
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple ketone as an efficient metal-free catalyst for visible-light-mediated Diels–Alder and aza-Diels–Alder reactions†
Jiri
Kollmann‡
,
Yu
Zhang‡
,
Waldemar
Schilling
,
Tong
Zhang
,
Daniel
Riemer
and
Shoubhik
Das
 *
*
Institut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: Shoubhik.Das@chemie.uni-goettingen.de
First published on 8th March 2019
Abstract
Diels–Alder reactions are highly effective between electron-rich dienes and electron-poor dienophiles. However, these reactions with electron-rich dienophiles are limited and require forcing conditions. Based on this, an efficient metal-free homogeneous system has been developed for the Diels–Alder reactions between electron-rich dienophiles and dienes under visible-light conditions. Additionally, this catalyst has shown excellent reactivity for aza-Diels–Alder reactions. This simple catalyst is commercially available, non-toxic, and cheap and has shown excellent reactivity which is comparable to and in some cases even better than the reported metal-based catalysts. Finally, the mechanism of this reaction has been proposed based on the experimental evidence.
Introduction
Diels–Alder (D–A) reactions are the fundamental strategy for the syntheses of versatile cyclohexene derivatives with the formation of new stereogenic centers.1 These reactions have been well-documented in organic syntheses, in the pharmaceutical industry and in biochemistry.2 It should be noted that D–A reactions are highly effective between electron-rich dienes and electron-poor dienophiles, which has been explained based on frontier molecular orbital (FMO) theory.3a To enhance the conversion as well as the stereoselectivity of thermally initiated organocatalytic Diels–Alder reactions, different strategies have been developed. Evans’ group established oxazolidinones as chiral organic auxiliaries which sterically hindered the attack of the diene on one face of the dienophile.3b MacMillan and co-workers developed imidazolidinones as highly enantioselective chiral organocatalysts which form iminium species with enones. The iminium species lower the LUMO of the dienophile and sterically block one face from being attacked by the diene.3c However, electronically mismatched coupling between dienes and dienophiles is highly challenging and requires strict conditions.1,2 These electronically mismatched D–A reactions can be achieved by the formation of radical cation intermediates from dienophiles via one electron oxidation by using stoichiometric oxidants such as aminium salts or by photoinitiated electron transfer (PET) with photosensitizers.4,5In contrast, visible-light-mediated radical cation generation from the electron-rich dienophile is more sustainable and does not require any stoichiometric oxidant (Fig. 1).6 Based on this, Yoon and co-workers developed a Ru complex which showed excellent reactivity towards electron-rich dienophiles under visible-light conditions.7 In this case the radical cation was generated by the Ru catalyst with the aid of visible-light and air/oxygen was utilized to turn over the catalyst. In fact, they also investigated metal-free photocatalysts, however their efforts were not successful.7a Then, Shores’ group and Ferreira's group developed Cr complexes under UV light irradiation.8 However, the Cr catalyst required a longer reaction time and an excess of diene (10 equiv.) to achieve full conversion.8b
