
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction and optimization of microbial cell factories for sustainable production of bioactive dammarenediol-II glucosides†
Zong-Feng
Hu‡
 ,
An-Di
Gu‡
,
Lan
Liang
,
Yan
Li
,
Ting
Gong
,
Jing-Jing
Chen
,
Tian-Jiao
Chen
,
Jin-Ling
Yang
* and
Ping
Zhu
*
,
An-Di
Gu‡
,
Lan
Liang
,
Yan
Li
,
Ting
Gong
,
Jing-Jing
Chen
,
Tian-Jiao
Chen
,
Jin-Ling
Yang
* and
Ping
Zhu
*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines & NHC Key Laboratory of Biosynthesis of Natural Products, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China. E-mail: yangjl@imm.ac.cn; zhuping@imm.ac.cn
First published on 24th April 2019
Abstract
Ginsenosides, the predominant bioactive components of Panax species, are biosynthesized by glycosylation at C3–OH and/or C20–OH of protopanaxadiol (PPD), and C6–OH and/or C20–OH of protopanaxatriol (PPT). Dammarenediol-II (DM), the direct precursor of PPD, has two hydroxyls at C3 and C20 positions, but DM glucosides have scarcely been identified from Panax species. Herein, we used two crude recombinant UDP-glycosyltransferases (UGTs), PgUGT74AE2 and UGTPg1 from Panax ginseng, to catalyze the glycosylation of DM with UDP-glucose (UDPG) as the sugar donor to produce DM glucosides 3-O-β-D-glucopyranosyl-dammar-24-ene-3β,20S-diol (3β-O-Glc-DM) and 20-O-β-D-glucopyranosyl-dammar-24-ene-3β,20S-diol (20S-O-Glc-DM), respectively. The in vitro and in vivo assays demonstrated that both 3β-O-Glc-DM and 20S-O-Glc-DM exhibited higher anti-colon cancer activities than natural ginsenosides. In order to produce DM glucosides in an economical, efficient and convenient way, we refactored the complete biosynthetic pathways of 3β-O-Glc-DM and 20S-O-Glc-DM by introducing the codon-optimized genes encoding DM synthase (DS) together with PgUGT74AE2 or UGTPg1 into Saccharomyces cerevisiae, respectively. Furthermore, multistep metabolic engineering strategies were applied, including optimization of chassis cell, multi-copy integration of heterologous genes via the CRISPR/Cas9 system, increase of precursor supply by overexpressing rate-limiting enzymes, down-regulation of the competitive pathway to redirect the metabolic flux towards the target products, and overexpression of the transcriptional activator. Finally, the titers of 2.4 g L−1 3β-O-Glc-DM and 5.6 g L−1 20S-O-Glc-DM were achieved through fed-batch fermentation in a 3 L bioreactor. This is the first study to demonstrate the anti-colon cancer activities of DM glucosides and to achieve the de novo biosynthesis of DM glucosides with high titers in microbial cell factories. This study has established a green and sustainable approach for the industrial production of DM glucosides, which provides promising candidates for new drug research and development.
Introduction
Panax ginseng has been extensively used as a tonic and antiaging agent for nearly 5000 years in traditional Chinese medicine. Ginsenosides, the predominant bioactive components of P. ginseng, have been demonstrated to exhibit versatile pharmacological properties such as anticancer, antiaging, antidiabetic, antihypertensive, immunomodulatory and neuroprotective activities.1,2 To date, more than 150 natural ginsenosides have been identified from Panax species.3 The structural and functional diversity of ginsenosides is attributed to not only their aglycone structure but also the types, numbers and positions of the sugar moieties. Based on the aglycone structure, ginsenosides are divided into two types: tetracyclic dammarane and pentacyclic oleanane. The dammarane-type ginsenosides are the major type and the main pharmacologically active components of ginsenosides, and involve the glycosides of protopanaxadiol (PPD) and protopanaxatriol (PPT). In the metabolic pathways of ginsenosides, PPD is formed by hydroxylation of dammarenediol-II (DM) at the C12 position, while PPT is formed by hydroxylation of PPD at the C6 position. The PPD-type ginsenosides are biosynthesized by glycosylation of PPD at C3–OH and/or C20–OH, while the PPT-type ginsenosides are biosynthesized by glycosylation of PPT at C6–OH and/or C20–OH (Fig. 1). However, DM glucosides have scarcely been identified from Panax species.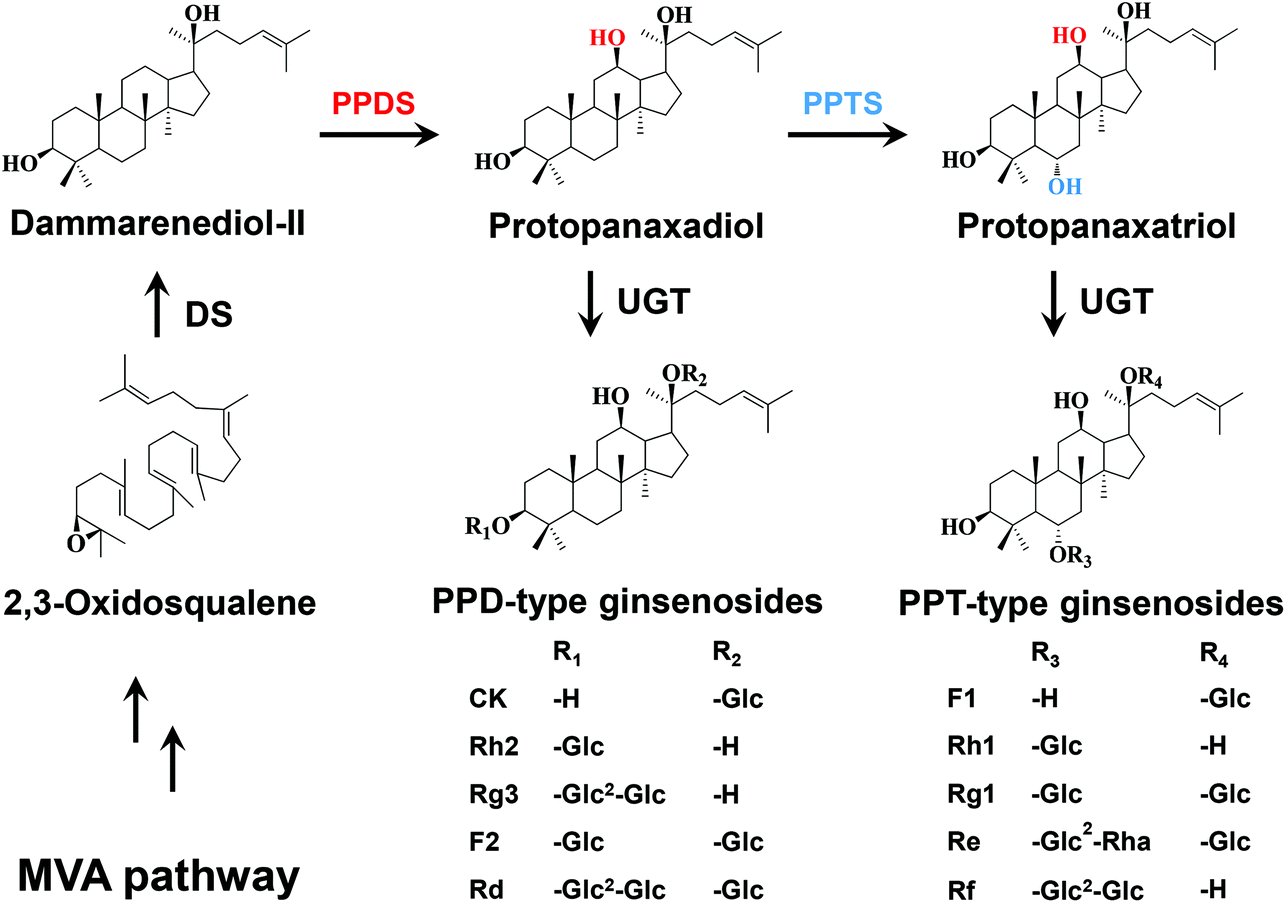 | ||
| Fig. 1 Biosynthetic pathway of ginsenosides in P. ginseng. MVA, mevalonate; DS, dammarenediol-II synthase; PPDS, protopanaxadiol synthase; PPTS, protopanaxatriol synthase; UGT, UDP-glycosyltransferase. | ||
It has been reported that the differences in the positions and numbers of hydroxyl groups and sugar moieties lead to the changes in bioactivities of ginsenosides.4,5 Moreover, the cytotoxicities of the dammarane-type ginsenosides are inversely proportional to the number of hydroxyl groups of aglycones, and to the number of sugar moieties linked to a specific aglycone.6 Since DM has fewer hydroxyl groups than PPD and PPT, it is spontaneously speculated that DM glucosides may exhibit higher cytotoxicities than PPD-type and PPT-type ginsenosides. However, DM glucosides can hardly be obtained from natural plant materials. On the other hand, it is expensive and environmentally unfriendly to produce DM glucosides by chemical synthesis methods.
Apart from enzyme catalysis,7,8 metabolic engineering as one of the green and sustainable biotechnologies has been extensively applied to not only refactor metabolic pathways of natural products in microbes for their heterologous synthesis but also generate unnatural products and even new compounds by artificially designing their metabolic pathways.9 The development of metabolic engineering allows the construction of more desirable microbial cell factories for the production of the target products.10Saccharomyces cerevisiae is the most commonly used eukaryotic host for metabolic engineering because it is well-characterized, genetically tractable and industrially robust, and there are multiple tools available for genetic manipulation.11 In particular, S. cerevisiae can inherently synthesize 2,3-oxidosqualene, which is the direct precursor for DM biosynthesis.12 Therefore, metabolic engineering of S. cerevisiae may provide an alternative approach for the production of DM glucosides.
In recent years, several UDP-glycosyltransferases (UGTs), which catalyze the glycosylation of C3–OH, C6–OH and C20–OH of PPD, PPT and DM, respectively, have been functionally characterized in P. ginseng.13–16 UGTPg1 is the first identified UGT for glycosylation of tetracyclic triterpenoids from P. ginseng, and glycosylates C20–OH of DM, PPD, Rh2, Rg3 and PPT to produce 20-O-β-D-glucopyranosyl-dammar-24-ene-3β,20S-diol (20S-O-Glc-DM), compound K, F2, Rd and F1, respectively.15,16 PgUGT74AE2 from P. ginseng selectively transfers a glucose moiety to C3–OH of PPD and compound K to form Rh2 and F2, respectively.14 UGTPg100 specifically glycosylates C6–OH of PPT and F1 to produce Rh1 and Rg1, respectively, while UGTPg101 catalyzes PPT to produce F1, followed by the generation of Rg1 from F1.15 These studies on UGTs and other related enzymes involved in ginsenoside biosynthesis lay a foundation for producing ginsenosides by metabolic engineering. As a result, metabolic engineering of S. cerevisiae has made great progress in efficient production of PPD,17 PPT,18 compound K,16 Rg3,13,14 Rh2,13,19 F1 and Rh1.15
In the present study, we aimed to obtain DM glucosides by enzymatic synthesis to demonstrate their pharmacological activities, and then construct microbial cell factories to produce them in a more economical way. Firstly, we synthesized or cloned the genes encoding dammarenediol-II synthase (DS), PgUGT74AE2 and UGTPg1 from P. ginseng. The two UGTs were heterologously expressed in Escherichia coli BL21 (DE3) and utilized to produce DM glucosides 3-O-β-D-glucopyranosyl-dammar-24-ene-3β,20S-diol (3β-O-Glc-DM) and 20S-O-Glc-DM by in vitro enzymatic glycosylation with UDP-glucose (UDPG) as the sugar donor and DM as the sugar acceptor. Furthermore, the anti-colon cancer activities of both 3β-O-Glc-DM and 20S-O-Glc-DM were demonstrated by in vitro and in vivo assays. In particular, in order to achieve the de novo biosynthesis of DM glucosides in microbial cell factories, we engineered S. cerevisiae by introducing the codon-optimized genes encoding DS together with PgUGT74AE2 or UGTPg1. The engineered strains were optimized to increase the production of DM glucosides by integrating multi-copy DS and UGT genes into yeast via the CRISPR/Cas9 system, overexpressing several key enzymes of the upstream biosynthetic pathway, down-regulating the competitive branch pathway and overexpressing the transcriptional activator HAC1. This study has established a green and sustainable approach to produce DM glucosides, which can enrich the resources for new drug research and development.
Experimental section
Strains and media
E. coli Trans1-T1 and BL21 (DE3) (TransGen Biotech, Beijing, China) were used for plasmid amplification and protein expression, respectively. E. coli strains were cultivated at 37 °C in LB medium with 100 mg L−1 ampicillin. S. cerevisiae INVSc1 and YPH499 (Invitrogen, USA) were used as the parent strains for the engineered yeast strains. The engineered yeast strains were cultivated at 30 °C either in SD medium lacking leucine, uracil, tryptophane and histidine where appropriate for selection of recombinants or in YPD medium for fermentation. All the yeast strains used in this study are listed in Table 1.| Strain | Genotype or characteristic | Source |
|---|---|---|
| YPH499 | MATa, ura3-52, lys2-801, ade2-101, trp1-Δ63, his3-Δ200, leu2-Δ1 | Invitrogen |
| INVSc1 | MATa, his3Δ1, leu2, trp1-289, ura3-52/MATα, his3Δ1, leu2, trp1-289, ura3-52 | Invitrogen |
| IN-ΔHXK2 | INVSC1, HXK2 Δ :: KanMX6 | This study |
| IN-ΔSER3 | INVSC1, SER3 Δ :: KanMX6 | This study |
| IN-ΔSOR1 | INVSC1, SOR1 Δ :: KanMX6 | This study |
| Y-ΔHXK2 | YPH499, HXK2 Δ :: KanMX6 | This study |
| Y-ΔSER3 | YPH499, SER3 Δ :: KanMX6 | This study |
| Y-ΔSOR1 | YPH499, SOR1 Δ :: KanMX6 | This study |
| Y6 | YPH499 with pESC-HIS-DS-GFP | This study |
| Y6-ΔHXK2 | Y-ΔHXK2 with pESC-HIS-DS-GFP | This study |
| Y1 | P TEF1 -synDS-GFP-TCYC1, PPGK1-tHMG1-TADH1, PTDH3-synPgUGT74AE2-TADH2 and HIS marker gene integrated into the δ1 site of Y-ΔHXK2 | This study |
| Y1C | P TEF1 -synDS-GFP-TCYC1, PPGK1-tHMG1-TADH1, PTDH3-synPgUGT74AE2-TADH2 and HIS marker gene integrated into the δ1 site of Y-ΔHXK2 with p-Cas9-gRNA | This study |
| Y1CS | P TDH3 -IDI1-TTPI1, PPGK1-ERG20-TADH1, PTEF1-ERG9-TCYC1, PPGK1-ERG1-TADH1, PTEF1-ERG7−-TCYC1 and LEU marker gene integrated into the δ4 site of Y1C | This study |
| Y1CSB | P TEF1 -BiP-TCYC1 and TRP marker gene integrated into the rDNA site of Y1CS | This study |
| Y1CSH | P TEF1 -HAC1-TCYC1 and TRP marker gene integrated into the rDNA site of Y1CS | This study |
| Y1CSP | P TEF1 -PDI1-TCYC1 and TRP marker gene integrated into the rDNA site of Y1CS | This study |
| Y2 | P TEF1 -synDS-GFP-TCYC1, PPGK1-tHMG1-TADH1, PTDH3-synUGTPg1-TADH2 and HIS marker gene integrated into the δ1 site of Y-ΔHXK2 | This study |
| Y2C | P TEF1 -synDS-GFP-TCYC1, PPGK1-tHMG1-TADH1, PTDH3-synUGTPg1-TADH2 and HIS marker gene integrated into the δ1 site of Y-ΔHXK2 with p-Cas9-gRNA | This study |
| Y2CS | P TDH3 -IDI1-TTPI1, PPGK1-ERG20-TADH1, PTEF1-ERG9-TCYC1, PPGK1-ERG1-TADH1, PTEF1-ERG7−-TCYC1 and LEU marker gene integrated into the δ4 site of Y2C | This study |
| Y2CSB | P TEF1 -BiP-TCYC1 and TRP marker gene integrated into the rDNA site of Y2CS | This study |
| Y2CSH | P TEF1 -HAC1-TCYC1 and TRP marker gene integrated into the rDNA site of Y2CS | This study |
| Y2CSP | P TEF1 -PDI1-TCYC1 and TRP marker gene integrated into the rDNA site of Y2CS | This study |
Plasmid construction
The genes encoding PgUGT74AE2 (GenBank: JX898529.1) and UGTPg1 (GenBank: KF377585.1) were amplified from the cDNA of P. ginseng prepared as described previously20 and subsequently cloned into pET-32a (Novagen, USA), resulting in pET-PgUGT74AE2 and pET-UGTPg1 for heterologous expression in E. coli BL21 (DE3). Additionally, the genes encoding PgUGT74AE2, UGTPg1 and DS (GenBank: AB265170.1) were also synthesized by GenScript (Nanjing, China) in Tianjin University (China) according to the codon bias of S. cerevisiae for improved expression while DS was fused with GFP through overlap extension PCR (OE-PCR). Yeast promoters (TEF1, TDH3 and PGK1), terminators (CYC1, ADH1, TPI1 and ADH2) and genes encoding truncated HMG-CoA reductase (tHMG1), isopentenyl diphosphate isomerase (IDI1), farnesyl diphosphate synthase (ERG20), squalene synthase (ERG9), squalene epoxidase (ERG1), immunoglobulin-binding protein (BiP), transcriptional activator HAC1 and protein disulfide isomerase (PDI1) were amplified from the genomic DNA isolated from S. cerevisiae INVSc1 using TIANamp Yeast DNA Kit (TianGen, Beijing, China). The antisense gene of lanosterol synthase (ERG7) was amplified from plasmid pESC-URA-ERG7− as the template.20 The synthesized genes and the amplified genes were fused with appropriate promoters and terminators to generate expression cassettes PTDH3-synPgUGT74AE2-TADH2, PTDH3-synUGTPg1-TADH2, PTEF1-synDS-GFP-TCYC1, PPGK1-tHMG1-TADH1, PTDH3-IDI1-TTPI1, PPGK1-ERG20-TADH1, PTEF1-ERG9-TCYC1, PPGK1-ERG1-TADH1, PTEF1-ERG7−-TCYC1, PTEF1-BiP-TCYC1, PTEF1-HAC1-TCYC1 and PTEF1-PDI1-TCYC1, and cloned into pEASY-Blunt (TransGen Biotech, Beijing, China), resulting in pEASY-PgUGT74AE2, pEASY-UGTPg1, pEASY-DS-GFP, pEASY-tHMG1, pEASY-IDI1, pEASY-ERG20, pEASY-ERG9, pEASY-ERG1, pEASY-ERG7−, pEASY-BiP, pEASY-HAC1 and pEASY-PDI1, respectively. The expression cassettes of BiP, HAC1 and PDI1 amplified from plasmids pEASY-BiP, pEASY-HAC1 and pEASY-PDI1 were ligated into the plasmid prDNA-TRP, resulting in prDNA-TRP-BiP, prDNA-TRP-HAC1 and prDNA-TRP-PDI1, respectively. The construction of the integration modules is described in detail in the ESI.† The overlapped order of expression cassettes for multi-copy integration is illustrated in Fig. S1.†To construct the Cas9/gRNA co-expression plasmid, the human codon-optimized Cas9 was amplified from plasmid FM-1 provided by Prof. Lu at College of Life Sciences of Nanjing Normal University.21 The cassette PTEF1-Cas9-TADH2 was generated by OE-PCR with TEF1 promoter, Cas9 and ADH2 terminator. The δ-specific gRNA sequence with SNR52 promoter and SUP4 terminator22 was amplified from plasmid pUC57-gRNA synthesized by Tianjin University. The plasmid backbone was amplified from pESC-URA. The above three fragments were assembled using In-Fusion Cloning Kit (Clontech, USA), resulting in the Cas9/gRNA co-expression plasmid p-Cas9-gRNA.
All the plasmids are summarized in Table S1,† and the primers used for plasmid construction are listed in Table S2.†
Enzymatic assays of PgUGT74AE2 and UGTPg1
The plasmids pET-PgUGT74AE2 and pET-UGTPg1 were transformed into E. coli BL21 (DE3), respectively. Heterologous expression, crude enzyme preparation and purification of PgUGT74AE2 and UGTPg1 were carried out as described previously.20 Enzymatic assays were conducted in 100 μL reaction mixtures containing 25 mM Tris-HCl (pH 8.0), 0.5 mM DM, 5 mM UDPG and crude recombinant PgUGT74AE2 or UGTPg1 at 40 °C for 24 h. The reaction mixture containing the lysate of E. coli BL21 harboring pET32a was used as the negative control. The reactions were terminated by adding 100 μL methanol. The target products were directly analyzed by HPLC-ESI-MS analysis after centrifugation and filtration, and further confirmed by 1H NMR, 13C NMR, HMBC and HSQC analyses after preparation by semi-preparative HPLC.For the kinetic study of PgUGT74AE2 and UGTPg1 towards DM and PPD (purity > 98%, Nanjing Spring & Autumn Biological Engineering Co., Ltd., China), reaction mixtures containing 25 mM Tris-HCl buffer (pH 8.0), a substrate (0.04–1.6 mM), 5 mM UDPG and 5 μg purified PgUGT74AE2 or UGTPg1 in a final volume of 100 μL were incubated at 40 °C for 20 min. The reactions were terminated by adding 100 μL methanol. The target products were quantified by HPLC analysis after centrifugation and filtration. All data of the Michaelis–Menten parameters are presented as means ± SD of three independent repeat experiments.
In vitro cytotoxicity tests of 3β-O-Glc-DM and 20S-O-Glc-DM
The anti-colon cancer activities of 3β-O-Glc-DM and 20S-O-Glc-DM were determined in human colon cancer cell lines (HCT15, HCT116 and SW48) by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay with Rg3 and compound K (purity > 98%, Nanjing Spring & Autumn Biological Engineering Co., Ltd., China) as positive controls. Cells were seeded in a 96-well plate. After incubation for 24 h, the cells were treated with various concentrations of DM glucosides or dimethyl sulfoxide (DMSO). After incubation for 120 h, 50 μL MTT stock solution (2 mg mL−1, Sigma Chemical) was added into each well, and the plates were incubated at 37 °C for an additional 4 h. The solution was then removed from each well, and 150 μL DMSO was added. Following gentle agitation, the absorbance was measured using an ELISA reader (Bio-Rad, USA) at 570 nm. Three parallel samples were measured each time. The absorbance values were normalized to the values obtained for vehicle-treated cells to determine the percentage of surviving cells. The median inhibitory concentration (IC50) was assessed based on the dose response curve.In vivo evaluation of the anti-colon cancer activities of 3β-O-Glc-DM and 20S-O-Glc-DM
BALB/c mice (males, 6–8 weeks old) were purchased from HFK Bioscience Co., Ltd. (Beijing, China). All animal protocols conformed to the Guidelines for the Care and Use of Laboratory Animals approved by the Animal Care and Use Committee of Chinese Academy of Medical Sciences and Peking Union Medical College. C26 murine colon cancer cells were counted and diluted to a density of 5 × 107 cells mL−1. BALB/c mice were subcutaneously implanted with 1 × 107 C26 murine colon cancer cells per mouse on the flank. 24 h after inoculation, BALB/c mice were randomly divided into 18 groups, and each group consisted of six mice. One group received p.o. 20% PEG400 as a model control. One group received an injection of 30.0 mg kg−1 of 5-FU (once every 3 days, the 1st, 4th and 7th day). 8 groups involved 10.0 mg kg−1 Rg3 and 10.0 mg kg−1 compound K used as positive controls, and 5.0 mg kg−1, 10.0 mg kg−1 and 20.0 mg kg−1 of 3β-O-Glc-DM or 20S-O-Glc-DM (dissolved in 20% PEG400) were continuously administered p.o. for 8 days, once a day. Other 8 groups were administered with 10.0 mg kg−1 Rg3, 10.0 mg kg−1 compound K and 5.0 mg kg−1, 10.0 mg kg−1 and 20.0 mg kg−1 3β-O-Glc-DM or 20S-O-Glc-DM in combination with 30.0 mg kg−1 5-FU, respectively (once every 3 days, the 1st, 4th and 7th day). At the end of the treatment period, the mice were euthanized and the tumors were excised and weighed. The inhibition rate (IR) of tumor growth was calculated using the following formula: IR (%) = [(A − B)/A]/100, where A is the average tumor weight of the model control and B is the average tumor weight of compound groups.Optimization of S. cerevisiae chassis cell
Genes encoding hexokinase II (HXK2), phosphoglycerate dehydrogenase (SER3) and sorbitol dehydrogenase (SOR1) were selected as the knockout targets to optimize S. cerevisiae INVSc1 and YPH499 for improving the synthesis of heterologous terpenoids.23 To construct gene deletion cassettes, the kanamycin resistance gene cassette was amplified from plasmid pUC6 and fused with about 500 bp upstream and downstream homologous sequences of the knockout target genes amplified from the genomic DNA of YPH499. The single gene-deficient mutants were individually constructed by integrating the deletion cassettes into the chromosomes of INVSc1 and YPH499 via homologous recombination, and selected on YPD plates supplemented with 300 mg L−1 G418. All the mutants were confirmed by PCR and the primers used for PCR are listed in Table S3.†The growth rates of the mutants were investigated in YPD medium with 2% glucose or YPG medium with 2% galactose as the sole carbon source, respectively. The mutants were inoculated into 100 mL shake flasks with an initial OD600 of 0.2 and incubated at 30 °C for 4 days. The OD600 values were measured at the scheduled time points using a UV-Vis spectrophotometer (Implen, NanoPhotometer Pearl, Germany). All data are presented as means ± SD of three independent repeat experiments.
Evaluation of the effect of HXK2 deletion on DM production
Transformation of S. cerevisiae was performed by a standard lithium acetate method.24 The engineered strains producing DM were used as a test model to investigate whether the carbon flux change of the yeast mutant was beneficial for the production of DM or not. Plasmid pESC-HIS-DS-GFP25 was transformed into YPH499 and the mutant Y-ΔHXK2, and then selected on SD-HIS plates to generate the engineered strains Y6 and Y6-ΔHXK2, respectively.To produce DM, the individual clones were first grown in 5 ml SD-HIS at 30 °C and 220 rpm for 24 h. Subsequently, the engineered strains were inoculated into 100 ml SG-HIS at an initial OD600 of 0.8 and incubated at 30 °C and 220 rpm for 6 days. The cell pellets were harvested, disrupted and extracted with n-hexane after being freeze-dried using a vacuum freeze-dryer. DM was quantified by HPLC analysis. All data are presented as means ± SD of three independent repeat experiments.
Construction of the engineered yeasts
To construct the engineered strain producing 3β-O-Glc-DM, the integration modules containing the expression cassettes of tHMG1, DS, PgUGT74AE2 and HIS together with plasmid p-Cas9-gRNA were co-transformed into the strain Y-ΔHXK2 and integrated into the δ1 site of Y-ΔHXK2. The biosynthetic pathway was assembled using DNA assembler and verified using PCR analysis of the junctions between each adjacent DNA fragment.26 Twenty single colonies were randomly picked from SD-HIS plates and verified using PCR. All right colonies were cultivated in YPD medium for 3 days to compare the titers of the target products. The strain Y1C with the highest titer of 3β-O-Glc-DM was selected for further engineering. The strain Y1 was constructed as described above without plasmid p-Cas9-gRNA. Similarly, the strain Y2C producing 20S-O-Glc-DM was also constructed by transforming the integration modules containing the expression cassettes of tHMG1, DS, UGTPg1 and HIS together with plasmid p-Cas9-gRNA into the strain Y-ΔHXK2. The strain Y2 was constructed just as Y2C without plasmid p-Cas9-gRNA.The strains Y1CS and Y2CS were constructed by integrating the following three DNA integration modules into the δ4 site of Y1C and Y2C, respectively. The three DNA integration modules contained IDI1, ERG20, ERG9, ERG1, the antisense fragment of ERG7 and LEU gene cassettes flanked by δ4 sequences. Twenty single colonies were randomly picked from SD-HIS-LEU plates and verified using PCR. All right colonies were cultivated in YPD medium for 3 days to compare the titers of the target products. The strains with the highest titers of the target products were used for further engineering.
Three DNA integration modules containing the expression cassettes of BiP, HAC1 and PDI1 were respectively obtained from plasmids prDNA-TRP-BiP, prDNA-TRP-HAC1 and prDNA-TRP-PDI1 digested with BamH I and Sac I. The three DNA modules were separately integrated into the rDNA sites of Y1CS and Y2CS, resulting in the strains Y1CSB, Y1CSH, Y1CSP, Y2CSB, Y2CSH and Y2CSP, respectively. Ten single colonies of each strain were picked from SD plates and then verified using PCR. All right colonies were cultivated in YPD medium for 3 days to screen the strains with the highest titers of the target products.
Relative quantification of the gene copy number by qPCR
The total genomic DNA of the engineered strains obtained by CRISPR/Cas9 directed homologous recombination was used to determine the relative gene dosages with the engineered strains obtained by conventional homologous recombination as the controls. The DS gene in the integration modules and the actin gene in the genomic DNA were selected as the target gene and reference gene, respectively. qPCR was performed using Ultra SYBR Mixture Kit (Cowin Biotech, China) on Roche LightCycler 480 II. The Ct values of the target gene DS were normalized to the reference gene actin. Data are presented as the relative gene dosages between the experimental and control strains. All reactions were performed in triplicate and analyzed as described previously.27 The primers used for qPCR are listed in Table S4.†Yeast cultivation and the fed-batch fermentation process
For shake flask fermentation, YPD medium was used to cultivate the engineered strains. All strains were first inoculated in 5 mL YPD medium and grown at 30 °C and 220 rpm for 12 h, and then transferred into 500 mL flasks containing 100 mL YPD medium with an initial OD600 of 0.2 at 30 °C and 220 rpm for 6 days. 5 mL fed solution (pH 5.5) containing 578 g L−1 glucose, 9 g L−1 KH2PO4, 5.12 g L−1 MgSO4·7H2O, 3.5 g L−1 K2SO4, 0.28 g L−1 Na2SO4, 2.1 g L−1 adenine, 2.5 g L−1 uracil, 5 g L−1 lysine, 10 mL L−1 trace element solution and 12 mL L−1 vitamin solution was fed into the medium at 48 h, 72 h and 96 h, respectively.28For the fed-batch fermentation process, the strains Y1CSH and Y2CSH were used for fermentation in a 3 L bioreactor (Shanghai Baoxing Bio-Engineering Equipment Co., Ltd., China). The single clones were firstly grown in 5 mL YPD medium at 30 °C and 220 rpm for 12 h, and then 1 mL suspensions were transferred into 100 mL YPD medium in 500 mL shake flasks and grown at 30 °C and 220 rpm for 24 h as seed cultures. 100 mL seed cultures were then transferred into 1 L YPD medium. Fermentation was carried out at 30 °C with an air flow rate of 3 L min−1. The dissolved oxygen (DO) was maintained at approximately 30% with an agitation cascade (300–900 rpm) and the pH was automatically controlled at 5.5 by addition of 5 M ammonium hydroxide. For the glucose fed-batch process, the above-mentioned fed solution was fed into the bioreactor and a simple exponential feed rate was used. The concentration of glucose was maintained below 1.0 g L−1 to keep the concentration of ethanol below 5.0 g L−1.28,29
Metabolite extraction and analysis
Cells were harvested by centrifugation (5000g, 10 min), freeze-dried and weighed. Then the cell pellets were lysed with 70% (v/v) ethanol at 70 °C for 60 min, and extracted with n-butanol three times. The extracts were dissolved in methanol and used for the analysis of the target products by HPLC. The cell pellets were also lysed with 20% (w/v) KOH and 50% (v/v) ethanol, and extracted with n-hexane for the analysis of squalene, DM and ergosterol by HPLC. The extracellular products in the supernatant were extracted directly with n-butanol or n-hexane and used for the analysis of extracellular target products, intermediates and by-products. All data are presented as means ± SD of three independent repeat experiments.HPLC, HPLC-ESI-MS and NMR analyses
HPLC analysis was performed on an Agilent 1200 system. A Cosmosil 5C18-MS-II column (5 μm, 4.6 mm × 150 mm) was used at 28 °C for chromatographic separation with a 1 mL min−1 flow rate and 203 nm UV wavelength. Semi-preparative HPLC was performed on a HPLC system equipped with a Shimadzu LC-6AD pump and a Shimadzu SPD-20A prominence UV-VIS detector (Shimadzu Corporation, Kyoto, Japan) using an Agilent C18 column (5 μm, 9.6 mm × 250 mm). The gradient elution system consisted of water and acetonitrile. The detailed conditions of analytic and semi-preparative HPLC for extracts of the enzyme reaction and yeast fermentation are described in Table S5.†ESI-MS data were acquired using an HPLC-LTQ/FTICR-MS system (Thermo Fisher Scientific, Massachusetts, United States) equipped with a Cosmosil 5C18-MS-II column (5 μm, 4.6 mm × 150 mm). NMR experiments were performed in CD3OD for all products on an INOVA 600 NMR spectrometer (Varian, California, USA) to obtain 1H NMR, 13C NMR, heteronuclear multiple-bond correlation spectroscopy (HMBC) and heteronuclear single quantum coherence (HSQC) spectra.
Results and discussion
Production of DM glucosides by PgUGT74AE2 and UGTPg1 catalysis
The UGT genes encoding PgUGT74AE2 and UGTPg1 were amplified from the cDNA of P. ginseng and subsequently heterologously expressed as N-terminal His-tagged proteins in E. coli BL21 (DE3), respectively. SDS-PAGE analysis showed that recombinant PgUGT74AE2 and UGTPg1 were both expressed in soluble forms, which could be easily purified through one-step Ni-NTA metal affinity chromatography (Fig. S2†).In order to identify the enzymatic activities of PgUGT74AE2 and UGTPg1 towards DM, the crude recombinant enzymes were incubated with UDPG as the sugar donor and DM as the sugar acceptor, respectively. After incubation at 37 °C for 24 h, the recombinant PgUGT74AE2 transferred a glucose moiety to C3–OH of DM to produce 3β-O-Glc-DM, which was confirmed by HPLC-ESI-MS, 1H NMR, 13C NMR, HMBC and HSQC (Fig. 2A, C, Fig. S3 and Table S6†). Previously, we confirmed that PgUGT74AE2 could glycosylate C3–OH of PPT.30 Moreover, PgUGT74AE2 was also reported to transfer a glucose moiety to C3–OH of PPD and compound K to form Rh2 and F2, respectively.14 Therefore, all the above-mentioned evidences revealed that PgUGT74AE2 could glycosylate not only C3–OH of PPD and CK but also C3–OH of DM and PPT. Similarly, incubation of UGTPg1 with UDPG and DM could produce 20S-O-Glc-DM confirmed by HPLC-ESI-MS, 1H-NMR, 13C-NMR, HMBC and HSQC (Fig. 2B, D, Fig. S4 and Table S7†), which was consistent with the result of a previous report.16
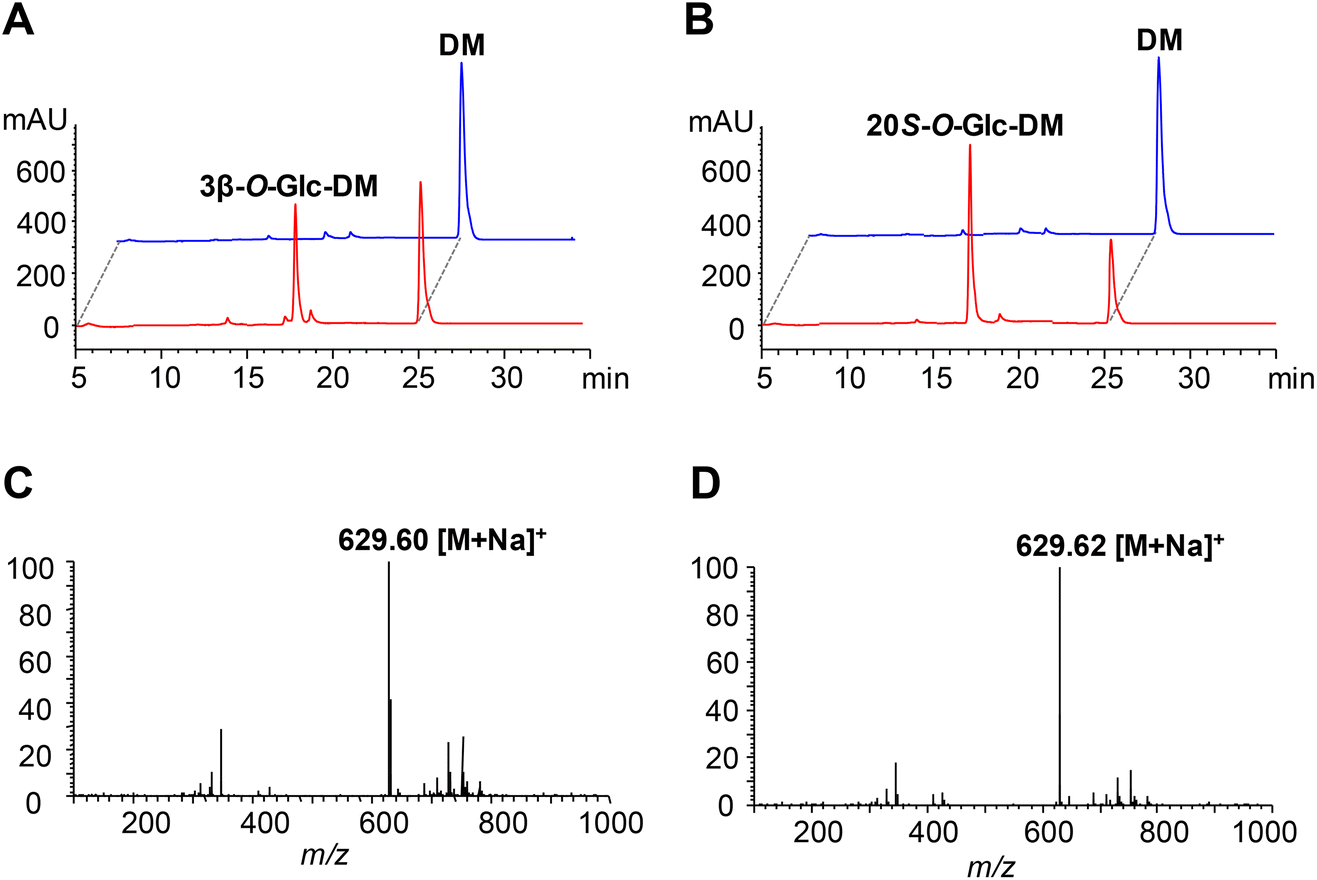 | ||
| Fig. 2 Functional characterization of PgUGT74AE2 and UGTPg1 towards DM. (A) HPLC analysis of the product from the incubation of PgUGT74AE2 with DM as the substrate for 24 h. Red, the cell extract of recombinant E. coli harboring pET-PgUGT74AE2; blue, negative control, the cell extract of recombinant E. coli harboring pET32a. (B) HPLC analysis of the product from the incubation of UGTPg1 with DM as the substrate for 24 h. Red, the cell extract of recombinant E. coli harboring pET-UGTPg1; blue, negative control, the cell extract of recombinant E. coli harboring pET32a. (C) The MS spectrum of the product from the reaction of PgUGT74AE2 catalyzing DM glycosylation. (D) The MS spectrum of the product from the reaction of UGTPg1 catalyzing DM glycosylation. | ||
Kinetic study of PgUGT74AE2 and UGTPg1
The kinetic parameters of PgUGT74AE2 and UGTPg1 towards DM and PPD were determined and the results are listed in Table 2. The Km of PgUGT74AE2 towards DM (0.38 mM) was almost equal to that of PgUGT74AE2 towards PPD (0.35 mM), while the turnover number (kcat) of PgUGT74AE2 towards PPD (1.38 s−1) was 1.33-fold that of PgUGT74AE2 towards DM (1.04 s−1). The catalytic efficiency (kcat/Km) of PgUGT74AE2 towards PPD (3.94 mM−1 s−1) was 1.43-fold that of PgUGT74AE2 towards DM (2.76 mM−1 s−1). In contrast, the Km of UGTPg1 towards PPD (0.44 mM) was 1.83-fold that of UGTPg1 towards DM (0.24 mM), while the turnover number of UGTPg1 towards PPD (2.34 s−1) was 2.46-fold that of UGTPg1 towards DM (0.95 s−1). The catalytic efficiency of UGTPg1 towards PPD (5.29 mM−1 s−1) was 1.36-fold that of UGTPg1 towards DM (3.88 mM−1 s−1). Taken together, the catalytic efficiencies of both PgUGT74AE2 and UGTPg1 towards PPD were higher than those of them towards DM. Furthermore, UGTPg1 was more efficient than PgUGT74AE2 on PPD or DM glycosylation. However, neither 3β-O-Glc-DM nor 20S-O-Glc-DM has been identified from Panax species. A possible explanation might be that DM was easy to be transformed into PPD and/or that both 3β-O-Glc-DM and 20S-O-Glc-DM were readily transformed into other ginsenosides via hydroxylation and extensive glycosylation. Consequently, little of 20S-O-Glc-DM and 3β-O-Glc-DM was produced and/or accumulated in Panax species.| Enzyme | Substrate | K m (mM) | k cat (s−1) | V max (nmol min−1 μg−1) | k cat/Km (mM−1 s−1) |
|---|---|---|---|---|---|
| PgUGT74AE2 | DM | 0.38 ± 0.01 | 1.04 ± 0.04 | 1.26 ± 0.03 | 2.76 ± 0.03 |
| PgUGT74AE2 | PPD | 0.35 ± 0.04 | 1.38 ± 0.01 | 1.66 ± 0.05 | 3.94 ± 0.01 |
| UGTPg1 | DM | 0.24 ± 0.03 | 0.95 ± 0.03 | 1.34 ± 0.07 | 3.88 ± 0.02 |
| UGTPg1 | PPD | 0.44 ± 0.06 | 2.34 ± 0.02 | 2.81 ± 0.03 | 5.29 ± 0.01 |
In vitro cytotoxicity tests of 3β-O-Glc-DM and 20S-O-Glc-DM
The DM glucosides 3β-O-Glc-DM and 20S-O-Glc-DM were tested for in vitro cytotoxicities against three human colon cancer cell lines. Rg3 and compound K were used as the positive controls because the two compounds were reported to be the most active ginsenosides against colon cancer.31,32 As shown in Table 3, 3β-O-Glc-DM inhibited the proliferation of HCT15, HCT116 and SW48 cells with IC50 values of 30.6, 63.7 and 56.9 μM while IC50 values of 20S-O-Glc-DM were 49.0, 46.8 and 94.6 μM, respectively. These results showed that both 3β-O-Glc-DM and 20S-O-Glc-DM exhibited higher cytotoxicities against human colon cancer cells than Rg3 and compound K. Compared with Rg3 and compound K, both 3β-O-Glc-DM and 20S-O-Glc-DM lack C12–OH. This implied that the existence of C12–OH might reduce the cytotoxicities of Rg3 and compound K against colon cancer cells. The results were in agreement with the previous findings that the cytotoxicities of the dammarane-type ginsenosides are inversely proportional to the number of hydroxyl groups of aglycones.6 These findings suggested that both 3β-O-Glc-DM and 20S-O-Glc-DM may be effective against human colon cancer.| Compound | IC50 (μM) | ||
|---|---|---|---|
| HCT15 | HCT116 | SW48 | |
| Rg3 | 268.4 | 231.8 | 93.2 |
| Compound K | 206.5 | 79.4 | 112.2 |
| 3β-O-Glc-DM | 30.6 | 63.7 | 56.9 |
| 20S-O-Glc-DM | 49.0 | 46.8 | 94.6 |
In vivo evaluation of the anti-colon cancer activities of 3β-O-Glc-DM and 20S-O-Glc-DM
Since both 3β-O-Glc-DM and 20S-O-Glc-DM displayed high cytotoxicities against colon cancer cell lines in vitro, we further evaluated their anti-colon cancer activities in the C26 colon cancer xenograft model. The results are shown in Table 4. In the group treated with 10 mg kg−1 3β-O-Glc-DM, the inhibition rate of the tumor weight was 36.9%, which was apparently higher than those of the groups treated with 10 mg kg−1 Rg3 (13.4%) and 10 mg kg−1 compound K (2.9%). In the group treated with 10 mg kg−1 20S-O-Glc-DM, the inhibition rate of the tumor weight was 14.0%, which was also higher than those of the groups treated with 10 mg kg−1 Rg3 and 10 mg kg−1 compound K. Furthermore, both 3β-O-Glc-DM and 20S-O-Glc-DM markedly increased the inhibition rate of the tumor weight when they were combined with 5-FU in treatment. The inhibition rates of 5 mg kg−1 3β-O-Glc-DM combined with 30 mg kg−1 5-FU and 20 mg kg−1 20S-O-Glc-DM combined with 30 mg kg−1 5-FU were 74.5% and 60.6%, respectively, which were much higher than that of 30 mg kg−1 5-FU alone (41.1%). Moreover, their synergistic effects were also better than those of 10 mg kg−1 Rg3 combined with 30 mg kg−1 5-FU (47.1%) and 10 mg kg−1 compound K combined with 30 mg kg−1 5-FU (51.6%). Therefore, whether administered alone or combined with 5-FU in treatment, the inhibitory effects of 3β-O-Glc-DM and 20S-O-Glc-DM were better than those of Rg3 and compound K, which corresponded to the results of the primary pharmacological tests in vitro. Particularly, it is encouraging that both 3β-O-Glc-DM and 20S-O-Glc-DM displayed higher anti-colon cancer activities even at a lower concentration than Rg3 when combined with 5-FU in treatment. To our knowledge, this is the first report to demonstrate the high anti-colon cancer activities of 3β-O-Glc-DM and 20S-O-Glc-DM, which promises the two compounds as new drug candidates.| Group | Dosage (mg kg−1) | Body weight (g) | Tumor weight (g) | Inhibition rate (%) | |
|---|---|---|---|---|---|
| Begin | End | ||||
| *p < 0.05, **p < 0.01, and ***p < 0.001, compared with the vehicle control; Δp < 0.05 and ΔΔΔp < 0.001, compared with 5-FU. | |||||
| Vehicle control | 16.3 ± 0.7 | 16.7 ± 0.4 | 1.26 ± 0.24 | ||
| 5-FU | 30.0 | 17.1 ± 0.6 | 18.4 ± 1.3 | 0.74 ± 0.16** | 41.1 |
| Rg3 | 10.0 | 16.7 ± 0.6 | 16.7 ± 1.2 | 1.09 ± 0.36 | 13.4 |
| Compound K | 10.0 | 16.7 ± 0.7 | 16.5 ± 0.8 | 1.22 ± 0.30 | 2.9 |
| 5.0 | 16.3 ± 0.8 | 16.5 ± 1.1 | 1.12 ± 0.29 | 10.9 | |
| 3β-O-Glc-DM | 10.0 | 16.8 ± 0.5 | 17.2 ± 0.6 | 0.80 ± 0.28* | 36.9 |
| 20.0 | 16.7 ± 0.7 | 16.3 ± 1.1 | 0.82 ± 0.20** | 35.3 | |
| 5.0 | 16.4 ± 0.5 | 16.7 ± 1.4 | 1.28 ± 0.25 | 1.7 | |
| 20S-O-Glc-DM | 10.0 | 16.4 ± 0.7 | 16.7 ± 1.2 | 1.08 ± 0.24 | 14.0 |
| 20.0 | 16.5 ± 0.9 | 16.9 ± 0.6 | 1.06 ± 0.27 | 16.0 | |
| 5-FU + Rg3 | 30.0 + 10.0 | 16.6 ± 1.0 | 17.2 ± 1.6 | 0.67 ± 0.18*** | 47.1 |
| 5-FU + compound K | 30.0 + 10.0 | 16.5 ± 0.8 | 17.5 ± 0.8 | 0.61 ± 0.14*** | 51.6 |
| 30.0 + 5.0 | 16.3 ± 0.8 | 15.5 ± 1.7 | 0.32 ± 0.05***ΔΔΔ | 74.5 | |
| 5-FU + 3β-O-Glc-DM | 30.0 + 10.0 | 16.9 ± 0.9 | 16.2 ± 1.2 | 0.50 ± 0.16***Δ | 60.6 |
| 30.0 + 20.0 | 16.8 ± 0.6 | 15.6 ± 1.2 | 0.49 ± 0.13***Δ | 61.4 | |
| 30.0 + 5.0 | 16.9 ± 0.7 | 17.6 ± 0.8 | 0.66 ± 0.15*** | 47.6 | |
| 5-FU + 20S-O-Glc-DM | 30.0 + 10.0 | 16.4 ± 1.0 | 17.1 ± 1.0 | 0.61 ± 0.17*** | 51.5 |
| 30.0 + 20.0 | 16.7 ± 0.9 | 16.6 ± 0.8 | 0.50 ± 0.17***Δ | 60.6 | |
Optimization of the chassis cell by deleting the primary metabolic genes in S. cerevisiae
The engineered pathways of ginsenosides involve the mevalonate (MVA) pathway from glucose to isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), which is intrinsic in the yeast host. It has been reported that deletion of some genes involved in primary metabolism, including glucose metabolism and amino acid biosynthesis, can improve acetyl-CoA supply to the MVA pathway in S. cerevisiae, which ultimately enhances the biosynthesis of downstream heterologous terpenoids.23 In the present study, we individually disrupted HXK2, SER3 and SOR1 genes in the yeast strains INVSc1 and YPH499, respectively. The growth rates of these yeast mutants were measured in YPD and YPG media, respectively. The results indicated that all mutants had a dramatically slower growth pattern than the wild-type strains except for Y-ΔHXK2 which grew slightly faster in both YPD and YPG media (Fig. 3A and B).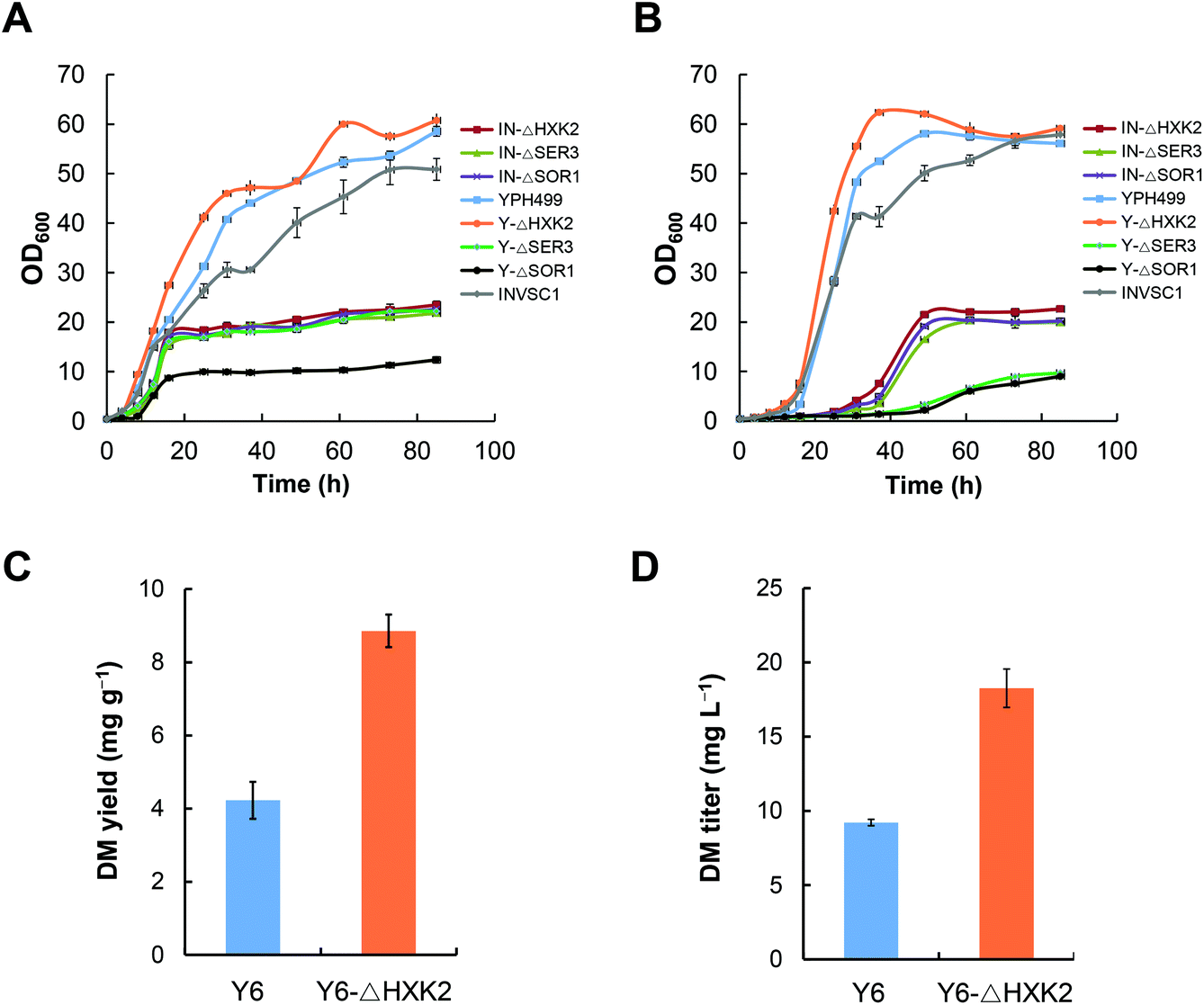 | ||
| Fig. 3 Effects of deleting the primary metabolic genes in S. cerevisiae on the cell growth and DM production. (A) The growth profiles of different mutants in YPD medium. (B) The growth profiles of different mutants in YPG medium. (C) A comparison of DM yields between Y6-ΔHXK2 and Y6. (D) A comparison of DM titers between Y6-ΔHXK2 and Y6. | ||
To evaluate the effect of HXK2 deletion on improving the biosynthesis of heterologous terpenoids, DM was chosen as the tested terpenoid. DS was expressed in the mutant Y-ΔHXK2 and the wild-type strain YPH499, resulting in DM producing strains Y6-ΔHXK2 and Y6, respectively. The DM production in Y6-ΔHXK2 (8.9 mg g−1, 18.26 mg L−1) was about 2-fold that in Y6 (4.2 mg g−1, 9.21 mg L−1) (Fig. 3C and D). We hypothesize that HXK2 deletion increased the production of DM probably by allowing more carbon flux to flow into the MVA pathway for IPP synthesis. Therefore, the mutant Y-ΔHXK2 was used as the chassis cell to produce DM glucosides by metabolic engineering.
Construction of the biosynthetic pathways of 3β-O-Glc-DM and 20S-O-Glc-DM via the CRISPR/Cas9 system in S. cerevisiae
In order to construct the biosynthetic pathway of 3β-O-Glc-DM in S. cerevisiae, the codon-optimized genes of PgUGT74AE2 and DS, together with tHMG1 from S. cerevisiae, were integrated into the chromosomes of Y-ΔHXK2 at the δ1 site. Accordingly, the biosynthetic pathway of 20S-O-Glc-DM was constructed in the same way by replacing the PgUGT74AE2 gene with the UGTPg1 gene (Fig. 4). Twenty colonies were randomly picked and verified using PCR. All right colonies were cultivated in YPD medium for 3 days for product identification and comparison of their titers. HPLC-ESI-MS analysis of intracellular and extracellular extracts confirmed the production of 3β-O-Glc-DM (Fig. 5A and C) and 20S-O-Glc-DM (Fig. 5B and D). The structures of the two products were then confirmed by 1H NMR, 13C NMR, HMBC and HSQC (Fig. S5 and S6, Tables S8 and S9†). The colonies with the highest titers of 3β-O-Glc-DM and 20S-O-Glc-DM were designated as Y1 and Y2, respectively. The titer of 3β-O-Glc-DM in the strain Y1 was 14.8 mg L−1 while the titer of 20S-O-Glc-DM in the strain Y2 was 18.6 mg L−1 after being cultivated in YPD medium for 6 days (Table 5). The successful biosynthesis of 3β-O-Glc-DM and 20S-O-Glc-DM in the engineered yeasts further verified that PgUGT74AE2 and UGTPg1 could catalyze the glycosylation of C3–OH and C20–OH of DM, respectively, not only in vitro but also in vivo.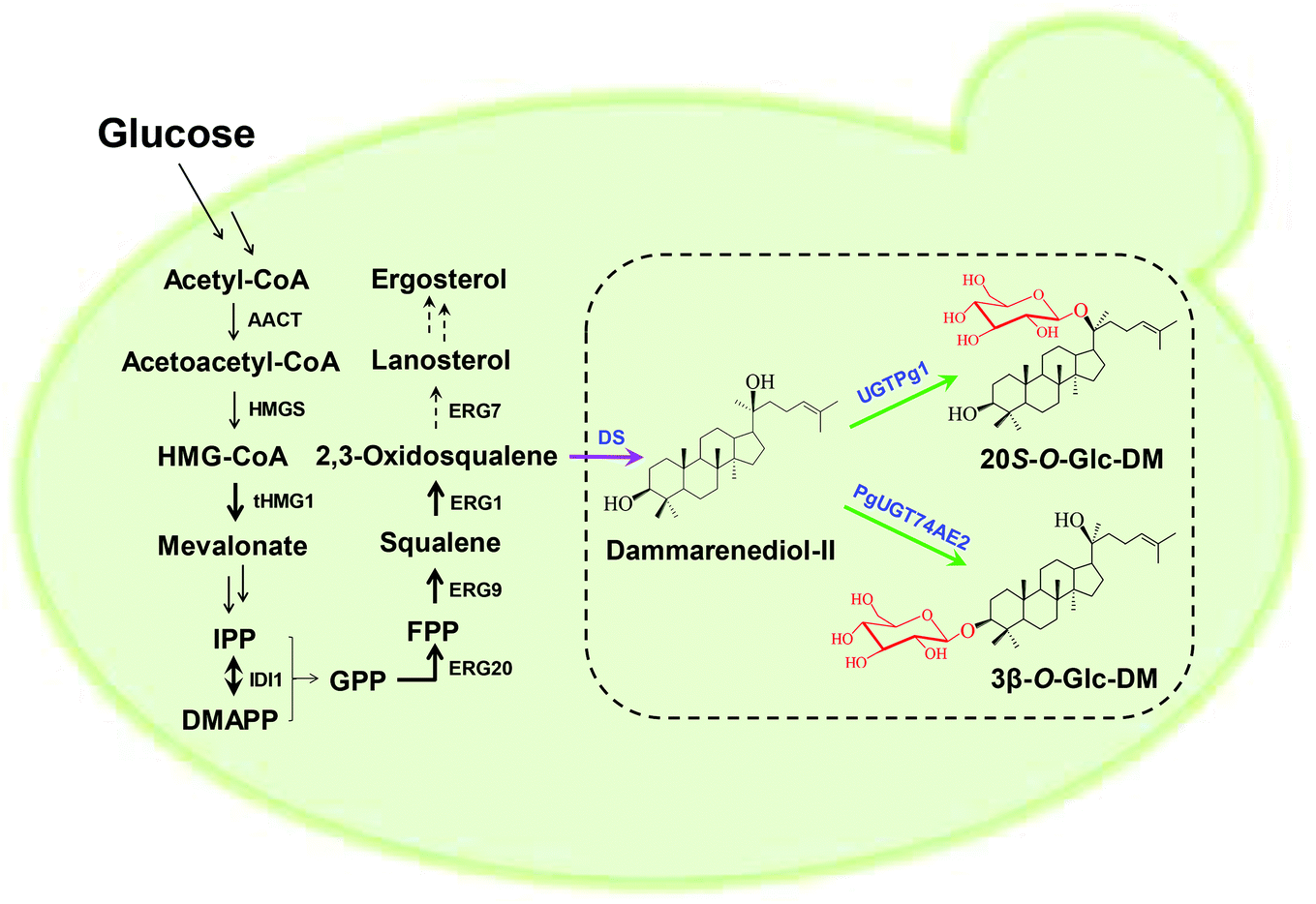 | ||
| Fig. 4 Refactored biosynthetic pathways of 3β-O-Glc-DM and 20S-O-Glc-DM in the engineered yeasts. Black arrows indicate the intrinsic S. cerevisiae pathway. HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; IPP, isopentenyl pyrophosphate; DMAPP, dimethylallyl pyrophosphate; GPP, geranyl diphosphate; FPP, farnesyl diphosphate; AACT, acetyl-CoA C-acetyltransferase; HMGS, 3-hydroxy-3-methylglutaryl-CoA synthase; tHMG1, truncated 3-hydroxy-3-methylglutaryl-CoA reductase; IDI1, isopentenyl diphosphate isomerase; ERG20, farnesyl diphosphate synthase; ERG9, squalene synthase; ERG1, squalene epoxidase; ERG7, lanosterol synthase; DS, dammarenediol-II synthase; UGT, UDP-glycosyltransferase. Multiple-step reactions are shown with double arrows; up-regulated reactions are shown with bold black arrows; and down-regulated reactions are shown with dashed arrows. The purple arrow represents the P. ginseng pathway. Green arrows represent the glycosylation reactions in this study. Black-colored genes are from S. cerevisiae; blue-colored genes are from P. ginseng. | ||
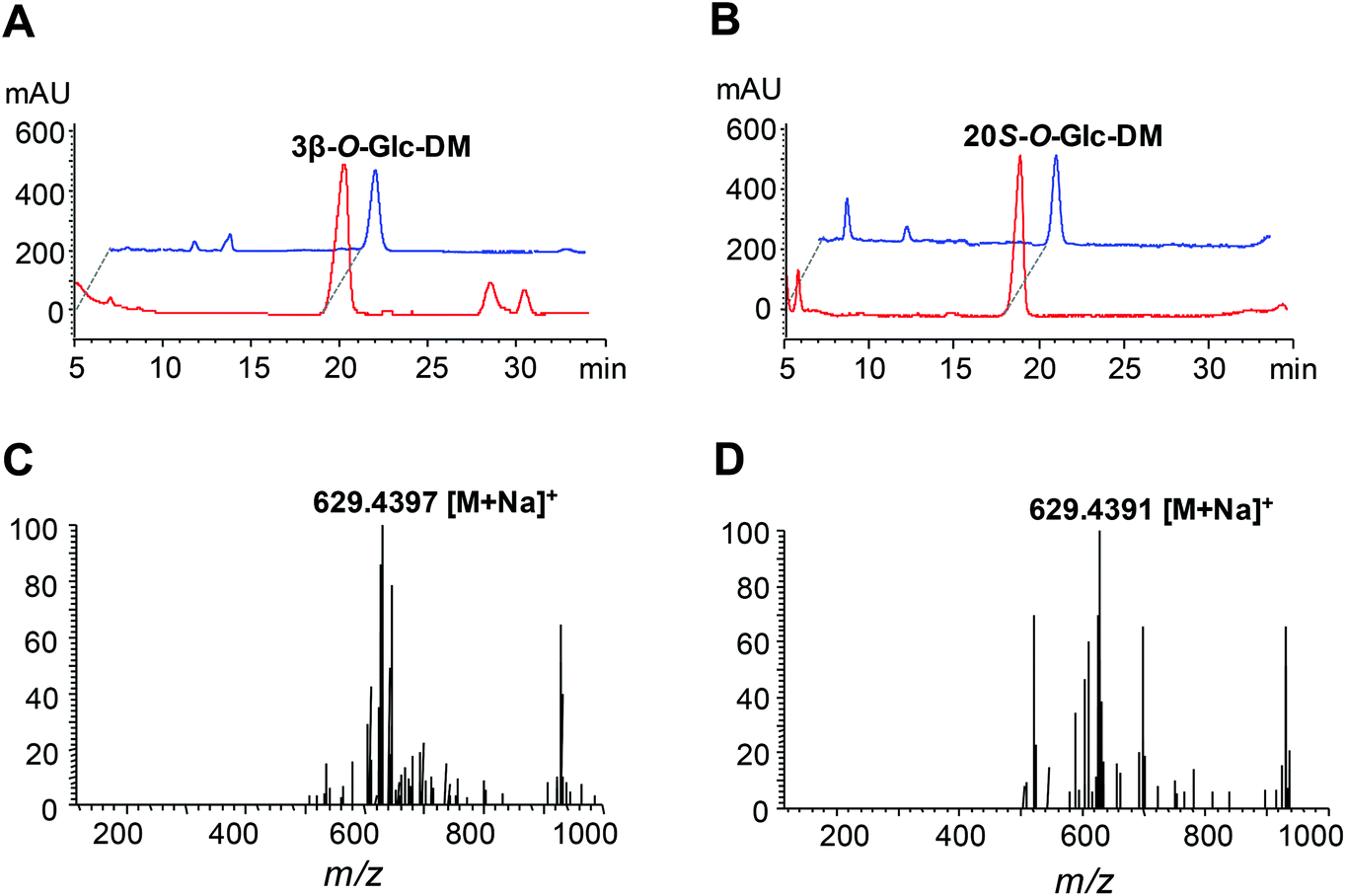 | ||
| Fig. 5 Production of 3β-O-Glc-DM and 20S-O-Glc-DM in the engineered yeasts. (A) HPLC analysis of the product of the strain Y1 extracted with n-butanol. Red, the product of the strain Y1; blue, authentic 3β-O-Glc-DM. (B) HPLC analysis of the product of the strain Y2 extracted with n-butanol. Red, the product of the strain Y2; blue, authentic 20S-O-Glc-DM. (C) The MS spectrum of the product of the strain Y1. (D) The MS spectrum of the product of the strain Y2. | ||
| Strain | DCW (g L−1) | Target products (mg L−1) | DM (mg L−1) | SQ (mg L−1) | ER (mg L−1) |
|---|---|---|---|---|---|
| a The titer of 3β-O-Glc-DM. b The titer of 20S-O-Glc-DM. All given data in this table represented mean values from three repeat experiments with corresponding standard deviations. The titers referred to the sum of intracellular and extracellular contents. DCW, dry cell weight; DM, dammarenediol-II; SQ, squalene; ER, ergosterol. | |||||
| Y1 | 12.8 ± 0.2 | 14.8 ± 0.4a | 34.7 ± 0.1 | 41.3 ± 0.8 | 26.1 ± 1.2 |
| Y1C | 15.1 ± 0.3 | 115.3 ± 1.0a | 56.0 ± 3.6 | 48.6 ± 0.1 | 33.0 ± 1.3 |
| Y1CS | 15.6 ± 0.3 | 261.9 ± 24.2a | 15.7 ± 0.7 | 56.5 ± 1.5 | 28.8 ± 0.9 |
| Y1CSB | 16.7 ± 0.6 | 264.0 ± 13.3a | 18.8 ± 0.2 | 72.4 ± 6.3 | 24.4 ± 0.8 |
| Y1CSH | 16.3 ± 0.7 | 414.8 ± 2.5a | 18.1 ± 0.8 | 75.8 ± 4.0 | 33.5 ± 0.5 |
| Y1CSP | 16.6 ± 0.3 | 374.6 ± 2.4a | 16.4 ± 0.4 | 55.1 ± 6.2 | 34.6 ± 0.9 |
| Y2 | 13.2 ± 0.3 | 18.6 ± 1.3b | 41.6 ± 2.6 | 43.5 ± 0.4 | 27.6 ± 0.1 |
| Y2C | 16.1 ± 0.1 | 265.1 ± 8.6b | 66.0 ± 1.6 | 48.0 ± 1.1 | 31.3 ± 0.1 |
| Y2CS | 17.8 ± 0.3 | 491.9 ± 4.3b | 16.5 ± 0.7 | 56.3 ± 1.5 | 29.8 ± 1.0 |
| Y2CSB | 19.3 ± 0.4 | 457.4 ± 12.5b | 21.2 ± 0.2 | 79.8 ± 0.6 | 35.4 ± 1.3 |
| Y2CSH | 18.7 ± 0.3 | 752.8 ± 38.4b | 16.8 ± 1.3 | 82.8 ± 0.4 | 32.2 ± 0.8 |
| Y2CSP | 19.0 ± 0.2 | 491.8 ± 51.2b | 23.2 ± 0.6 | 85.1 ± 1.3 | 43.6 ± 0.6 |
In general, a higher copy number of biosynthetic genes in the engineered strain may lead to higher production of the target product. δ site-specific integration is efficient in multi-copy integration of a single gene, while multi-copy integration of a long biosynthetic pathway involving several genes still remains a significant challenge.33,34 The CRISPR/Cas9 system has been demonstrated to be effective in improving the integration efficiency and integrated copy number of a long biosynthetic pathway and is thus widely used in genome engineering and metabolic engineering.21,22,35,36 In order to enhance the production of the target products, we introduced the CRISPR/Cas9 expression plasmid p-Cas9-gRNA together with the integration modules comprising the two biosynthetic pathways of 3β-O-Glc-DM and 20S-O-Glc-DM into Y-ΔHXK2, respectively. The colonies with the highest titers of 3β-O-Glc-DM and 20S-O-Glc-DM were selected from twenty colonies, respectively, and designated as Y1C and Y2C correspondingly, which were used for further engineering (Fig. S7†). As expected, the titer of 3β-O-Glc-DM in the strain Y1C remarkably increased 7.8-fold (from 14.8 to 115.3 mg L−1) and the titer of 20S-O-Glc-DM in the strain Y2C increased 14.3-fold (from 18.6 to 265.1 mg L−1) over their parent strains Y1 and Y2, respectively. Moreover, the production of precursors (including DM and squalene) and ergosterol was also boosted in Y1C and Y2C. In addition, the cell biomass of Y1C and Y2C in fermentation was unexpectedly higher than that of Y1 and Y2, suggesting that the high production of 3β-O-Glc-DM and 20S-O-Glc-DM did not interfere with the growth of the engineered strains at all (Table 5).
In order to investigate the relationship between the integrated gene dosages and the production of the target products, the gene copy numbers were compared among the recombinants with different titers of the target products. As shown in Fig. 6, the titers of both 3β-O-Glc-DM and 20S-O-Glc-DM were closely correlated with the gene dosages integrated into the engineered yeasts, and they were enhanced gradually with the increase of the gene copy number to a certain extent. The strain with the highest gene copy number produced the highest titer of 3β-O-Glc-DM. Strains 3 and 7 had similar gene copy numbers, but their titers of 3β-O-Glc-DM were obviously different. It was probably because the similar copies of genes were integrated into the different δ1 sites of S. cerevisiae chromosomes, which led to different expression levels of recombinant enzymes and thus resulted in the different titers of 3β-O-Glc-DM (Fig. 6A). However, the titer of 20S-O-Glc-DM was not always positively correlated with the integrated gene copy number (Fig. 6B). The highest gene copy number might not result in the highest titer of the target product probably due to the fact that excessive overexpression of the heterologous pathway might exert too much metabolic burden on the engineered strain.
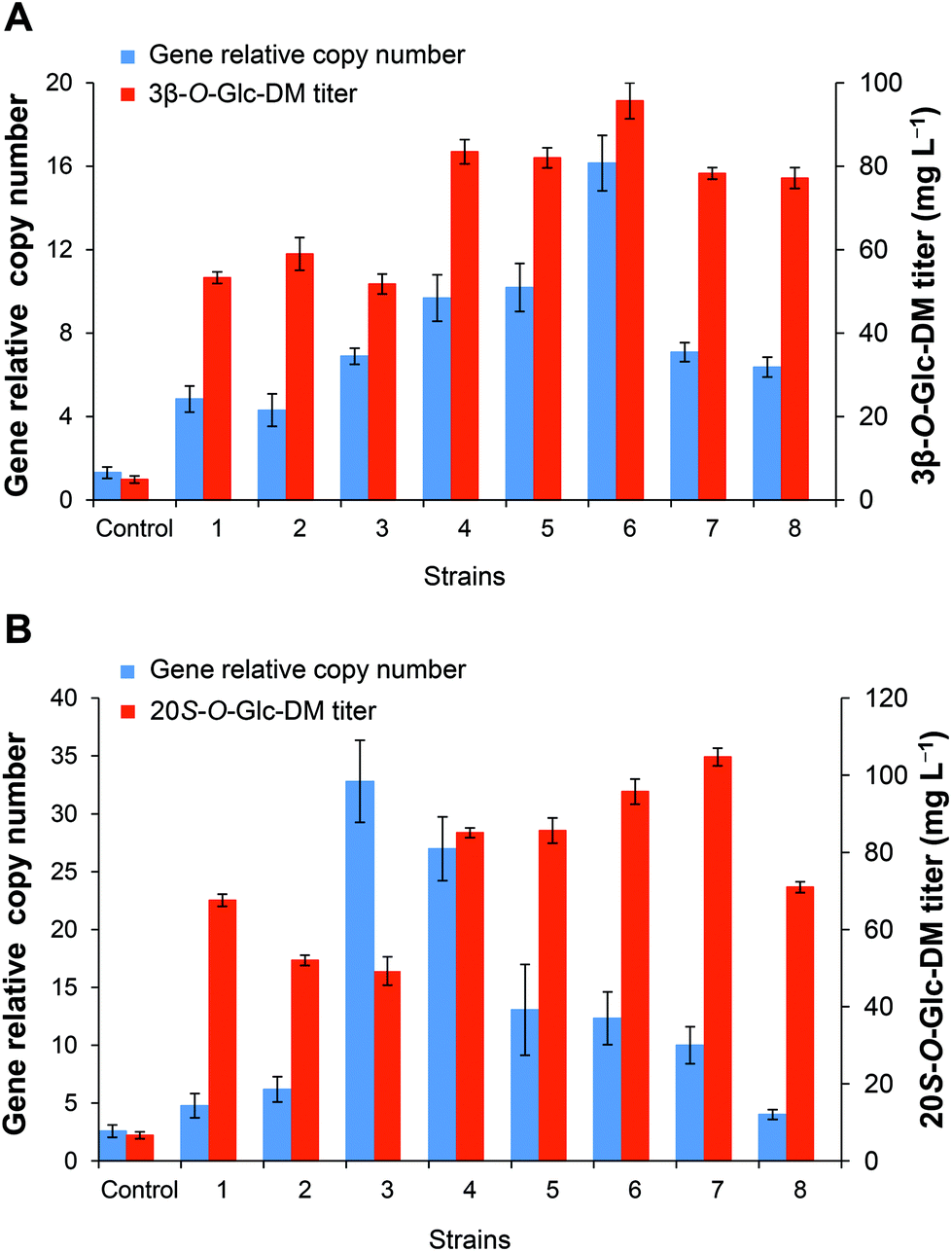 | ||
| Fig. 6 The relationship between the copy numbers of the DS gene and the titers of DM glucosides. (A) The engineered strains producing 3β-O-Glc-DM. Control, the engineered strain obtained by conventional homologous recombination; 1–8, the engineered strains obtained by CRISPR/Cas9 directed homologous recombination. (B) The engineered strains producing 20S-O-Glc-DM. Control, the engineered strain obtained by conventional homologous recombination; 1–8, the engineered strains obtained by CRISPR/Cas9 directed homologous recombination. All strains were cultivated in YPD medium for 3 days. | ||
Improving the precursor supply to increase the production of 3β-O-Glc-DM and 20S-O-Glc-DM in the engineered yeasts
The engineered pathways of 3β-O-Glc-DM and 20S-O-Glc-DM in S. cerevisiae involved the endogenous MVA pathway from glucose to IPP and DMAPP, the pathway from IPP and DMAPP to 2,3-oxidosqualene, and heterologous pathways from 2,3-oxidosqualene to 3β-O-Glc-DM or 20S-O-Glc-DM, respectively (Fig. 4). The increased precursor supply may be able to further increase the production of the target products. HMG-CoA reductase is a rate-limiting enzyme of the MVA pathway in S. cerevisiae, and overexpression of tHMG1 has been employed to increase the production of many terpenoids, such as artemisinic acid,28,37 taxadiene,38 gisnenosides13,17,20 and oleanolic acid.39 Accordingly, we overexpressed tHMG1 in the engineered strains to increase the carbon flux through the MVA pathway. Moreover, the cyclization of 2,3-oxidosqualene is the key branch point of endogenous ergosterol and heterogeneous triterpenoid biosynthesis. Consequently, several enzymes responsible for 2,3-oxidosqualene biosynthesis, including IDI1, ERG20, ERG9 and ERG1, were also overexpressed to improve the 2,3-oxidosqualene supply for increasing triterpenoid production. Meanwhile, ERG7 responsible for ergosterol biosynthesis was down-regulated by antisense technology to redirect the metabolic flux towards the triterpenoid biosynthetic pathway in S. cerevisiae (Fig. 4).40We overexpressed IDI1, ERG20, ERG9 and ERG1 by integrating these genes into the chromosomes of Y1C and Y2C at the δ4 site while suppressing the expression of ERG7 with the antisense fragment to improve the production of 3β-O-Glc-DM and 20S-O-Glc-DM. The colonies with the highest titers of 3β-O-Glc-DM and 20S-O-Glc-DM were selected from twenty colonies, respectively, and designated as Y1CS and Y2CS correspondingly (Fig. S8†). As shown in Table 5, compared with the parent strains Y1C and Y2C, the titers of 3β-O-Glc-DM in the strain Y1CS and 20S-O-Glc-DM in the strain Y2CS increased from 115.3 to 261.9 mg L−1 and from 265.1 to 491.9 mg L−1, while the titers of squalene increased from 48.6 to 56.5 mg L−1 and from 48.0 to 56.3 mg L−1, respectively. However, the titers of ergosterol were reduced slightly from 33.0 to 28.8 mg L−1 and from 31.3 to 29.8 mg L−1 in Y1CS and Y2CS, respectively. These results indicated that overexpression of IDI1, ERG20, ERG9 and ERG1 greatly enhanced the production of 3β-O-Glc-DM and 20S-O-Glc-DM by opening up the source of their precursor squalene, and ergosterol biosynthesis was suppressed by down-regulation of ERG7 expression. In contrast, the titers of DM in Y1CS and Y2CS were decreased sharply from 56.0 to 15.7 mg L−1 and from 66.0 to 16.5 mg L−1, respectively, which might be because DM was rapidly converted into the target products by UGTs and thus rarely accumulated.
Effect of overexpressing of chaperone and transcriptional activator genes on the production of 3β-O-Glc-DM and 20S-O-Glc-DM
The high production of the target products in the engineered strains depends on the high heterologous expression levels of their biosynthetic enzymes. In order to further increase the production of DM glucosides in the engineered strains, several chaperones and transcriptional activators were overexpressed to up-regulate the expression of related enzymes. Among them, PDI1 is a chaperone for facilitating appropriate folding of proteins on the endoplasmic reticulum, while BiP is another endoplasmic reticulum resident protein directly involved in the process of protein folding and assembling. Moreover, HAC1, a transcription factor of the unfolded protein response pathway, can induce the expression of chaperones and enzymes involved in the secretion process to increase the expression of heterologous proteins.In this study, BiP, HAC1 and PDI1 genes were integrated into the chromosomes of Y1CS and Y2CS at the rDNA site, respectively. The colonies with the highest titers of 3β-O-Glc-DM were selected from ten colonies, respectively, and designated as Y1CSB, Y1CSH and Y1CSP correspondingly. Similarly, the colonies with the highest titers of 20S-O-Glc-DM were also selected and designated as Y2CSB, Y2CSH and Y2CSP correspondingly (Fig. S9†). As shown in Table 5, the titers of 3β-O-Glc-DM and 20S-O-Glc-DM increased from 261.9 to 414.8 mg L−1 and from 491.9 to 752.8 mg L−1 in Y1CSH and Y2CSH, by overexpression of HAC1, which were 1.6-fold and 1.5-fold those of Y1CS and Y2CS, respectively. In the meantime, the titers of squalene in Y1CSH and Y2CSH also increased from 56.5 to 75.8 mg L−1 and from 56.3 to 82.8 mg L−1, respectively. However, overexpression of BiP and PDI1 had no observable effect on the production of 3β-O-Glc-DM and 20S-O-Glc-DM except that overexpression of PDI1 increased the titer of 3β-O-Glc-DM from 261.9 to 374.6 mg L−1.
In summary, the titers of 3β-O-Glc-DM and 20S-O-Glc-DM in the engineered yeasts greatly increased 28.0-fold (from 14.8 to 414.8 mg L−1) and 40.5-fold (from 18.6 to 752.8 mg L−1), respectively, by integrating multi-copy metabolic pathway genes into yeasts via the CRISPR/Cas9 system, increasing the precursor supply through overexpressing key enzymes, and improving the expression of heterologous enzymes through overexpressing transcriptional activator HAC1 (Fig. 7). We speculated that the reason for the lower titer of 3β-O-Glc-DM than that of 20S-O-Glc-DM in the engineered yeasts was that the catalytic efficiency of PgUGT74AE2 towards DM was lower than that of UGTPg1. Besides, the engineered pathways of 3β-O-Glc-DM and 20S-O-Glc-DM involve several redox enzymes such as tHMG1 and ERG9. In order to further increase the production of 3β-O-Glc-DM and 20S-O-Glc-DM, the cofactor engineering strategies can be used for solving the problematic redox imbalance in metabolic modification in the future.41,42
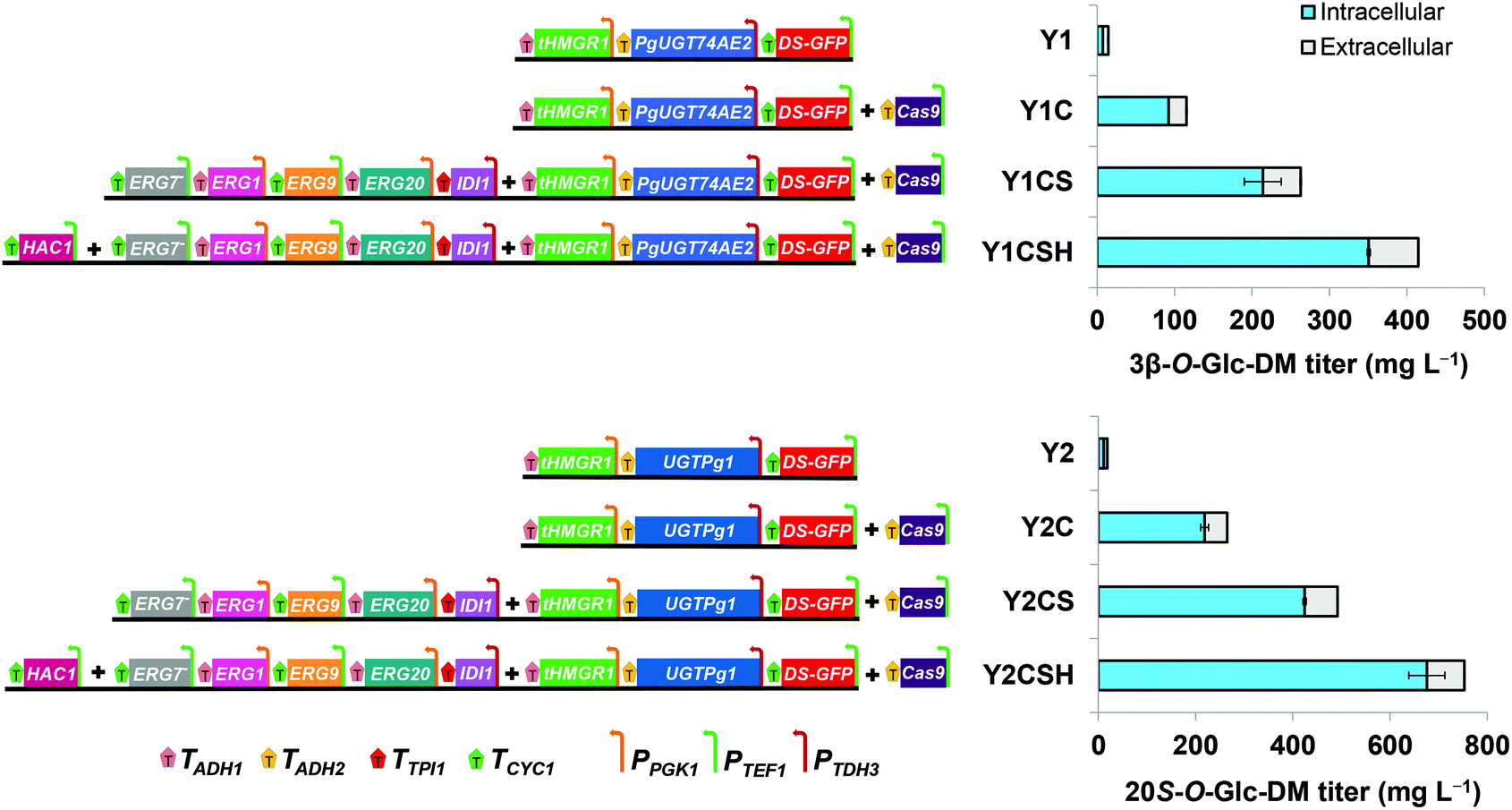 | ||
| Fig. 7 Production of 3β-O-Glc-DM and 20S-O-Glc-DM in the engineered yeasts. | ||
Production of 3β-O-Glc-DM and 20S-O-Glc-DM through fed-batch fermentation
In order to achieve higher production of 3β-O-Glc-DM and 20S-O-Glc-DM, we chose the engineered strains Y1CSH and Y2CSH for fed-batch fermentation which was performed in a 3 L bioreactor with the pH controlled at 5.5. As shown in Fig. 8, the fermentation batch phase began with 20 g L−1 glucose, and exponential feeding was initiated after glucose was almost depleted at about 20 h after inoculation.29 The cell biomass of Y1CSH continued to increase rapidly until 96 h, then maintained a steady state and reached the maximum OD600 of 1522 at 168 h. After that, the cell biomass gradually decreased. Production of 3β-O-Glc-DM was detected both in cell and medium from 24 h to 192 h. The total titer of 3β-O-Glc-DM increased rapidly with continued cell growth until the highest titer was up to 2.4 g L−1 at 144 h, while it decreased to 2.3 g L−1 at 192 h (Fig. 8A). Similarly, the cell biomass of Y2CSH continued to increase rapidly until 72 h and reached the maximum OD600 of 1125 at 168 h. The titer of 20S-O-Glc-DM continued to increase with continued cell growth until the highest titer reached 5.6 g L−1 at 144 h, and remained basically constant until 192 h (Fig. 8B). These results confirmed that the process of fed-batch fermentation adopted in this study had great potential for the industrial production of DM glucosides using the engineered yeasts.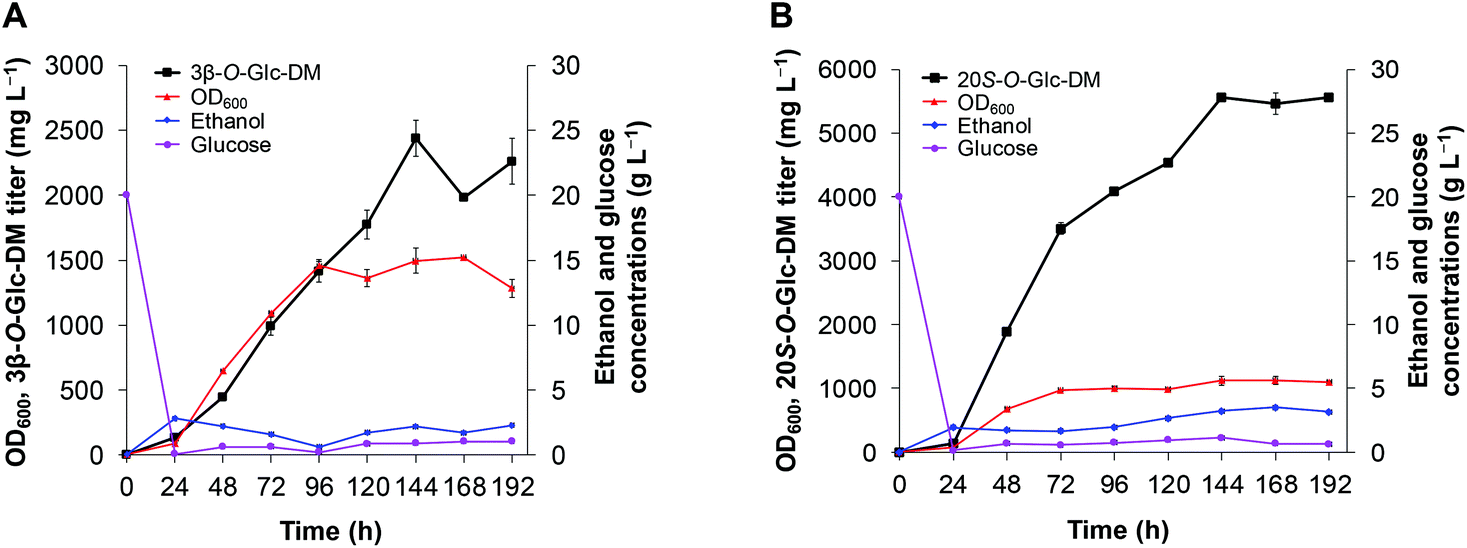 | ||
| Fig. 8 Production of DM glucosides through fed-batch fermentation in a 3 L bioreactor. (A) 3β-O-Glc-DM production using the engineered strain Y1CSH. (B) 20S-O-Glc-DM production using the engineered strain Y2CSH. | ||
Conclusion
In this study, we used PgUGT74AE2 and UGTPg1 from P. ginseng to catalyze the glycosylation of DM to produce DM glucosides 3β-O-Glc-DM and 20S-O-Glc-DM, respectively. We demonstrated that both 3β-O-Glc-DM and 20S-O-Glc-DM exhibited higher anti-colon cancer activities than Rg3 and compound K by in vitro and in vivo assays. For more efficient and sustainable production of 3β-O-Glc-DM and 20S-O-Glc-DM, we successfully refactored their biosynthetic pathways by introducing P. ginseng DS together with PgUGT74AE2 or UGTPg1 genes into S. cerevisiae and further optimized them by multistep metabolic engineering strategies. Furthermore, we also established the fed-batch fermentation process for the two engineered yeasts. The titers of 3β-O-Glc-DM and 20S-O-Glc-DM reached up to 2.4 g L−1 and 5.6 g L−1 in a 3 L bioreactor, respectively. This is the first study to report the high anti-colon cancer activities of DM glucosides and the de novo biosynthesis of DM glucosides with high titers by a green and sustainable approach. The engineered yeast strains developed in this work can serve as microbial cell factories for the industrial production of DM glucosides. Our work has laid a foundation for the research and development of DM glucosides.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Prof. L. Lu (College of Life Sciences of Nanjing Normal University) for providing plasmid FM-1 and Y. J. Yuan (Tianjin University, China) for synthesizing DS, PgUGT74AE2 and UGTPg1 genes. This research was financially supported by the grants from the National Natural Science Foundation of China (81673341), CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-3-012), the Drug Innovation Major Project (2018ZX09711001-006) and the Fundamental Research Funds for the Central Universities (2017PT35001).References
- S. Shibata, J. Korean Med. Sci., 2001, 16, 28–37 CrossRef PubMed.
- L. P. Christensen, Adv. Food Nutr. Res., 2009, 55, 1–99 CAS.
- Y. J. Kim, D. B. Zhang and D. C. Yang, Biotechnol. Adv., 2015, 33, 717–735 CrossRef CAS PubMed.
- C. F. Chen, W. F. Chiou and J. T. Zhang, Acta Pharmacol. Sin., 2008, 29, 1103–1108 CrossRef CAS PubMed.
- J. F. Xue, Z. J. Liu, J. F. Hu, H. Chen, J. T. Zhang and N. H. Chen, Brain Res., 2006, 1106, 91–98 CrossRef CAS PubMed.
- L. N. Atopkina, G. V. Malinovskaya, G. B. Elyakov, N. I. Uvarova and H. J. Woerdenbag, Planta Med., 1999, 65, 30–34 CrossRef CAS PubMed.
- B. J. Li, H. Wang, T. Gong, J. J. Chen, T. J. Chen, J. L. Yang and P. Zhu, Nat. Commun., 2017, 8, 15544 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, I. Llarena, A. Sánchez-Iglesias and F. López-Gallego, ACS Synth. Biol., 2018, 7, 875–884 CrossRef.
- Y. Z. Luo, B. Z. Li, D. Liu, L. Zhang, Y. Chen, B. Jia, B. X. Zeng, H. M. Zhao and Y. J. Yuan, Chem. Soc. Rev., 2015, 44, 5265–5290 RSC.
- H. Y. Wu, Y. J. Li, Q. Ma, Q. Li, Z. F. Jia, B. Yang, Q. Y. Xu, X. G. Fan, C. L. Zhang, N. Chen and X. X. Xie, Metab. Eng., 2018, 49, 248–256 CrossRef CAS PubMed.
- A. Krivoruchko and J. Nielsen, Curr. Opin. Biotechnol., 2015, 35, 7–15 CrossRef CAS.
- P. Tansakul, M. Shibuya, T. Kushiro and Y. Ebizuka, FEBS Lett., 2006, 580, 5143–5149 CrossRef CAS PubMed.
- P. P. Wang, Y. J. Wei, Y. Fan, Q. F. Liu, W. Wei, C. S. Yang, L. Zhang, G. P. Zhao, J. M. Yue, X. Yan and Z. H. Zhou, Metab. Eng., 2015, 29, 97–105 CrossRef CAS PubMed.
- S. C. Jung, W. Kim, S. C. Park, J. Jeong, M. K. Park, S. Lim, Y. Lee, W.-T. Im, J. H. Lee, G. Choi and S. C. Kim, Plant Cell Physiol., 2014, 55, 2177–2188 CrossRef CAS PubMed.
- W. Wei, P. P. Wang, Y. J. Wei, Q. F. Liu, C. S. Yang, G. P. Zhao, J. M. Yue, X. Yan and Z. H. Zhou, Mol. Plant, 2015, 8, 1412–1424 CrossRef CAS PubMed.
- X. Yan, Y. Fan, W. Wei, P. P. Wang, Q. F. Liu, Y. Wei, L. Zhang, G. P. Zhao, J. M. Yue and Z. H. Zhou, Cell Res., 2014, 24, 770–773 CrossRef CAS PubMed.
- Z. B. Dai, Y. Liu, X. N. Zhang, M. Y. Shi, B. B. Wang, D. Wang, L. Q. Huang and X. L. Zhang, Metab. Eng., 2013, 20, 146–156 CrossRef CAS PubMed.
- Z. B. Dai, B. B. Wang, Y. Liu, M. Y. Shi, D. Wang, X. N. Zhang, T. Liu, L. Q. Huang and X. L. Zhang, Sci. Rep., 2014, 4, 3698 CrossRef PubMed.
- Y. Zhuang, G. Y. Yang, X. H. Chen, Q. Liu, X. L. Zhang, Z. X. Deng and Y. Feng, Metab. Eng., 2017, 42, 25–32 CrossRef CAS PubMed.
- H. C. Liang, Z. F. Hu, T. T. Zhang, T. Gong, J. J. Chen, P. Zhu, Y. Li and J. L. Yang, Metab. Eng., 2017, 44, 60–69 CrossRef CAS.
- C. Zhang, X. H. Meng, X. L. Wei and L. Lu, Fungal Genet. Biol., 2016, 86, 47–57 CrossRef CAS PubMed.
- C. Ronda, J. Maury, T. Jakočiūnas, S. A. B. Jacobsen, S. M. Germann, S. J. Harrison and A. T. Nielsen, Microb. Cell Fact., 2015, 14, 97 CrossRef PubMed.
- Z. Q. Sun, H. L. Meng, J. Li, J. F. Wang, Q. Li, Y. Wang and Y. S. Zhang, PLoS One, 2014, 9, e112615 CrossRef PubMed.
- D. Burke, D. Dawson and T. I. M. Stearns, Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, Cold Spring Harbor Laboratory Press, New York, 2000 Search PubMed.
- H. C. Liang, L. L. Gao, Z. F. Hu, T. Gong, J. J. Chen, J. L. Yang and P. Zhu, Acta Pharm. Sin., 2016, 51, 998–1003 Search PubMed.
- Z. Y. Shao, H. Zhao and H. M. Zhao, Nucleic Acids Res., 2009, 37, e16 CrossRef PubMed.
- Q. H. Wang, L. Liang, W. C. Liu, T. Gong, J. J. Chen, Q. Hou, J. L. Yang and P. Zhu, J. Asian Nat. Prod. Res., 2017, 19, 581–594 CrossRef CAS PubMed.
- P. J. Westfall, D. J. Pitera, J. R. Lenihan, D. Eng, F. X. Woolard, R. Regentin, T. Horning, H. Tsuruta, D. J. Melis, A. Owens, S. Fickes, D. Diola, K. R. Benjamin, J. D. Keasling, M. D. Leavell, D. J. McPhee, N. S. Renninger, J. D. Newman and C. J. Paddon, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E111–E118 CrossRef CAS PubMed.
- J. R. Lenihan, H. Tsuruta, D. Diola, N. S. Renninger and R. Regentin, Biotechnol. Prog., 2008, 24, 1026–1032 CrossRef CAS PubMed.
- T. T. Zhang, T. Gong, Z. F. Hu, A. D. Gu, J. L. Yang and P. Zhu, Acta Pharm. Sin., 2018, 53, 1565–1570 Search PubMed.
- H. D. Yuan, H. Y. Quan, Y. Zhang, S. H. Kim and S. H. Chung, Mol. Med. Rep., 2010, 3, 825–831 CAS.
- M. K. Choo, H. Sakurai, D. H. Kim and I. Saiki, Oncol. Rep., 2008, 19, 595–600 CAS.
- J. Yuan and C. B. Ching, Biotechnol. Bioeng., 2013, 111, 608–617 CrossRef.
- J. Yuan and C. B. Ching, ACS Synth. Biol., 2014, 4, 23–31 CrossRef PubMed.
- S. B. Shi, Y. Y. Liang, M. M. Zhang, E. L. Ang and H. M. Zhao, Metab. Eng., 2016, 33, 19–27 CrossRef CAS.
- T. Jakočiūnas, M. K. Jensen and J. D. Keasling, Metab. Eng., 2016, 34, 44–59 CrossRef.
- C. J. Paddon, P. J. Westfall and D. J. Pitera, Nature, 2013, 496, 528–532 CrossRef CAS PubMed.
- B. Engels, P. Dahm and S. Jennewein, Metab. Eng., 2008, 10, 201–206 CrossRef CAS PubMed.
- Y. J. Zhao, J. J. Fan, C. Wang, X. D. Feng and C. Li, Bioresour. Technol., 2018, 257, 339–354 CrossRef CAS.
- Q. H. Wang, L. L. Gao, H. C. Liang, G. H. Du, T. Gong, J. L. Yang and P. Zhu, Acta Pharm. Sin., 2015, 50, 118–122 CAS.
- J. E. Kim, I. S. Jang, B. H. Sung, S. C. Kim and J. Y. Lee, Sci. Rep., 2018, 8, 15820 CrossRef.
- J. Liu, H. Li, G. Zhao, Q. Caiyin and J. Qiao, J. Ind. Microbiol. Biotechnol., 2018, 45, 313–327 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc04066d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |