic products as the primary source of fuel and automotive body construction, respectively. Among the artificial hydrocarbon synthesis methods, the Fischer–Tropsch (CO and H2)1 and Sabatier (CO2 and H2)2 reactions are well known in the field. However, H2 is generally produced through methane reformation and using syngas (CO and H2), which, in turn, are primarily synthesised by coal gasification. Therefore, in the long term,3 as an ideal system, it is desirable to convert abundant carbon (e.g., CO2/biomass) and H2 (e.g., water/seawater/biomass) sources into energy/basic chemicals using renewable energy by a rare metal-free method.As shown by Calvin's experiment4 and astrochemistry,5 diverse organic materials are formed from H2, CO and CO2 by the radiation radical reaction in a complex history after prolonged radiation exposure and thermal metamorphism under diverse conditions, such as radiation (particle, electron and gamma-ray), gas atmosphere (H2, N2, O2, etc.), temperature (10 K (space)—several thousand degree celsius (meteorite at atmosphere-entry)), and interaction with various mineral contents and special events (e.g., gamma-ray bursts and superflares). Although the reaction conditions are not clear enough, CO2/CO, H2 and H2O have been considered as the starting materials for CH4 or CH3OH formation, and such extraterrestrial organic matter production, including hydrocarbons, has attracted attention in the chemistry field.6
In radiation chemistry,7 H2 formation8 from H2O as well as CO formation9 from CO2-saturated water have been confirmed to involve the generation of radical intermediates in the presence of inorganic additives such as metal ions, raw metal, and metal oxides. However, the catalytic effect was significantly small. It is considered that the metal catalyst does not work enough as the reaction is carried out in water under ambient atmosphere, and the intermediate radical concentration formed by irradiation is very low. Therefore, we thought of the addition of other reactive radicals. The radical reaction easily happens in water under ambient atmosphere. Formaldehyde,10 formic acid11 and acetic acid12 have been detected in the gamma-ray (GR) radiolysis of the CO2/H2O mixture. Unfortunately, GR has a low dose rate (∼10 kGy h−1). Consequently, the produced H/OH/CO radical concentration is extremely low. Such short-lived radicals immediately undergo recombination, which results in low conversion yield/energy efficiency. To date, the yield of CO2-to-CO gas conversion has been reported in only one study (as 0.1%),13 while no data are available on the CO2 conversion yield/efficiency for organic matter production in the CO2/H2O system, as described earlier. Moreover, a long reaction time is necessary for high-dose irradiation, and the repeating irradiation results in product decomposition by radiation and the radicals.
A similar decomposition reaction has also been known in the study of wastewater treatment by an electron beam (EB),14 that is, an application using the character as the decomposition method. Therefore, chemical production processes using GR/EB have not been applied in the energy/chemical industry. However, in the case of the EB, irradiation of up to 100 kGy s−1 is currently possible. That is, such a high dose rate of EB irradiation in the presence of an organic long-lived reactive radical15 is expected to allow simple reaction mechanisms caused by decreasing the irradiation times to produce energy substances/basic chemicals from CO2 through radical trapping.
We propose herein a CO2 conversion approach with an organic additive by high-dose rate EB (Fig. 1a). This can be performed metal-free in one step within a few seconds under normal atmospheric conditions. Under a high-dose rate EB radiation in the presence of a radical source additive, the generated H2, CO and additive radicals form an organic matter. To the best of our knowledge, no previous study has reported CO2 conversion with organic additives by high-dose rate EB.
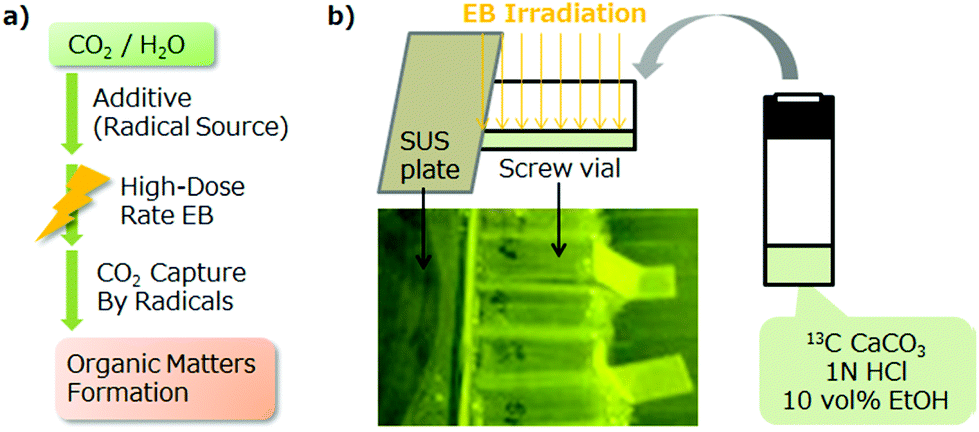 | ||
| Fig. 1 (a) Proposed CO2 conversion approach by high-dose rate EB irradiation with the organic additive. (b) EB irradiation overview of the glass vials used in the experiments. | ||
The present study uses 13C-CaCO3, 1 N HCl and additive EtOH (natural isotope ratio, 13C/12C <0.01) as the starting materials to identify the carbon origin of products. 13C-CaCO3 and 1 N HCl are used not for the CO2 sources, but for experimental convenience and reproducibility of the CO2 concentration in the irradiation experiments. EtOH is considered as the simplest structure model of biomass because the OH and ethyl groups are the partial structure of biomass components, such as lignin, cellulose, sugars, starches, oils, and proteins, known as radical sources under the radiation irradiation conditions.7 Therefore, various biomasses would have potential as organic additives to capture CO2. EtOH is also a better H2 generator than water;18 hence, the acceleration of the CO2 reduction/trapping reaction by the H2 generated is also expected, except for CO2 trapping caused by alkyl radicals formed by EB irradiation from EtOH. The products and origins are studied based on organic analysis data (nuclear magnetic resonance (NMR) spectroscopy, UV spectroscopy, capillary electrophoresis (CE), and gas chromatography-mass spectroscopy (GC-MS)). In addition, the conversion yield/energy efficiency and the energy input for 100% conversion yield (GJ per t-CO2) are estimated to assess the potential for use in sustainable energy technology and novel reaction development.
Results and discussion
We set up the EB irradiation reaction system as shown in Fig. 1b. In a 7 mL screw cap vial with a septum for an analysis syringe, 50 mg (0.5 mmol) of 13C-CaCO3 was dissolved in 1 mL of 1 N HCl (1 mmol) corresponding to 12.2 mL 13C-CO2 gas at SATP (standard ambient temperature and pressure). EtOH (0.1 mL, 1.72 mmol) was added, and the sample was irradiated with a 3 MeV EB at desired doses/dose rates in a polyethylene (PE)-sealed pack. We also planned to use sealed glass tubes; however, we cancelled this to avoid the risk of explosion from the inner pressure increase with the temperature increase. The reaction temperature was checked using a temperature label (range: 50–120 °C) placed on the vial surface.The power (3 MeV) of the EB was decided by our actual dose–depth curve data that EB can reach in 100% relative dose at a water depth of approximately 7 mm. In the preliminary experiment using 800 keV EB at 100 kGy (10 kGy s−1 ×10), the EB seemed to be almost stopped by a glass vial with 1 mm thickness, and the products had a trace amount.§ The screw cap was shielded by a steel use stainless (SUS) plate during irradiation for reproducibility and the prevention of contamination from the cap material.
Reproducibility was confirmed by performing three replicates of preliminary experiments (CaCO3/1 N HCl/EtOH at 100 kGy s−1 ×1, 3 MeV EB, n = 3) for irradiation method establishment. After EB irradiation, the obtained gas and aqueous phases were analysed using GC with thermal conductivity detector/mass spectrometry (GC-TCD/MS) and CE, respectively (Fig. 2, Table S1†). H2, CO2, 12C-CO, 13C-CO, 12C-CH4 and 12C-C2H6 were detected from the gas phase, whilst formic and acetic acids were obtained from the aqueous phase. Each content concentration almost had the same value in the three experiments. The analysis results indicated that the present experiment set-up has enough reproducibility.
 | ||
| Fig. 2 Reproducibility confirmation under the same conditions (CaCO3/1 N HCl/EtOH, 100 kGy s−1 ×1, n = 3). Bar graph with the error bar of (a) H2 and CO, (b) 12C-CH4, 13C-CO and 12C-CO, (c) 13C-CH4 and 12C-C2H6, (d) formate and acetate. | ||
We then examined the irradiation experiments at different doses (25, 100 and 300 kGy) and dose rates (25 kGy s−1 for 25/100 kGy and 100 kGy s−1 for 100/300 kGy). Given that a temperature of roughly 80 °C was employed in previous 100 kGy irradiation experiments, the maximum irradiation dose was limited to 300 kGy. Several samples (e.g., EtOH aqueous solution, CaCl2 aqueous solution and CO2 bubbling solution) were prepared as references. The reference samples were irradiated at only 100 kGy (100 kGy s−1 ×1).
The temperatures recorded after irradiation were roughly less than 50 °C for 25 kGy (25 kGy s−1 ×1), 100 kGy (25 kGy s−1 ×4), 80 °C for 100 kGy (100 kGy s−1 ×1) and 120 °C for 300 kGy (100 kGy s−1 ×3). These values indicate that the glass vials with small specific heat capacities were heated by EB and might have led to high temperature- or pressure-accelerated reactions. Moreover, the above-mentioned temperature was not directly proportional to the dose rate. It took approximately 1 min for a 1 s irradiation. Therefore, in repeating irradiation, the sample vials were cooled by air among the interval time between the first and next irradiation.
Fig. 3 shows the GC analysis results, revealing that H2, CO2, 12C-CO, 13C-CO, 12C-CH4, and 12C-C2H6 were detected (Fig. S1†). Notably, 13C-C2H6 could not be clearly detected. Product yields increased until 100 kGy; however, the yield at 300 kGy was smaller than that at 100 kGy. The yield of organic acids at 300 kGy significantly exceeded that at 100 kGy, as will be described later. Therefore, gaseous products formed at 300 kGy were used for organic acid formation. The organic matter with a 13C/12C isotope ratio higher than that of the corresponding natural one can be regarded as the products derived partly from 13C-CaCO3. The 13C/12C isotope ratio for CO was larger than the natural ratio (0.01%), whereas those of CH4 were similar to the natural 0.01% (Table S2,† entries 1–4). Consequently, 13C-CO can be deduced to have originated from 13C-CaCO3, whereas the 12C-CO and 12C-hydrocarbons were products originating from EtOH.
 | ||
| Fig. 3 (a) GC analysis results of the irradiated samples for H2 and CO2. (b) GC-MS analysis results for CO, CH4 and C2H6. | ||
Regarding the origin of the major product (H2), the relative concentration of H2 produced by H2O decomposition was under 1% (Table S2,† entries R1, R5, and R7), while that of hydrogen produced by decomposition of EtOH equalled 6–8% (Table S2,† entries R2–4 and R6). Therefore, EtOH decomposition can be considered to be the major origin of H2, as expected. The CO, CH4 and C2H6 concentrations at 100 kGy s−1 were higher than those at 25 kGy s−1, indicating an increase in the radical species generated. The other gas contents would be mainly N2, O2, H2O or EtOH, although we do not have analytical data on these. Interestingly, the 13C/12C isotope ratio of CH4 produced in the reaction with 13C-CO2 as the reference (Table S2,† entries R5 and R7) was over 0.01%, which suggested that CH4 was formed by the reduction of 13C-CO (produced from 13C-CO2) with H2 formed from EtOH. Detectability depends on the total amount of 13C atoms in the molecule and not only the isotope ratio. This will further be validated in a future study.
The change of the chemical species in the aqueous phase was monitored using the 13C NMR and UV spectra. NMR spectra were recorded using 128 scans. At this number of scans, no 13C carbon signals of natural products (except for the solvent) are typically observed, i.e., only high-isotope-ratio compounds are detected. Consequently, the 13C peaks observed under these conditions would be derived from 13CO2. For the aqueous phase, the peaks of carboxylic acids (166 and 178 ppm) as the main product were observed (Fig. 4a), which increased with the dose increase. In the same dose of 100 kGy, the peaks at 100 kGy s−1 were also higher than those at 25 kGy s−1. Other species, such as aldehydes (180–200 ppm) and ketones (190–220 ppm), were not observed. Regarding carbonate ions, we could not obtain the data for CaCO3 because of the significantly low solubility; however, the peak for Ca(HCO3)2 is 161.19 ppm, and such a peak was not detected. Overall, the spectra were simple and met our expectation that the high-dose rate irradiation will accelerate and simplify the reaction. In the reference samples, these carboxylic acid peaks were only observed when both CO2 and EtOH were present (Fig. S2,† entries R4 and R6).
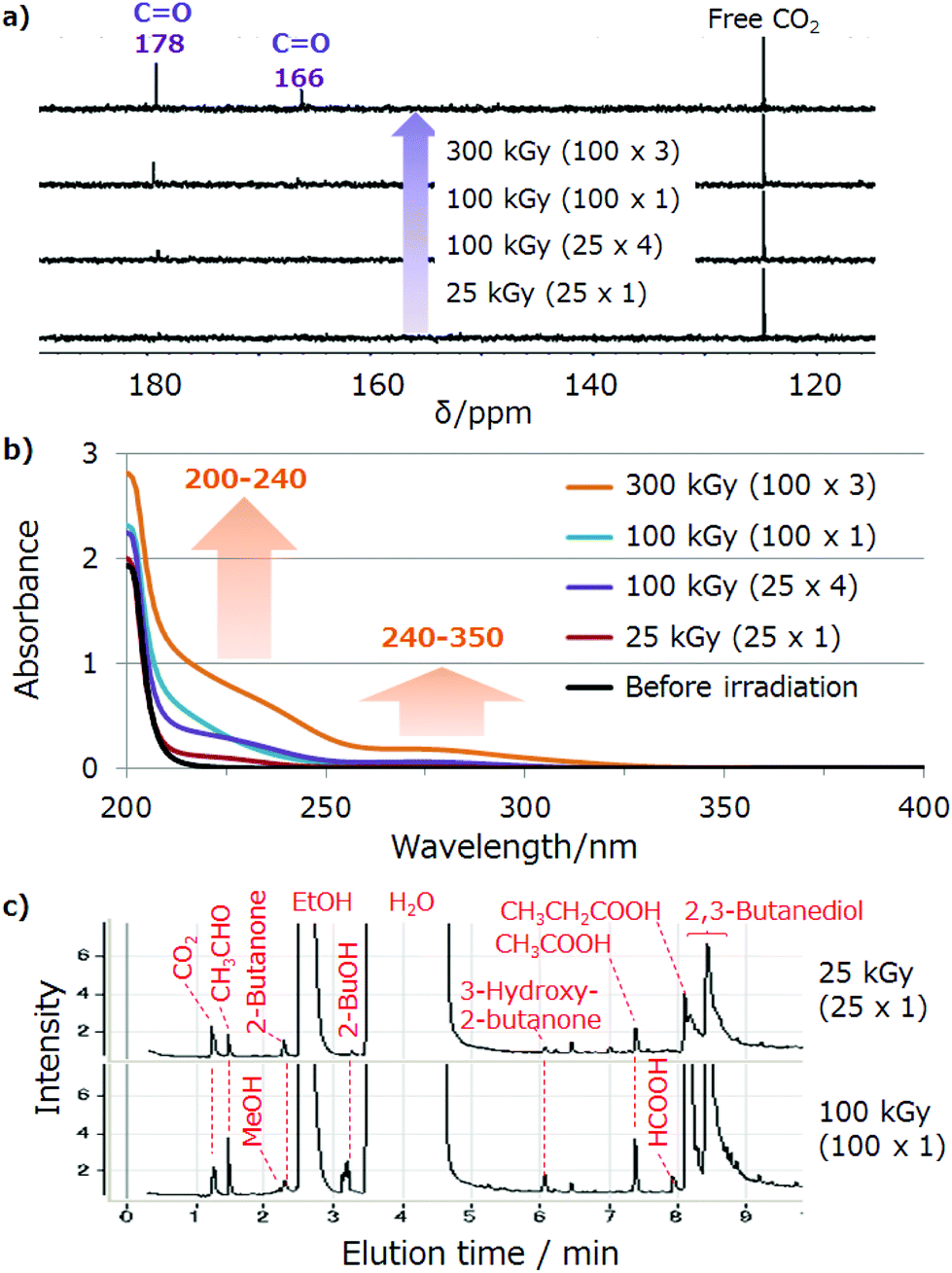 | ||
| Fig. 4 (a) Dose-dependent 13C NMR spectra (400 MHz, in D2O, scan: 128 times). (b) UV spectra of the aqueous phase after EB irradiation. (c) GC-MS chart of the selected aqueous phase (25 and 100 kGy). Reaction solution: CaCO3 50 mg/1 N HCl 1 mL/10 vol% EtOH. NMR sample: 0.4 mL irradiated solution/0.2 mL D2O mixture. UV sample: 0.9 mL irradiated solution/0.9 mL H2O mixture. | ||
Similarly, the UV spectra also changed with increasing dose (Fig. 4b). Absorbances corresponding to carboxylic acids (200–240 nm) and aldehydes/ketones (240–350 nm) were observed, with the major absorbance for formate, acetate and propionate lying under ∼240 nm (Fig. S3†). Peaks at 166 and 178 ppm in the 13C NMR spectra were ascribed to formate and acetate, respectively.
Product speciation was further clarified by GC-MS analysis of the aqueous phase (Fig. 4c and S3†). The major product was identified as 2,3-butanediol, and its formation was ascribed to the previously described dimerisation of excess EtOH (Fig. S4a†).19 Therefore, the UV absorbance change was mainly attributed to 2,3-butanediol formation (Fig. S5†). Acetaldehyde, MeOH, 2-butanone, 2-BuOH, 3-hydroxy-2-butanone and organic (formic, acetic and propanoic) acids were detected as minor products (Fig. 4c). Notably, HCHO was not detected, and MeOH was the only 13C-enriched product (Fig. S4b†). Although no peaks other than those of carboxylic acids were observed in the 13C NMR spectra, these spectra were believed to feature a peak of 13C-MeOH overlapped with that of EtOH. Therefore, non-carboxylic-acid species except for 13C-MeOH were concluded to originate from EtOH and products of its subsequent transformation such as 2,3-butanediol. Quantitative GC-MS and LC-MS analyses of products were hard to carry out because the solution to be analysed contained large amounts of CaCl2. Then, the concentration of the organic acids, including all the isotopes in the aqueous phases, was analysed by using CE for the selected samples (Table S2,† entries 1, 3 and 4).
Formic, acetic and propionic acids were detected as the primary components in the aqueous phase (Table S3 and Fig. S6†). Furthermore, the aqueous phase was subjected to methyl esterification (methyl group = 12C), and the distribution of carbon isotopes in the thus obtained products was measured using GC-MS. As a result, the 13C isomer as formic acid, 12–13C and 13–13C isomers as acetic acid and 12–12–13C and 12–13–13C isomers as propionic acid were observed (Fig. 5). The 13C/12C isotope ratio of the obtained organic acids was clearly larger than their natural abundance (Table S4†). These results also indicate that CO2 generated by acid decomposition or CO generated by the radiolysis of CO2 reacted with the radical species from EtOH.
 | ||
| Fig. 5 Typical GC-MS result for the irradiated sample (300 kGy). (a) Chromatogram chart after methyl esterification (methyl group = 12C): red line: m/z = 61 (13C-HCOOMe and 12C-COOMe ion); blue line; m/z = 74 (12–12C-CH3COOMe); and green line: m/z = 89 (12–12–13C-CH3CH2COOMe). MS spectra of (b) HCOOMe, (c) CH3COOMe and (d) CH3CH2COOMe. | ||
Fig. 6 shows the plot of the concentration of each 13C product estimated using CE (Table S5†) and the GC-MS isotope ratio for each dose. The organic acid concentrations increased with an increase in the radiation dose. The formation of 13–13C-acetic acid and 12–13–13C-propionic acid suggests methyl radical formation from 13C-MeOH. The concentration of propionic acid at 300 kGy was larger than that of acetic acid, indicating the relative stability of alkyl radicals (ethyl > methyl) and the chain reaction from acetic acid.
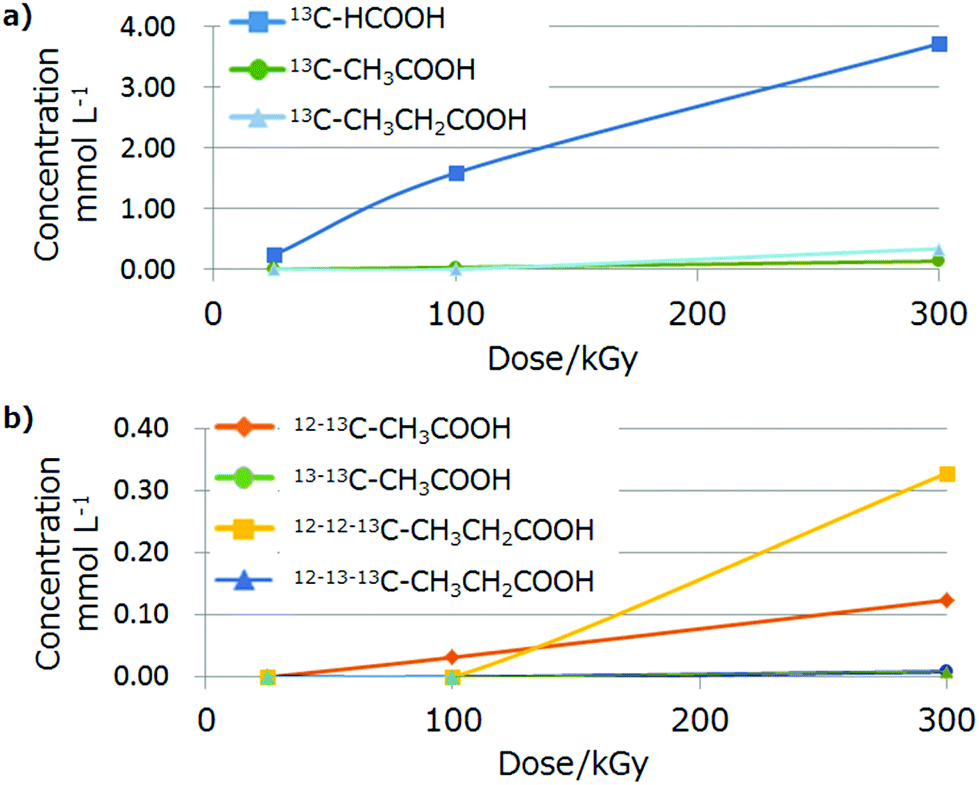 | ||
| Fig. 6 Dose-dependency of the 13C organic acid concentration estimated by CE and GC-MS analyses. (a) Total concentration of each 13C compound and (b) isotope ratio for acetic or propionic acids. Dose (dose rate × pass): 25 (25 × 1), 100 (100 × 1) and 300 (100 × 3) kGy. | ||
Fig. 7 summarises the formation mechanisms of products in the gas phase and 13C-products in the aqueous phase deduced from the above observations and known reactions. EB irradiation of CO2 produces CO,9,13 whereas that of EtOH produces hydrogen as well as hydroxy and hydrocarbon radical species,16 which can form H2, CH4 and C2H6 in the gas phase. In the aqueous phase, CO is converted into formic acid and MeOH. 12C-Methyl radicals produced from EtOH react with CO2 to form 12–13C-acetic acid, and the corresponding reaction of ethyl radicals yields 12–12–13C-propionic acid. Alternatively, 12C-methyl radicals can react with 13C-formic or 12–13C-acetic acids. Furthermore, 13C-methyl radicals derived from 13C-MeOH afford 13–13C-acetic acid by reacting with CO2, while the reaction of 12C-methyl radicals with 13–13C-acetic acid yields 12–13–13C-propionic acid.
 | ||
| Fig. 7 Possible formation mechanisms of products in the gas phase and 13C-products in the aqueous phase. Undetected intermediates are enclosed within brackets. e− denotes EB, and red bold C denotes the 13C carbon of 13C-CO2. | ||
Fig. 8 shows the proposed formation mechanisms of 12C-products detected by direct GC-MS analysis of the aqueous phase. Briefly, oxidation of EtOH by OH radicals or H2O2 affords acetaldehyde and acetic acid, while EB irradiation of EtOH affords 1-hydroxyethyl radicals that undergo dimerisation to yield 2,3-butanediol.17 Partial oxidation of 2,3-butanediol gives 2-hydroxy-3-butanone, and EB irradiation-mediated decomposition of the latter affords 2-butanone. It is worth noting that 2-butanone can also be produced by the oxidation of 2-BuOH formed by the EB irradiation-mediated decomposition of 2,3-butanediol. MeOH is produced through the reaction of methyl radicals with OH radicals and is further oxidised to give HCOOH. Finally, ethyl radicals react with HCOOH under EB irradiation to give propanoic acid.
 | ||
| Fig. 8 Possible formation mechanisms of 12C-products detected by GC-MS analysis of the aqueous phase. Undetected intermediates are enclosed by brackets. e− denotes EB-derived electrons, while [O] denotes OH radicals or H2O2. | ||
As seen in the organic compound decomposition in wastewater by EB-induced OH radical and carbonate ion,14 such a decomposition reaction might competitively exist. Moreover, the reaction temperature is considered as one of the factors in the present reaction system. In addition, the interaction with the Cl ion/radical may occur in the aqueous phase as the acceleration of H2 production by Br/Cl ions from seawater has been reported.18 In the photo-induced radical reaction of CO with alcohol and alkyl halide,19 the alkyl radical formed from the alkyl halide, whilst the present EB reaction directly provided the alkyl/hydroxyalkyl radical from EtOH. Although the presence of numerous species made the understanding of the reaction mechanism complicated, almost all products were identified, and a plausible reaction mechanism was suggested. In our future work, we plan to confirm the formation of radicals (not detected herein) by ESR measurements and pulse radiolysis experiments and clarify the origin of hydrogen/oxygen atoms by performing isotope labelling experiments (e.g., by replacing H2O with D2O and 18O-H2O or EtOH with C2D6OD and 18O-EtOH, respectively).
The conversion yield/energy efficiency was estimated to assess the potential for use in sustainable energy and novel reaction development. The conversion yields for CO and organic acids were estimated based on initial CO2 and each product amount in the unit of mol, whilst those of others were obtained from the initial EtOH amount.
Energy conversion efficiency (EE) was estimated based on the heat of combustion (HC, kJ mol−1) of each product and the electric power (kJ mol−1) per 1 vial at 300 kGy irradiation of the EB irradiation equipment as follows:
| EE (%) = product energy (J)/input electrical energy (J) × 100 |
| Product energy (J) = molar amount of product × C (kJ mol−1) |
The above input electrical energy, that is, the electric power per vial, was calculated as ∼327 J from the specification of the EB irradiation equipment (scanning width: 180 cm, scanning speed: 100 cm min−1 and electric power: 150 kWh).
The above EE was the calculation based on the product energy and electric energy for whole contents, including H2O and CaCl2, except for CO2/EtOH. As a radiation reaction, the conversion efficiency of radiation energy (RE) for only CO2 or EtOH is interesting matters from a fundamental viewpoint. The dose can be converted into energy using the relationship of 10 kGy(kJ kg−1) = 10 kJ. The sample weight was 11.6 g; thus, the RE is estimated to be 11.6 J per 10 kGy. RE is absorbed by the entire target molecule. As the absorbed RE should be proportional to the electron number (EN, i.e. H2O:18 electrons), only the RE for CO2, that is, net RE of CO2 can be estimated by calculating all electrons and the electron ratio from the molar amount. The net RE for CO2 should be smaller than the total RE. The energy loss of 12% by the glass vial was applied for RE before calculating REE. The radiation energy conversion efficiency (REE) was estimated using the above product energy and net RE for CO2 or EtOH considering the electron number ratio in the starting material (H2O, EtOH, CaCl2 and CO2) as follows:
| REE (%) = product energy (J)/net RE (J) × 100 |
| Net radiation energy (J) = RE (J) × (CO2 EN/total EN) |
| RE (J) = total irradiation dose (300 kGy)/10 × 11.6 |
Thus, the EE and the REE of CO and organic acids were estimated from CO2, whilst those of others were from EtOH. Both energy losses caused by the PE pack or glass vial and energy addition caused by reflecting EB from the sample set board or other sample surfaces were omitted for simplification.
According to the above mathematical formulae, the present total conversion yield/EE at 300 kGy is 4.99%/6.22% (H2: 3.48/5.72, CO: 0.03/0.01%, formic acid: 1.31/0.29%, acetic acid: 0.05/0.04% and propionic acid: 0.12/0.16%). H2, CH4 and C2H6 were derived from EtOH; hence, the conversion yield/EE of CO2 alone (CO, formic acid, acetic acid, and propionic acid) is 1.51/0.50% in total. The only CO2 conversion yield of CO in the gas phase has been reported as 0.1%.13 In our case, that was 1.51%, indicating a 15 time larger conversion yield. Also, the CO2-to-CO conversion yield/EE of CO2 in bubbled aqueous solution as reported in a previous research (Table S2,† R7) was only 0.006/0.002%, i.e., ∼3.1 times lower than the values of 0.018/0.006% obtained at 100 kGy with EtOH (Table S2,† R6).
In contrast, the total REE (%) of CO2 or EtOH at 300 kGy irradiation is estimated as follows: 51.5% (CO: 0.90%, formic acid: 30.3%, acetic acid: 3.71% and propionic acid: 16.6%) and EtOH: 76.9% (H2: 76.4, CH4: 0.44 and C2H6: 0.06). The conversion efficiency for RE itself is moderate, further improvement and new reaction development are expected. Comparing the product concentration at 100 kGy (Table S2,† entries 3 and R1), in the absence of EtOH, the yield/efficiency is expected to be ∼15 times lower than the condition in the presence with EtOH. This finding indicated that CO2 conversion is accelerated by radical sources such as EtOH.
The energy input for 100% yield is estimated as 38000 GJ per t-CO2. Although we could not find other data, the value would be insufficient compared to the 1.3 GJ per t-CO2 in the CO2 recovery process by amine solution.20 Nevertheless, the present work is the first report assessing radiation-induced CO2 conversion yield/EE. Further improvement of reaction conditions and the experimental procedure as well as irradiation equipment development are expected to bring about further advances in this field. For instance, the EE becomes roughly three times by decreasing the electric power (50 kWh) if we only use the 500 keV EB equipment. Moreover, the present samples contained ∼90% H2O, which can absorb the corresponding RE. Consequently, one should be able to increase REE by decreasing H2O volume. Excess EtOH causes the occurrence of side reactions, which implies that the removal of this excess should improve the conversion yield, EE and REE by discouraging the occurrence of these reactions. Also, the use of H2 or hydrocarbon would become a reductant candidate to obtain a transformed FT process. Although the solubility of H2 gas in H2O is roughly 1/60 against that of CO2, the reaction of CO2 under H2 in H2O has not been known to date. We intend to study this in a future study for further analysis of reaction mechanism and conversion yield improvement. Thus, it is expected that the CO2 conversion yield, EE and REE can be maximised by optimising irradiation conditions and equipment.
Conclusions
This study was inspired by extraterrestrial organic matter formation and proposed a CO2 conversion approach based on high-dose rate EB irradiation of an acid-decomposed CaCO3/additive EtOH mixture. The proposed conversion reaction is a simple one-step process that can be performed without metal catalysts under normal atmospheric conditions within a few seconds. Except for the 2,3-butanediol formation from excess EtOH as the major product, the reaction employing 13C-CaCO3 and EtOH as starting materials produced a mixture of H2, CO, CH4, C2H6 and organic acids. CO and organic acids partially included 13C-carbon from 13C-CaCO3, whilst the others were products from EtOH. The high-dose rate EB provided increased CO2 conversion products compared to the low-dose rate EB. The total conversion yield/EE of CO2 at 300 kGy was obtained as 1.51/0.50% (CO: 0.03/0.01%, formic acid: 1.31/0.29%, acetic acid: 0.05/0.04% and propionic acid: 0.12/0.16%). The CO2 conversion yield is ∼15 times larger than that of only known CO2 gas radiolysis (0.1%, CO only). The total REE of CO2 at 300 kGy was 51.5% (CO: 0.90%, formic acid: 30.3%, acetic acid: 3.71% and propionic acid: 16.6%). The yield/efficiency in the presence of EtOH at 100 kGy was ∼15 times higher than that obtained in the absence of EtOH. The energy input for a 100% conversion yield was 38000 GJ per t-CO2. This value is still unacceptably high for the practical use of the suggested CO2 utilisation/conversion process. Nevertheless, the combination of high-dose rate EB irradiation with organic additives facilitated CO2 capture by radicals to obtain an improved CO2 conversion efficiency/yield. Selectivity control and yield enhancement are the next important issues to be addressed. In our future work on this topic, we plan to deal with mechanism analysis, process/system improvement, conversion yield/efficiency modification and new reaction exploration.
Experimental
Materials and methods
13C-CaCO3 (13C-99%) was purchased from Taiyo Nippon Sanso Co. 13C-CO2 (13C-99%) was from SHOKO Science Co. Ethanol (99.5%), 1 N HCl solution and water (ultrapure) were purchased from FUJIFILM Wako Pure Chemical Co. The screw vials with septum for a syringe (pierce vial CV-70, 7 mL, glass thickness 1 mm, cap: phenol resin, septum: silicon rubber, vial: borosilicate glass) and temperature label (THERMO LABEL 8E-50, temperature range: 50–120 °C) were purchased from AS ONE Co. All products were used as received without further purification or cleaning.The electron beam was irradiated by using a NHV Corporation EPS-3000 (scanning type, acceleration voltage: 3 MeV, EB current: 50 mA, scanning width: 180 cm, scanning speed: 100 cm min−1 and electric power: 150 kWh).
The 13C NMR spectra were measured with JEOL JMM-EX400. The UV spectra were obtained using JASCO V-570. CE analysis was performed using Agilent Technology 7100CE. GC-MS analysis was conducted using Shimadzu GC-2014 for H2 and CO2 and Agilent Technology 6890GC-5973MSD for CO, hydrocarbons, aqueous phase and organic acids.
General procedure for EB irradiation§
In a 7 mL screw cap vial with a temperature label and a septum for a syringe, 50 mg (0.5 mmol) of 13C-CaCO3 was dissolved in 1 mL of 1 N HCl (1 mmol) corresponding to 11.2 mL of CO2 gas. After CO2 generation, 0.1 mL (1.72 mmol) of 12C-EtOH was added into the resulting solution at the end of CO2 generation to avoid loss due to the CO2 bubble and the screw vial was closed with the cap with a septum. The final volume of the gas phase in the vial was 5.9 mL, corresponding to 0.53 mg (0.26 mmol) of CO2. CO2 in the aqueous phase was estimated as 0.15 mg (0.07 mmol) from solubility to water. Finally, the initial CO2 amount used as the starting material was estimated as 0.68 mg (0.33 mmol). The capped vial was sealed with a polyethylene pack (50 μm thickness) for safety. The sealed sample pack with the vial cap protected by an SUS plate was then irradiated with the EB at desired doses/dose rates under air at room temperature. Each analysis was performed after air cooling for more than 1 day.Product analysis procedure by GC and CE
The gas phase was analysed using GC (CO) and GC with a TCD detector (H2 and CO2). The aqueous phase was purified by filtration through a NANOSEP 3K OMEGA filter and then directly analysed by GC-MS without additional pre-treatment. Additionally, the irradiated solution was diluted using 10 mL of ultrapure water and then analysed using CE. Subsequently, 180 mg NaCl was added to 0.5 mL of the irradiated solution, then stirred for 2 min using Voltex at 2000 rpm. Next, 0.5 mL of acetonitrile was added to the solution and stirred for another 2 min. After centrifugation (2000 rpm, 5 min), 100 μL of the organic phase was added along with 10 μL of 0.2 M phenyltrimethylammonium hydroxide for methylation. This solution was processed through GC-MS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank T. Okazaki, K. Nakai, N. Hirai, T. Nishikimi, K. Nishida, T. Kano and Y. Noguchi of NHV Corporation for their kind cooperation in the EB irradiation experiments.Notes and references
- F. Fischer and H. Tropsch, Brennst.-Chem., 1926, 7, 97 Search PubMed; D. Leckel, Energy Fuels, 2009, 23, 2342 CrossRef CAS.
- (a) A. Sternberg and A. Bardow, ACS Sustainable Chem. Eng., 2016, 4, 4156 CrossRef CAS; (b) M. Roiaz, E. Monachino, C. Dri, M. Greiner, A. Knop-Gericke, R. Schlögl, G. Comelli and G. E. Vesselli, J. Am. Chem. Soc., 2016, 138, 4146 CrossRef CAS PubMed.
- (a) J. Leclaire and D. J. Heldebrant, Green Chem., 2018, 20, 5058 RSC; (b) N. Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982 RSC; (c) A. Goeppert, M. Czaun, J.-P. Jones, G. K. S. Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995 RSC; (d) R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884 RSC; (e) E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112 RSC.
- (a) W. M. Garrison, D. C. Morrison, J. G. Hamilton, A. A. Benson and M. Calvin, Science, 1951, 114, 416 CrossRef CAS PubMed; (b) C. Ponnamperuma, R. M. Lemmon, R. Mariner and M. Calvin, Proc. Natl. Acad. Sci. U. S. A., 1963, 49, 737 CrossRef CAS PubMed.
- (a) K. Kobayashi, T. Kaneko, T. Saito and T. Oshima, Origins Life Evol. Biospheres, 1998, 28, 155 CrossRef CAS; (b) P. Ehrenfreund and S. B. Charnley, Annu. Rev. Astron. Astrophys., 2000, 28, 427 CrossRef; (c) K. Yamada, Interstellar Molecules: Their Laboratory and Interstellar Habitat, Springer, Heidelberg, 2011 CrossRef; (d) S. Kwok, Organic Matter in the Universe, Wiley-VCH, Weinheim, 2011 CrossRef; (e) S. Vinatier, B. Schmitt, B. Bézard, P. Rannou, C. Dauphin, R. de Kok, D. E. Jennings and F. M. Flasar, Icarus, 2018, 310, 89 CrossRef CAS; (f) L. R. Dartnell, Astrobiology, 2011, 11, 551 CrossRef CAS PubMed; (g) F. Postberg, N. Khawaja, B. Abel, G. Choblet, C. R. Glein, M. S. Gudipati, B. L. Henderson, H.-W. Hsu, S. Kempf, F. Klenner, G. Moragas-Klostermeyer, B. Magee, L. Nölle, M. Perry, R. Reviol, J. Schmidt, R. Srama, F. Stolz, G. Tobie, M. Trieloff and J. H. Waite, Nature, 2018, 558, 564 CrossRef CAS PubMed.
- G. A. Olah, T. Mathew and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 6905 CrossRef CAS PubMed.
- (a) E. Collinson and A. J. Swallow, The Radiation Chemistry of Organic Substances, Pergamon Press, London, 1955, p. 471 Search PubMed; (b) C. D. Jonah, Radiation Chemistry: Present Status and Future Trends (Studies in Physical and Theoretical Chemistry), Elsevier, Amsterdam, 2001 Search PubMed; (c) B. I. Kharisov, Radiation Synthesis of Materials and Compounds, CRC Press, New York, 2013 Search PubMed.
- (a) S. Seino, A. Yamamoto, R. Fujimoto, K. Hashimoto, M. Katsura, S. Okuda and K. Okitsu, J. Nucl. Sci. Technol., 2001, 38, 633 CrossRef CAS; (b) C. Fourdrin, H. Aarrachi, C. Latrille, S. Esnouf, F. Bergaya and S. LeCaër, Environ. Sci. Technol., 2013, 47, 9530 CrossRef CAS PubMed; (c) R. Yamada and Y. Kumagai, Int. J. Hydrogen Energy, 2012, 37, 13272 CrossRef CAS.
- X.-Z. Wu, M. Hatashita, Y. Enokido and H. Kakihana, Chem. Lett., 2000, 572 CrossRef CAS.
- (a) M. Oshima, K. Kitamura, A. Tani, T. Sugahara and K. Ohgaki, J. Phys. Chem. B, 2014, 118, 13435 CrossRef CAS PubMed; (b) Z. Cai, X. Li, Y. Katsumura and O. Urabe, Nucl. Technol., 2001, 136, 231 CrossRef CAS.
- G. Albarrfin, K. E. Collins and C. H. Collins, J. Mol. Evol., 1987, 25, 12 CrossRef.
- N. Getoff, Int. J. Hydrogen Energy, 1994, 19, 667 CrossRef CAS.
- P. Hartech and S. Dondes, J. Chem. Phys., 1957, 26, 1727 CrossRef.
- (a) M. G. Nickelsen, W. J. Cooper, K. Lin, C. N. Kurucz and T. D. Waite, Water Res., 1994, 28, 1227 CrossRef CAS; (b) D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Water Res., 2018, 134, 118 CrossRef PubMed.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed.
- E. Collinson and A. J. Swallow, The Radiation Chemistry of Organic Substances, Pergamon Press, London, 1955, p. 492 Search PubMed.
- J. T. Allan, E. Hayon and J. J. Weiss, J. Chem. Soc., 1959, 3913 RSC.
- Y. Kumagai, A. Kimura, M. Taguchi, R. Nagaishi, I. Yamagishi and T. Kimura, J. Nucl. Sci. Technol., 2013, 50, 138 Search PubMed.
- K. Nagahara, I. Ryu, M. Komatsu and N. Sonoda, J. Am. Chem. Soc., 1997, 119, 5465 CrossRef CAS.
- S. Yamamoto, H. Machida, Y. Fujioka and T. Higashii, Energy Procedia, 2013, 37, 505 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, CE, MS, UV analysis data (pdf). See DOI: 10.1039/c9gc00525k |
| ‡ Present address: Research Institute for Sustainable Humanosphere Kyoto University Uji, Kyoto 611-0011, Japan. |
| § The product yield is expected to depend on the reaction vessel material and the thickness used. According to the web page on the stopping power for electrons of National Institute of Standards and Technology (NIST) USA (https://physics.nist.gov/PhysRefData/Star/Text/ESTAR.html), for example, in the case of the present vial (borosilicate glass, 1 mm thickness), the EB energy loss (absorption by vial) at 3 MeV EB irradiation is calculated as approximately 12%. |
| This journal is © The Royal Society of Chemistry 2019 |