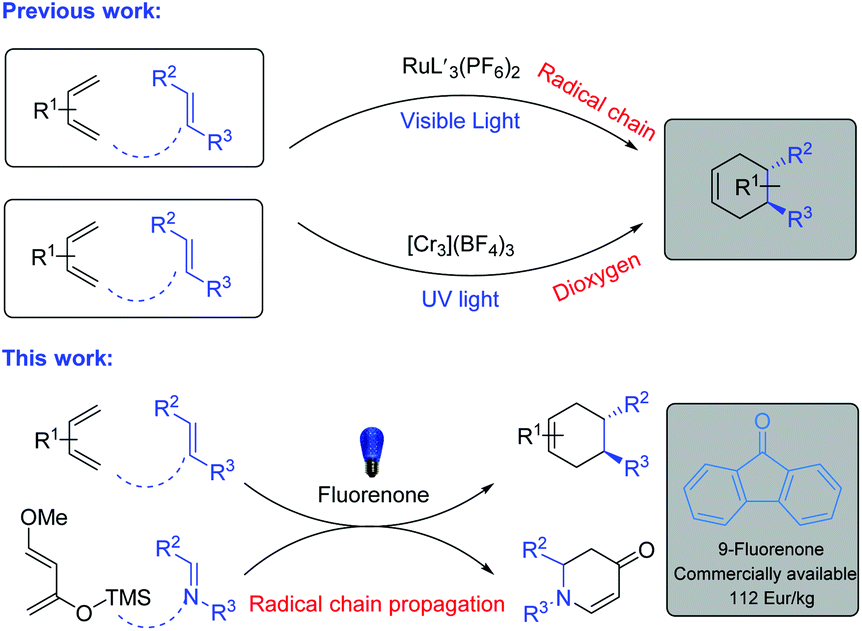 | ||
| Fig. 1 Homogeneous catalysts for photocatalytic D–A reactions of electron-rich dienophiles. | ||
Parallel to the D–A reactions, aza-Diels–Alder reactions are also efficient and powerful approaches for the syntheses of N-heterocycles in a straightforward way.9 With the same concept of the generation of radical cation intermediates from electron-rich dienophiles, iminium ions can be generated from imines with the aid of visible light.6 Based on this, Zhang, Arai and Ohkuma groups developed visible-light-promoted aza-Diels–Alder reactions with Ru and Cr catalysts.10 It should be noted that in all the cases of visible-light-mediated D–A or aza-D–A reactions, only transition metal based photocatalysts exist. In contrast, organic dyes are commercially available, cheaper in price, non-toxic in nature and are an interesting avenue to explore.11 Based on all the above information and in continuation of our interest in metal-free catalysis, herein we describe an efficient metal-free homogeneous catalyst for visible-light-mediated Diels–Alder and aza-Diels–Alder reactions.12
Results and discussion
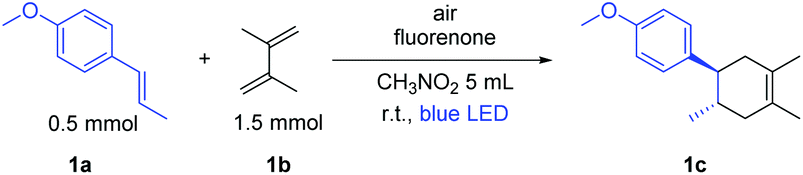
At the outset of our reaction, we focused on organic dyes which have a high oxidation potential (e.g. in the case of fluorenone +1.7 V vs. SCE)11a to generate radical cations from trans-anethole (1a) (+1.1 V vs. SCE).7a We started the optimization using trans-anethole (1a) as a dienophile and 2,3-dimethyl-1,3-butadiene (1b) as a diene under air (Table S1; see the ESI†). Eight different organic dyes were applied using nitromethane (MeNO2) as the solvent and MgSO4 as the desiccant under the irradiation of 12 W blue LED for 4 h. Among them, fluorenone showed 81% yield of the corresponding Diels–Alder product. Subsequently, other aprotic polar solvents such as THF, DMF, DMSO and ACN were investigated but did not show any improvement of the yield. Nitromethane was the best solvent in this reaction as aprotic polar solvents with a higher dipole moment at comparable polarities can solvate the formed radical cation intermediate which enhances the product formation.13With these optimized reaction conditions in hand, we extended the scope of this metal-free system to other electron-rich dienophiles and dienes (Scheme 1; entries 1c–11c). The formation of [4 + 2] cycloadducts proceeded smoothly using 2,3-dimethyl-1,3-butadiene as a diene and other electron-rich dienophiles such as myrcene, isoprene etc. (Scheme 1; entries 2c–4c). In addition to these, 1,2-dimethoxy-4-propenylbenzene was also effectively explored as a dienophile with a series of dienes (Scheme 1; entries 5c–7c). Furthermore, 2,5-furandione was also suitable in our system and generated corresponding cyclohexene derivatives in excellent yields in 2 h (Scheme 1; entries 8c–9c). It should be noted that for 2,5-furandione, expensive metal-based catalysts are required to obtain the desired product.14 However, with our cheap and metal-free fluorenone catalyst, these cyclohexene derivatives can be achieved with excellent yields in only 2 h. Additionally, 9-vinylcarbazole was also capable of acting as a dienophile and gave 11c with 2,3-dimethyl-1,3-butadiene as a diene.
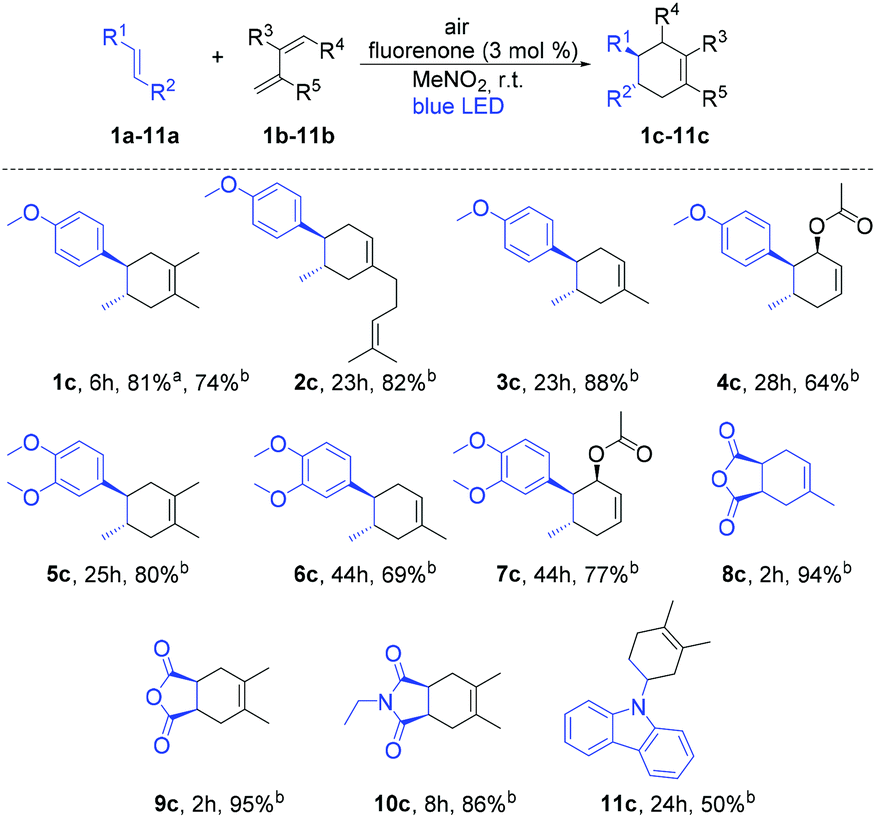 | ||
Scheme 1 Scope of the fluorenone-catalyzed Diels–Alder reactions under visible-light irradiation. Reaction conditions: Air, 12 W blue LED, dienophiles (0.5 mmol, 1 eq.), dienes (3 eq.), photocatalyst (3 mol%), 5 mL solvent, room temperature. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yield was determined by NMR yield using iodoform as an internal standard. bIsolated yields. Yield was determined by NMR yield using iodoform as an internal standard. bIsolated yields. | ||
Inspired by the excellent reactivity of our catalyst in [4 + 2] cycloaddition reactions, we sought to apply these reaction conditions to aza-D–A reactions. Our main target was to synthesize important N-heterocycles in a straightforward way. For this purpose, we aimed to synthesize 2,3-dihydropyridin-4(1H)-one derivatives as these have wide applications for the treatment of emergent infectious diseases.15 Additionally, these compounds have been utilized for the syntheses of piperidine containing natural products and bioactive molecule synthesis.16 In general, syntheses of 2,3-dihydropyridin-4(1H)-one derivatives require either expensive metal based catalysts or a longer reaction time.17 Gratifyingly, fluorenone showed an excellent yield within 2 h when (E)-1-(4-methoxyphenyl)-N-phenylmethanimine (1aa) as a dienophile and Danishefsky's diene (1ab) were applied to synthesize 2-(4-methoxyphenyl)-1-phenyl-2,3-dihydropyridin-4(1H)-one (1ac) as the product (Table S2†). Different ratios of diene and dienophile were also investigated and to our delight, with ratio 2:1 the reaction proceeded smoothly with the formation of 96% of the desired product in 2 h.
With these optimized reaction conditions, we applied the fluorenone catalyst to other imines to obtain diverse 2,3-dihydropyridin-4(1H)-one derivatives (Scheme 2). Various electron-rich imines showed high reactivities with the formation of desired products in 2 h up to 96% yield. The catalyst showed high tolerance towards alkyl, aryl and heteroaryl substituents (Scheme 2; entries 2ac–10ac). Additionally, six new 2,3-dihydropyridin-4(1H)-one derivatives have been synthesized whose biological activities should be further investigated considering the promising lead compounds against human infectious diseases (Scheme 2; entries 3ac–4ac, 7ac–10ac).16 Expediently, with this fluorenone catalyst, we have been able to synthesize previously reported bioactive derivatives which were evaluated in vitro against a wide spectrum of viruses (Scheme 3; entries 1ac, 11ac–12ac).15a The scope of the fluorenone-catalyzed aza-Diels–Alder reaction was not only limited to the synthesis of 2,3-dihydropyridin-4(1H)-one derivatives but also applied to synthesize other important heterocycles (Scheme 4). For example, when the Danishefsky's diene was replaced by 1-methoxy-1,3-butadiene, it generated a 1,2,3,6-tetrahydropyridine derivative (Scheme 4; entry 13ac). In addition to this, 2,3-dimethyl-1,3-butadiene reacted with 1,4-benzoquinone to generate 3,4-dimethyl-1-oxaspiro[5.5]undeca-3,7,10-trien-9-one (Scheme 4; entry 14ac).
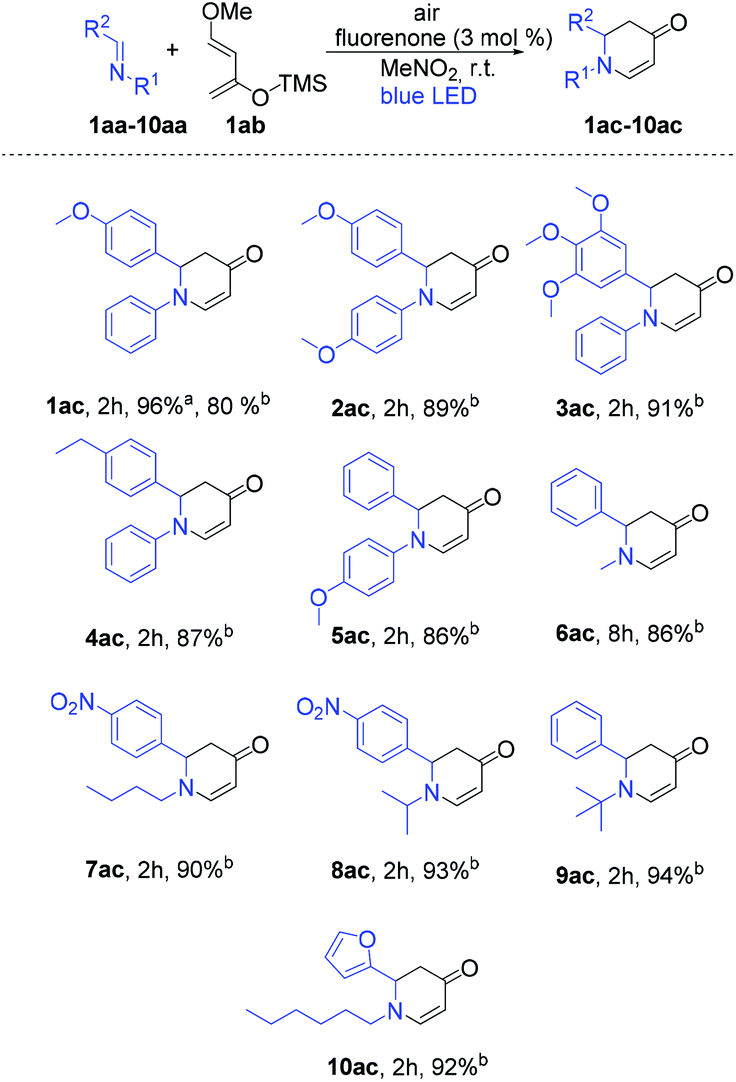 | ||
| Scheme 2 Scope of the fluorenone-catalyzed aza-Diels–Alder reactions under visible light irradiation. Reaction conditions: Air, 12 W blue LED, imines (0.10 mmol, 1 eq.), diene (2 eq.), photocatalyst (3 mol%), 5 mL solvent, room temperature. aYield was determined by NMR yield using iodoform as an internal standard. bIsolated yields. | ||
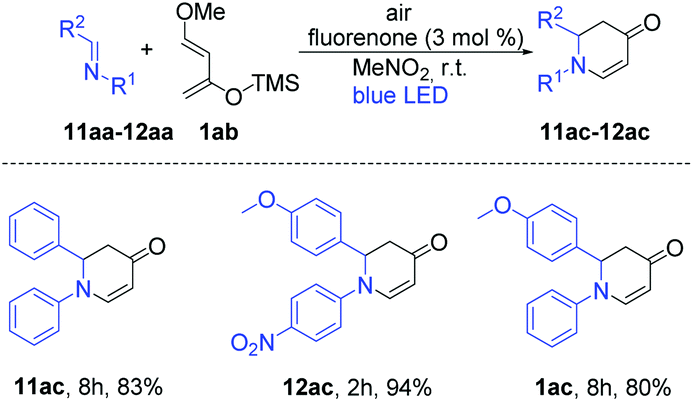 | ||
| Scheme 3 Syntheses of bioactive derivatives using the fluorenone catalyst. Reaction conditions: Air, 12 W blue LED, imines (0.10 mmol, 1 eq.), diene (2 eq.), photocatalyst (3 mol%), 5 mL solvent, room temperature. | ||
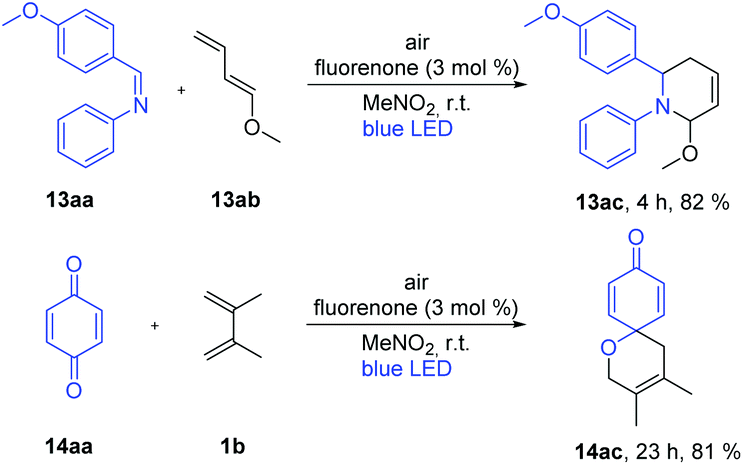 | ||
| Scheme 4 Fluorenone-catalyzed hetero-Diels–Alder reactions under visible light irradiation. Reaction conditions: Air, 12 W blue LED, imine or 1,4-benzoquinone (0.10 mmol, 1 eq.), dienes (2 eq.), photocatalyst (3 mol%), 5 mL solvent, room temperature. | ||
Considering the broad scope of this catalyst, we sought to enquire the role of the catalyst, air, and light source in this reaction. Control experiments showed no product formation in the absence of light or the photocatalyst (see the ESI†). Notably, 60% yield was achieved under nitrogen, which clearly indicates that there is a chain propagation mechanism similar to the earlier report on Ru complexes.7a Singlet oxygen in the air acts as an indispensable electron mediator which further increases the yield. Furthermore, the effect of different quenchers was investigated to recognize the reactive oxygen species from air and possible intermediates (Table 1).11c–e When 2,6-di-tert-butyl-4-methylphenol (BHT) or 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) were added to the reaction mixture, the reaction was completely inhibited, which proved a radical pathway. Furthermore, the addition of CuCl2 to the reaction mixture showed lower yields, which showed the involvement of single electron processes in this photocatalytic system. The addition of sodium azide also showed a decreased yield, indicating the involvement of singlet oxygen, or activated oxygen species.
|
|
|||
|---|---|---|---|
| Quencher | Equivalents | Yield [%] | Scavenger for |
| Reaction conditions: Air, 12 W blue LED, 0.5 mmol dienophiles (1 eq.), 1.5 mmol dienes (3 eq.), photocatalyst (3 mol%), 5 mL solvent, room temperature. | |||
| BHT | 1.0 | 0 | Radical |
| TEMPO | 1.0 | 0 | Radical |
| CuCl2 | 1.0 | 73 | Single electron |
| NaN3 | 1.0 | 63 | Singlet oxygen |
To acquire further information about the reaction mechanism, Stern–Volmer quenching experiments were carried out (see the ESI†). The excited state of the photocatalyst was quenched by trans-anethole.18 As shown in Fig. S3,† a clear decrease of light emission was observed with the increasing concentration of trans-anethole (1a) and no change was observed with different concentrations of 2,3-dimethyl-1,3-butadiene.
Combining all the mechanistic experiments, we have been able to propose the mechanism for visible-light-mediated D–A and aza-D–A reactions in Fig. 2. At first, the photocatalyst was excited to the photo-excited state by the irradiation of visible light and underwent a single electron transfer (SET) with trans-anethole (1a). In fact the redox potential value of the catalyst (+1.7 V vs. SCE)11a is enough to oxidize 1a (+1.1 V vs. SCE).7a The resulting radical cation 1a˙+ reacted with the diene via [4 + 2] cycloaddition to afford the radical cation product 1c˙+. This product 1c˙+ accepted one electron mainly from another equivalent of 1a in a chain propagation step to form the final product (1c).7a Meanwhile, oxygen will also be involved partly in electron donation. Here the role of air/oxygen was to turn over the reduced photocatalyst to the original state. The reported value for the reduction potential of the excited-state fluorenone resides at −0.61 V vs. SCE,11d which is sufficient for the reduction of molecular oxygen to its superoxide radical form (O2/O2˙−) with the reduction potential residing at −0.56 V vs. SCE.19
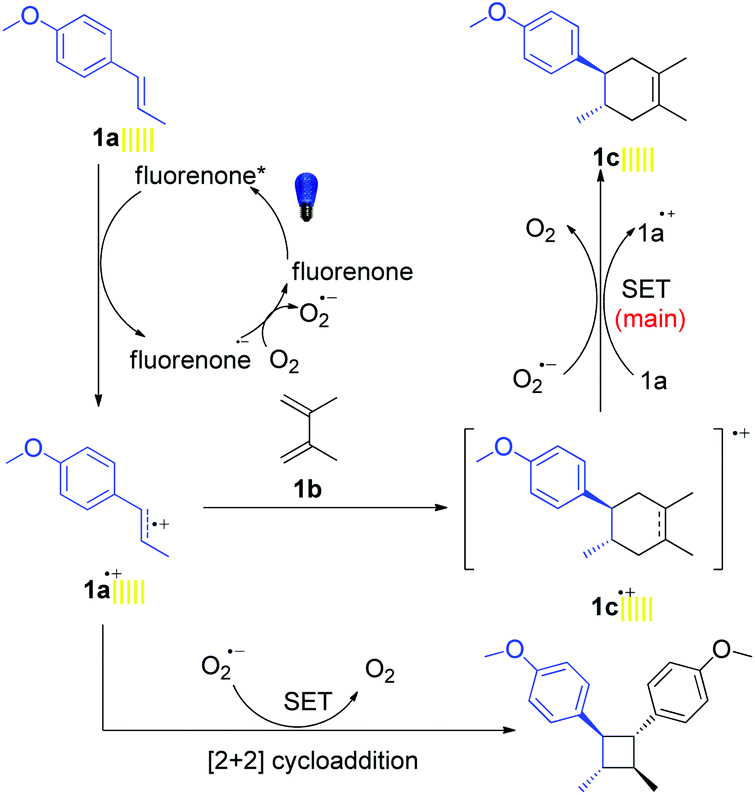 | ||
| Fig. 2 Proposed reaction mechanism for the fluorenone catalyzed Diels–Alder reactions. | ||
The [2 + 2] cycloaddition also proceeded well via SET with the product 1a˙+ in the absence of diene.20 In fact, cyclobutane derivatives have wide applications in natural product syntheses and many of them already have become promising candidates as anticancer, antiviral and antifungal drugs.20b Additionally, lignan and neolignan type compounds also contain a cyclobutane motif (Scheme 5a).20b Based on all this information, we applied the fluorenone catalyst to promote [2 + 2] cycloaddition reactions to obtain cyclobutane derivatives (Scheme 5b; entries 1d–2d). To our delight, the catalyst not only showed the activity for the homodimerization of alkenes (1d) but also showed an excellent reactivity towards the heterodimerization of alkene (2d).
 | ||
| Scheme 5 (a) Examples of bioactive cyclobutane lignan natural products. (b) Our work on [2 + 2] cycloaddition reactions. Reaction conditions: Air, 12 W blue LED, dienophiles (0.5 mmol, 1 eq.), dienes (3 eq.), photocatalyst (3 mol%), 5 mL solvent, room temperature. | ||
Conclusions
In summary, we have developed fluorenone as a metal-free catalyst for visible-light-mediated cycloaddition reactions. This catalyst is very cheap and commercially available and has shown excellent substrates scope for [4 + 2] and [2 + 2] cycloaddition reactions with electron-rich dienophiles. In addition, we have been able to synthesize bioactive molecules via aza-Diels Alder reactions. We believe this protocol can be further applied and extended for the syntheses of pharmaceuticals and natural products. Additionally, detailed mechanistic studies revealed the role of the catalyst and oxygen and led to the proposed mechanism of this reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Fonds der Chemischen Industrie (FCI, Liebig-Fellowship to S. D.) and Chinese Scholarship Council (CSC funding to Y. Z.) for the financial support. We are highly thankful to Prof. Dr Lutz Ackermann for his support behind our work.Notes and references
- (a) X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515 CrossRef CAS PubMed; (b) C. K. Nicolaou, A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef.
- (a) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2015, 115, 9922 CrossRef CAS PubMed; (b) T. Gatzenmeier, M. van Gemmeren, Y.-W. Xie, D. Höfler, M. Leutzsch and B. List, Science, 2016, 351, 949 CrossRef CAS PubMed; (c) G.-L. Li, T. Liang, L. Wojtas and J. C. Antilla, Angew. Chem., Int. Ed., 2013, 52, 4628 CrossRef CAS PubMed; (d) R. F. Higgins, S. M. Fatur, S. G. Shepard, S. M. Stevenson, D. J. Boston, E. M. Ferreira, N. H. Damrauer, A. K. Rappe and M. P. Shores, J. Am. Chem. Soc., 2016, 138, 5451 CrossRef CAS PubMed.
- (a) K. N. Houk, Acc. Chem. Res., 1975, 8, 361 CrossRef CAS; (b) D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1984, 106, 4261 CrossRef CAS; (c) K. A. Ahrendt, C. J. Borths and D. W. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- (a) D. J. Bellville, D. W. Wirth and N. L. Bauld, J. Am. Chem. Soc., 1981, 103, 718 CrossRef CAS; (b) N. L. Bauld, D. J. Bellville, R. Pabon, R. Chelsky and G. Green, J. Am. Chem. Soc., 1983, 105, 2378 CrossRef CAS; (c) D. J. Bellville, N. L. Bauld, R. Pabon and S. A. Gardner, J. Am. Chem. Soc., 1983, 105, 3584 CrossRef CAS.
- (a) C. R. Jones, B. J. Allman, A. Mooring and B. Saphic, J. Am. Chem. Soc., 1983, 105, 652 CrossRef CAS; (b) R. A. Pabon, D. J. Bellville and N. L. Bauld, J. Am. Chem. Soc., 1983, 105, 5158 CrossRef CAS; (c) G. C. Calhoun and G. B. Schuster, J. Am. Chem. Soc., 1984, 106, 6870 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) Y.-B. Zhao and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 9336 CrossRef CAS PubMed.
- (a) S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350 CrossRef CAS PubMed; (b) A. E. Hurtley, M. A. Cismesia, M. A. Ischay and T. P. Yoon, Tetrahedron, 2011, 67, 4442 CrossRef CAS PubMed.
- (a) S. M. Stevenson, R. F. Higgins, M. P. Shores and E. M. Ferreira, Chem. Sci., 2017, 8, 654 RSC; (b) S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506 CrossRef CAS PubMed; (c) R. F. Higgins, S. M. Fatur, S. G. Shepard, S. M. Stevenson, D. J. Boston, E. M. Ferreira, N. H. Damrauer, A. K. Rappe and M. P. Shores, J. Am. Chem. Soc., 2016, 138, 5451 CrossRef CAS PubMed.
- (a) J. M. Eagan, M. Hori, J.-B. Wu, K. S. Kanyiva and S. A. Snyder, Angew. Chem., Int. Ed., 2015, 54, 7842 CrossRef CAS PubMed; (b) G. Masson, C. Lalli, M. Benohouda and G. Dagousseta, Chem. Soc. Rev., 2013, 42, 902 RSC; (c) H. Mandai, K. Mandai, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 17961 CrossRef CAS PubMed.
- (a) N. Arai and T. Ohkuma, J. Org. Chem., 2017, 82, 7628 CrossRef CAS PubMed; (b) W.-H. Wang, Y. Yuan, X.-S. Gao, B. Hu, X.-M. Xie and Z.-G. Zhang, ChemCatChem, 2018, 10, 2878 CrossRef.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) M. Majek and A. J. Von Wangelin, Acc. Chem. Res., 2016, 49, 2316 CrossRef CAS PubMed; (c) Y. Zhang, D. Riemer, W. Schilling, J. Kollmann and S. Das, ACS Catal., 2018, 8, 6659 CrossRef CAS; (d) W. Schilling, D. Riemer, Y. Zhang, N. Hatami and S. Das, ACS Catal., 2018, 8, 5425 CrossRef CAS; (e) D. Riemer, W. Schilling, A. Goetz, Y. Zhang, S. Gehrke, I. Tkach, O. Holloczki and S. Das, ACS Catal., 2018, 8, 11679 CrossRef CAS; (f) M. Majek and A. J. Von Wangelin, Angew. Chem., Int. Ed., 2015, 54, 2270 CrossRef CAS PubMed.
- (a) D. Riemer, B. Mandaviya, W. Schilling, A. C. Götz, T. Kühl, M. Finger and S. Das, ACS Catal., 2018, 8, 3030 CrossRef CAS; (b) D. Riemer, P. Hirapara and S. Das, ChemSusChem, 2016, 9, 1916 CrossRef CAS PubMed; (c) P. Hirapara, D. Riemer, N. Hazra, J. Gajera, M. Finger and S. Das, Green Chem., 2017, 19, 5356 RSC.
- V. Santacroce, R. Duboc, M. Malacria, G. Maestri and G. Masson, Eur. J. Org. Chem., 2017, 2095 CrossRef CAS.
- (a) K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2003, 125, 11460 CrossRef CAS PubMed; (b) Y. Ogasawara, S. Uchida, K. Yamaguchi and N. Mizuno, Chem. – Eur. J., 2009, 15, 4343 CrossRef CAS PubMed.
- (a) A. Peduto, A. Massa, A. D. Mola, P. de Caprariis, P. L. Colla, R. Loddo, S. Altamura, G. Maga and R. Filosa, Chem. Biol. Drug Des., 2011, 77, 441 CrossRef CAS PubMed; (b) J. Sun, E.-Y. Xia, Q. Wu and C.-G. Yan, ACS Comb. Sci., 2011, 13, 421 CrossRef CAS PubMed; (c) A. R. Ranade and G. I. Georg, J. Org. Chem., 2014, 79, 984 CrossRef CAS PubMed; (d) S. Fustero, J. Miro, M. Sanchez-Rosello and C. Del Pozo, Chem. – Eur. J., 2014, 20, 14126 CrossRef CAS PubMed.
- (a) M. Ali, S. H. Ansari and J. S. Qadry, J. Nat. Prod., 1991, 54, 1271 CrossRef CAS; (b) J. Matsuo, R. Okado and H. A. Ishibashi, Org. Lett., 2010, 12, 3266 CrossRef CAS PubMed.
- (a) N. S. Josephsohn, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 4018 CrossRef CAS PubMed; (b) S. Kobayashi, S. Komiyama and H. Ishitani, Angew. Chem., Int. Ed., 1998, 37, 979 CrossRef CAS; (c) S. Kobayashi, M. Ueno, S. Saito, Y. Mizuki, H. Ishitani and Y. Yamashita, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5476 CrossRef CAS PubMed; (d) S. Kaneko, Y. Kumatabara, S. Shimizu, K. Maruoka and S. Shirakawa, Chem. Commun., 2017, 53, 119 RSC; (e) Y. Takeda, D. Hisakuni, C.-H. Lin and S. Minakata, Org. Lett., 2015, 17, 318 CrossRef CAS PubMed.
- D. M. Arias-Rotondoa and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803 RSC.
- W. Huang, B.-C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438 CrossRef CAS.
- (a) M. A. Ischay, M. E. Anzovino, J.-N. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886 CrossRef CAS PubMed; (b) M. Riener and D. A. Nicewicz, Chem. Sci., 2013, 4, 2625 RSC; (c) R. Li, B. C. Ma, W. Huang, L. Wang, D. Wang, H. Lu, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 3097 CrossRef CAS; (d) M. A. Ischay, M. S. Ament and T. P. Yoon, Chem. Sci., 2012, 3, 2807 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc00485h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |