
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis and polymerisation of β-mannosyl acrylates produced from renewable hemicellulosic glycans†
Anna
Rosengren‡
a,
Samuel J.
Butler‡
a,
Monica
Arcos-Hernandez
b,
Karl-Erik
Bergquist
b,
Patric
Jannasch
 b and
Henrik
Stålbrand
*a
b and
Henrik
Stålbrand
*a
aDepartment of Biochemistry and Structural Biology, Department of Chemistry, Lund University, PO Box 124, S-221 00 Lund, Sweden. E-mail: Henrik.Stalbrand@biochemistry.lu.se; Fax: +46-46-222 4116; Tel: +46-46-222 8202
bCentre for Analysis and Synthesis, Department of Chemistry, Lund University, PO Box 124, S-221 00 Lund, Sweden
First published on 21st March 2019
Abstract
We show that glycoside hydrolases can catalyse the synthesis of glycosyl acrylate monomers using renewable hemicellulose as a glycosyl donor, and we also demonstrate the preparation of novel glycopolymers by radical polymerisation of these monomers. For this, two family 5 β-mannanases (TrMan5A from Trichoderma reesei and AnMan5B from Aspergillus niger) were evaluated for their transglycosylation capacity using 2-hydroxyethyl methacrylate (HEMA) as a glycosyl acceptor. Both enzymes catalysed conjugation between manno-oligosaccharides and HEMA, as analysed using MALDI-ToF mass spectrometry (MS) as an initial product screening method. The two enzymes gave different product profiles (glycosyl donor length) with HEMA, and with allyl alcohol as acceptor molecules. AnMan5A appeared to prefer saccharide acceptors with lower intensity MS peaks detected for the desired allyl and HEMA conjugates. In contrast to AnMan5A, TrMan5A showed pronounced MS peaks for HEMA-saccharide conjugation products. TrMan5A was shown to catalyse the synthesis of β-mannosyl acrylates using locust bean gum galactomannan or softwood hemicellulose (acetyl-galactoglucomannan) as a donor substrate. Evaluation of reaction conditions using galactomannan as a donor, HEMA as an acceptor and TrMan5A as an enzyme catalyst was followed by the enzymatic production and preparative liquid chromatography purification of 2-(β-manno(oligo)syloxy) ethyl methacrylates (mannosyl-EMA and mannobiosyl-EMA). The chemical structures and radical polymerisations of these novel monomers were determined using 1H and 13C NMR spectroscopy and size-exclusion chromatography. The two new water soluble polymers have a polyacrylate backbone with one or two pendant mannosyl groups per monomeric EMA unit, respectively. These novel glycopolymers may show properties suitable for various technical and biomedical applications responding to the current demand for functional greener materials to replace fossil based ones.
Introduction
Significant research efforts are currently devoted to establishing novel routes for the utilisation of major renewable resources in the form of plant biomass. The major motivation is to replace fossil resources in the production of energy, chemicals and materials. Glycans build up the major part of plants, in which hemicellulose constitutes a large hitherto unused resource.1 Simultaneously there is increased interest to utilise defined glycan moieties in novel glycopolymer structures, in which the backbone carries pendant saccharide groups.2 Here the term glycopolymer refers to polymers synthesised by the polymerisation of glycomonomers which are saccharides conjugated to a reactive group, for example acrylates.By attachment of functional groups to saccharides, creating functionalised glycomonomers, novel glycopolymers can be synthesised, e.g., through thiol–ene click chemistry or radical polymerisation.3–6 The polar carbohydrate groups in glycopolymers can be expected to facilitate water solubility and decrease backbone association. Many application areas can be foreseen and examples include colloidal stabilisers and coatings.7 The glycan moieties in glycopolymers can also provide powerful biological recognition for biomedical and biocontrol applications.2
The complexity of carbohydrate structures leads to challenges in the synthesis of glycomonomers for polymerisation reactions. Synthesis of glycopolymers such as polyacrylates with pendant saccharides requires well defined saccharide acrylate monomers as starting materials. The chemical synthesis of such glycomonomers is very demanding and includes multiple protection and deprotection steps.8 There is increasing interest in utilising enzymatic synthesis for these applications. Enzymatic synthesis is a greener alternative compared to chemical synthesis.9 Advantages of enzymatic synthesis include high specificity, operation at ambient temperature, and avoiding harsh chemicals and toxic by-products.10 Furthermore, regio- and stereo-selective synthesis of glycoconjugates can be achieved with enzyme catalysis, without several steps of protection chemistry.11 These advantages make it attractive to use renewable and abundant glycans as glycosyl donor substrates in enzyme catalysed reactions with functional glycosyl acceptors. Such reactions result in novel reactive glycomonomers, which in turn can be polymerised into glycopolymers. Glycoside hydrolases (GHs) with the retaining mechanism are of particular interest since they may, in addition to bond cleavage, catalyse the synthesis of new glycoside bonds via kinetically controlled transglycosylation12–14 (Scheme 1). During the reaction, the glycosyl donor substrate is conjugated with a glycosyl acceptor. The reactive part of the acceptor is a hydroxyl group and alcohols can be used in the synthesis of alkyl glycosides (green surfactants).13,15
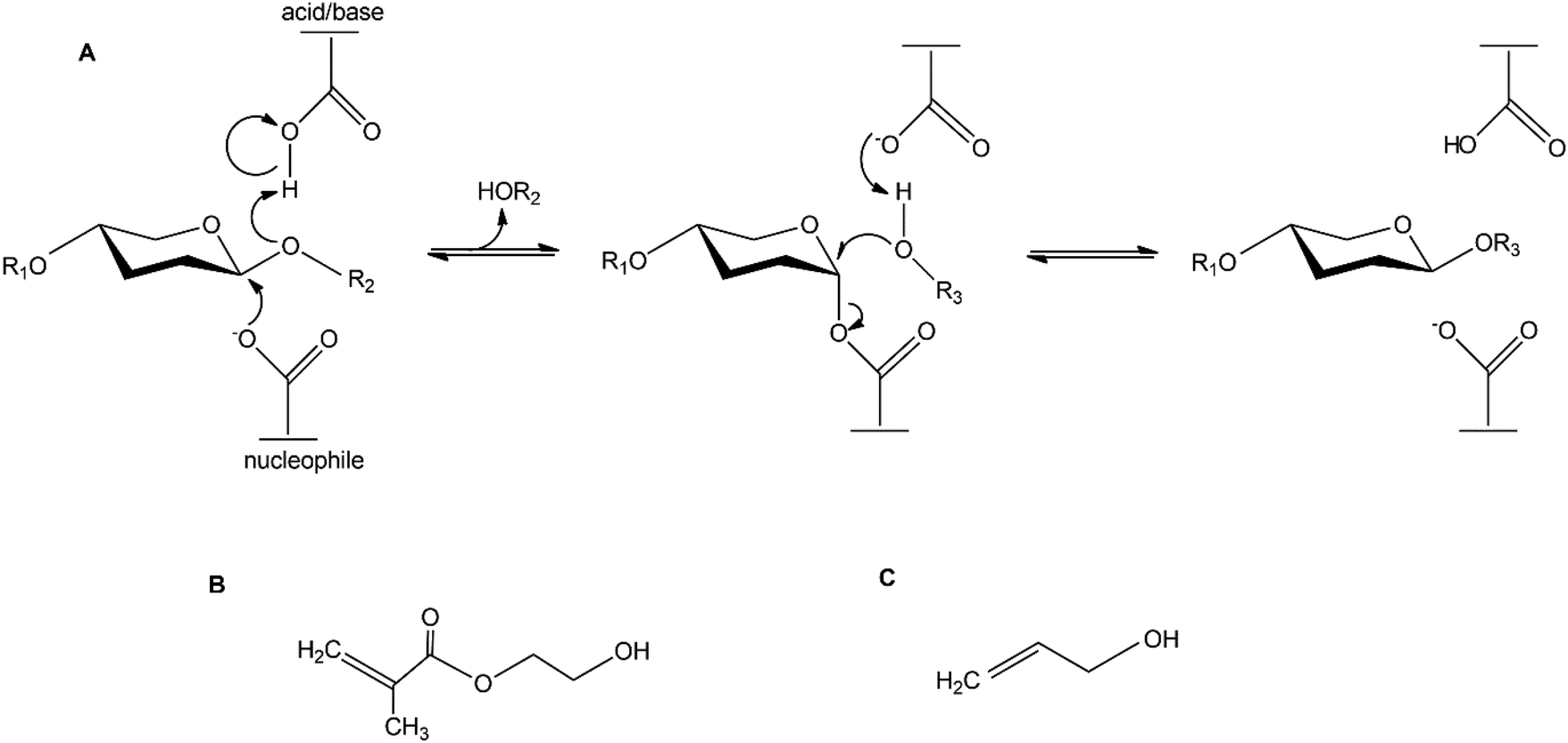 | ||
| Scheme 1 (A) Retaining mechanism of glycoside hydrolases. R1 and R2 represents the continuation of the saccharide chain. R3 represents the continuation of the acceptor molecule The acceptor molecules used in this study are: (B) 2-hydroxy ethyl methacrylate (HEMA) and (C) allyl alcohol. | ||
A few studies on enzymatic synthesis of glucosyl acrylates, catalysed by amylases or β-glucosidases, with starch5 or cellobiose16 as donor substrates have previously been reported. In this study we investigate the possibility of using abundant and renewable β-mannan as the donor substrate for the synthesis of mannosyl acrylates. There are different types of polymeric β-mannans (Scheme 2). Galactomannan is found in plant seeds in the form of industrially applied locust bean gum (LBG) and the more complex polysaccharide O-acetyl galactoglucomannan (AcGGM) is the main hemicellulose in softwoods.1 Constituting up to 25% of the dry wood weight, AcGGM is a major renewable resource which can be obtained from forest industry waste streams.17–19
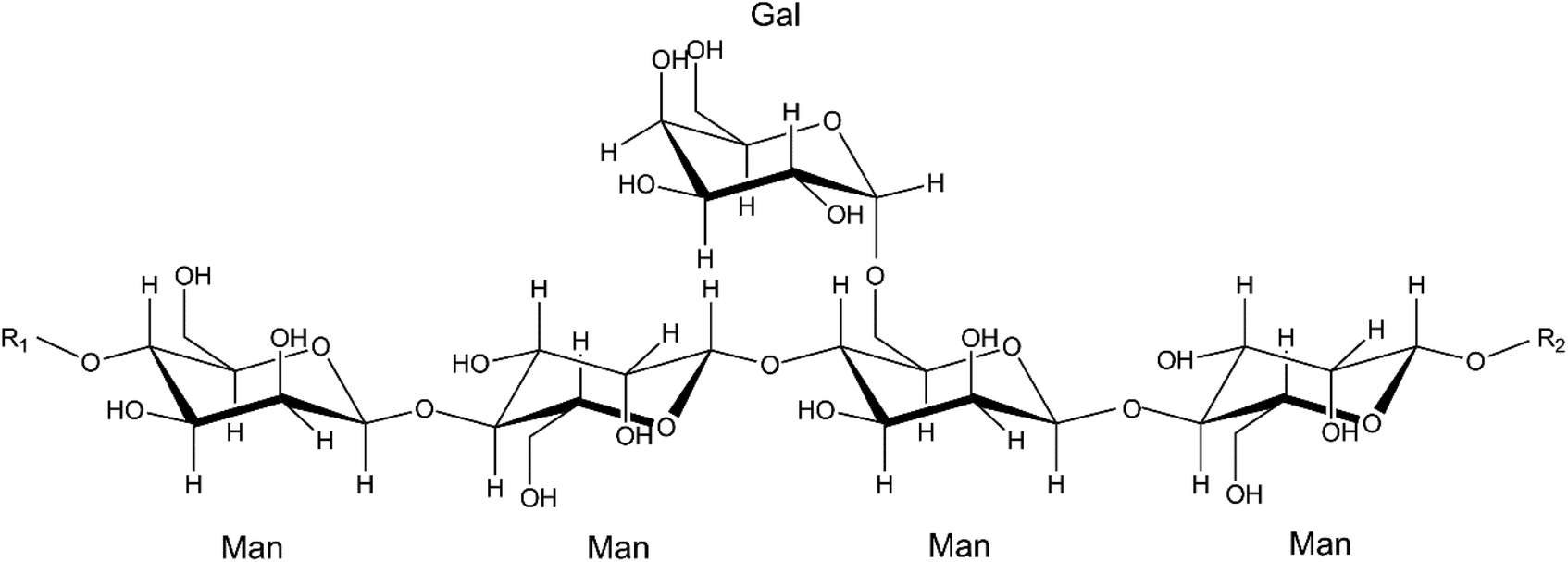 | ||
Scheme 2 Molecular structure of locust bean gum galactomannan (LBG) with a galactose![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mannose ratio of 1:4. Man = mannose, Gal = galactose. In O-acetyl galactoglucomannan (AcGGM), the mannosyl units can be acetylated at positions C2 and C3 to various extents, and the polysaccharide backbone also contains glucose moieties. R1 and R2 represent the continuation of the glycan chain. :mannose ratio of 1:4. Man = mannose, Gal = galactose. In O-acetyl galactoglucomannan (AcGGM), the mannosyl units can be acetylated at positions C2 and C3 to various extents, and the polysaccharide backbone also contains glucose moieties. R1 and R2 represent the continuation of the glycan chain. | ||
Among the enzymes responsible for efficient degradation of AcGGM and other β-mannans are the GH family 5 β-mannanases. β-Mannanases catalyse the cleavage of β-mannans and due to their catalytic mechanism being retained they may have the ability to catalyse transglycosylation, i.e. the synthesis of β-mannosidic bonds, fusing donor saccharides and acceptor molecules as described above. This can be utilised for enzymatic synthesis of novel glycoconjugates. The active site of β-mannanases is often located in a cleft where two catalytic amino acids are situated. During the catalytic event, a covalent intermediate is formed between the enzyme and the saccharide. This intermediate is then disrupted by either a water molecule (in hydrolysis) or another acceptor molecule (in transglycosylation), the latter leading to the synthesis of a new glycoconjugate (Scheme 1). We have previously shown that the β-mannanase TrMan5A from Trichoderma reesei possesses the ability to perform transglycosylation, linking donor mannosides to various acceptor alcohols.20–22 We have also previously studied the synthetic capacity of β-mannanases from Aspergillus nidulans and found one β-mannanase, AnMan5B, with exceptionally high transglycosylation capacity with saccharide acceptors compared to other β-mannanases.21
Here we expand our previous approaches on enzymatic synthesis, with a focus on investigating the potential use of a polymerisable acceptor molecule, hydroxyethyl methacrylate (HEMA) (Scheme 1B), in transglycosylation reactions with β-mannanases (TrMan5A and AnMan5B) and different β-mannans. The primary aim is to produce novel mannosyl acrylates to be used as glycomonomers in polymerisation reactions.
This research supports the potential for finding a biotechnological route for using renewable mannans as starting materials for enzymatic synthesis of novel functional building blocks for preparation of glycopolymers. AcGGM is indeed an interesting renewable resource with great potential, and we show that it can act as the donor substrate for enzymatic synthesis of mannosyl acrylates. However, in this first study of enzymatic synthesis of mannosyl acrylates we choose to use the less complex and commercially available LBG as the donor substrate in scaled up synthetic reactions. Enzymatic reactions with TrMan5A, LBG and HEMA generated the least heterogenic product profile. Through β-mannanase catalysed transglycosylation, two mannosyl acrylate monomers were produced. These were purified in sufficient amounts to allow for structural determination by NMR spectroscopy, and their respective homopolymers were prepared by conventional radical polymerisations.
Experimental
Materials
Azo-Carob galactomannan, low viscosity carob (locust bean gum) galactomannan (LBG) with a mannose:galactose ratio of 1:0.28, mannobiose (M2), mannotriose (M3), mannotetraose (M4), mannopentaose (M5) and mannohexaose (M6) were supplied by Megazyme (Bray, Ireland). Locust bean gum galactomannan with a mannose:galactose ratio of 1:0.25 was from Sigma-Aldrich (St Louis, MO, USA). Dextran (50 kDa, 150 kDa, 270 kDa, 410 kDa and 1400 kDa) was from Fluka Chemie AG (Buchs, Switzerland). O-Acetyl galactoglucomannan (AcGGM) was prepared and analysed according to Lundqvist et al.17 (average molecular weight of 5900 Da and molar monomer composition, mannose:glucose:galactose:acetyl being 1:0.4:0.15:0.7). Mannose (M1) (microbiology grade), sodium acetate (molecular biology grade), acetic acid, 2-hydroxyethyl methacrylate (HEMA) 97% containing 200 ppm hydroquinone (HQ), 2,5-dihydroxy benzoic acid (DHB), hydroquinone (HQ, ≥99%), diethyl ether (≥97%) containing 1 ppm BHT as an inhibitor, ethanol, butanol, acetonitrile (ACN, anhydrous 99.9%, HPLC gradient grade), N-(1-naphthyl) ethylenediamine dihydrochloride, sulphuric acid, ammonium persulphate (APS), and potassium disulfite were all obtained from Sigma-Aldrich (St Louis, MO, USA). All aqueous solutions were prepared with MilliQ water.
β-Mannanases
Trichoderma reesei Man5A (TrMan5A) was obtained by production in Trichoderma reesei as described previously.23 An aliquot of the freeze-dried enzyme powder was dissolved in 50 mM Na-acetate buffer at pH 5.3. The solution was centrifuged thrice and refilled with buffer in Vivaspin columns with a 10 kDa cut-off (Sartorius, Göttingen, Germany). Aspergillus nidulans Man5B (AnMan5B) was expressed in Pichia pastoris and purified as previously reported.21 The protein concentration was determined by measuring the absorbance at 280 nm and using the extinction coefficients 123145 M−1 cm−1 for TrMan5A and 118510 M−1 cm−1 for AnMan5B, calculated using ProtParam (http://web.expasy.org/protparam/).
Enzymatic reactions with M4 and HEMA or allyl alcohol
Reactions were set up with either TrMan5A or AnMan5B (2 μM), mannotetraose (M4) (5 mM) as the glycosyl donor substrate and either HEMA (25 vol%) or allyl alcohol (25 vol%) as the glycosyl acceptor (Scheme 1), in 20 mM Na-acetate buffer at pH 5.3. The total reaction volume was 50 μl. Incubation was done in sealed 0.2 ml PCR tubes at 37 °C, samples (10 μl) were taken at 5 min, and 1, 3 and 5 h and reactions were terminated by boiling for 5 min, after which no enzyme activity remained.Matrix-assisted laser desorption ionisation – time of flight mass spectrometry (MALDI-ToF MS)
MALDI-ToF MS was used for the analysis of reactions with oligosaccharide and polysaccharide donor substrates incubated with the acceptor and either TrMan5A or AnMan5B. Samples (0.5 μl) were co-crystallised with 0.5 μl DHB (10 g L−1 in H2O) on a stainless steel MALDI plate, using warm air to dry the spots. MALDI-ToF MS data were collected using a MALDI-ToF 4700 (Applied Biosystems) in positive reflector mode. In general, a laser intensity of 5500–6000 was used and 20 sub-spectra with 50 shots per spectrum were collected from each sample spot. The software “Data Explorer version 4.5” was used for the analysis of mass spectrometry data. MALDI-ToF-ToF MS/MS data were collected in positive mode with a laser intensity of 6500 and a precursor window of ±0.100 Da and 75 sub-spectra with 15 shots per spectrum for each sample spot were collected.Thin layer chromatography
For reactions with TrMan5A and HEMA, samples were also analysed by thin layer chromatography (TLC) using aluminium plates covered with silica gel 60 (Merck, Darmstadt, Germany). 1 μl of manno-oligosaccharide standards (M1–M6, 10 mM) and 2 μl of samples were loaded on the plate. A mobile phase of butanol–ethanol–water in a volume ratio of 10:8:7 was used for separation for about 8 h at room temperature. Visualisation of sugars was done by soaking the plate in a solution containing N-(1-naphthyl) ethylenediamine dihydrochloride, ethanol and sulphuric acid24 followed by drying and incubation at 105 °C for 10 min.
Determination of retained β-mannanase activity in aqueous HEMA solution
TrMan5A was incubated at 37 °C in aqueous solutions of HEMA ranging from 0 to 25 vol%. Analysis of residual enzyme activity was performed at 0, 1, 5 and 24 h (for 20% HEMA also at 48 h). For analysis of β-mannanase activity, an assay with azo-carob galactomannan was used. The assay was performed according to recommendations from the manufacturer (Megazyme, Bray, Ireland).Enzymatic reactions with polymeric β-mannan and HEMA
To assess the ability of TrMan5A to use HEMA as the acceptor in reactions with polysaccharides, locust bean gum galactomannan (LBG, Sigma-Aldrich) was firstly tested as the glycosyl donor substrate. Initial reactions (50 μL) were performed with TrMan5A (2 μM), LBG (Sigma-Aldrich) (0.25% (w/v)) and HEMA (0–25 vol%) in 30 mM Na-acetate buffer at pH 5.3. Samples were incubated at 37 °C for 24 h, followed by boiling for 5 min to terminate the reactions. The more complex polysaccharide AcGGM (Scheme 2) was also tested as the glycosyl donor substrate in reactions with HEMA as the glycosyl acceptor. The spruce AcGGM was extracted and analysed according to Lundqvist et al.17 An aliquot of freeze dried AcGGM was dissolved in milliQ water. Reactions (50 μl) were performed with TrMan5A (2 μM) and AcGGM (0.35% (w/v)) in 20 mM Na-acetate buffer at pH 5.3 with or without HEMA (25 vol%). Incubation was done at 37 °C for 24 h followed by 5 min boiling to terminate the reactions. These initial reactions with polysaccharide substrates were analysed with MALDI-ToF MS.To assess the effect of different reaction parameters on the synthesis of mannosyl acrylates, time-course studies were carried out on reactions with TrMan5A (0.2–2 μM), LBG (Megazyme) (0.25–3% (w/v)) and HEMA (10–20 vol%) in 30 mM Na-acetate buffer at pH 5.3. Incubation was done at 37 °C for up to 48 h. Sample aliquots were collected after <5 min, 0.5 h, 1 h, 2 h, 5 h, 8 h, 24 h and 48 h, terminating the reaction through boiling for 5 min. Reactions with 2 μM TrMan5A were terminated after 24 h, while reactions with 0.2 μM TrMan5A were terminated after 48 h. Reaction volumes ranged from 0.75 to 5 ml and the impact of altered reaction parameters was evaluated by HPLC analysis (see below).
HPLC analysis
After initial screening for products using MALDI-ToF MS, as described above, samples were analysed by HPLC. Unreacted HEMA was partially extracted with diethyl ether, to diminish the void peak and allow for accurate HPLC analysis of the analytes. Equal volumes of the reaction sample and diethyl ether were mixed thoroughly, the organic phase was removed, and the extraction was performed three times. Prior to injection on the column, the samples were diluted with ACN to be compatible with the starting conditions for the HPLC analysis. Samples from 0.25–1% (w/v) LBG incubation were diluted 1:1 in ACN, while samples from 2–3% (w/v) LBG incubation were diluted 1:3 in ACN. All samples were filtered through a 0.2 μm polytetrafluoroethylene (PTFE) filter prior to loading. An Ultimate 3000 system from Thermo Scientific was used for HPLC analysis. Data were collected using UV-detection at 205 nm, which has previously been used for the detection of glucosyl acrylates.5 Samples were run on an NH2 column (LUNA amino, 250 × 4.6 mm, 3 μm, Phenomenex, Torrance, CA, USA) using hydrophilic interaction liquid chromatography (HILIC) conditions. H2O and ACN were used as the mobile phase at 1 ml min−1. A gradient from 5 to 60% H2O over 30 min was used for separation. The column was then washed with 90% H2O for 10 min followed by equilibration with 5% H2O for 10 min. The injection volume was 10 μl and the column temperature was maintained at 40 °C. Fractions of eluting peaks were collected and analysed by MALDI-ToF MS and MS/MS, as described above.
Preparative scale production of 2-(β-manno(oligo)syloxy) ethyl methacrylates (MnEMAs)
Based on the detected product peak areas from the HPLC analysis, reaction conditions that gave the largest product peak areas of 2-(β-mannosyloxy) ethyl methacrylate (M1EMA) and 2-(β-mannobiosyloxy) ethyl methacrylate (M2EMA) were chosen for preparative production of these compounds. In a total reaction volume of 50 ml, 0.2 μM TrMan5A was incubated with 3% LBG (w/v) (Megazyme) and 20 vol% HEMA in 30 mM sodium acetate buffer at pH 5.3, for 48 h at 37 °C. The reaction vessels were occasionally mixed through inversion, to ensure homogeneous conditions. Unreacted HEMA was extracted three times with diethyl ether, in a volume proportion of 1:1. The organic phase was discarded. 50 μl of the polymerisation inhibitor HQ (10 g L−1) was added to the remaining, aqueous fraction, which was then concentrated in an RVC 2–18 SpeedVac (Martin Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany) maintaining temperatures <40 °C. To condition the sample for preparative HPLC, the resulting concentrated syrup (∼2.5 ml) was diluted first in 2.5 ml H2O and then in increments of ACN to a final concentration of 95% ACN. The incremental increase of ACN precipitated saccharide moieties with DP > 3 (as analysed using MALDI-ToF MS), which were separated from the soluble fraction, by decantation and filtration, before loading the sample onto the HPLC column.
Preparative HPLC purification of MnEMAs
Preparative HPLC was performed with a preparative HPLC system (2545 Quaternary gradient module, 2707 Autosampler, Fraction collector III, Prep degasser, 2998 Photodiode array detector and Column oven) (Waters, Milford, MA, USA). UV-data were collected at 205 nm. Prior to loading samples were filtered through a 0.2 μm PTFE filter. Samples, equivalent to 22.5 ml of the original reaction volume, in 95% ACN, were purified using a preparative NH2 column (LUNA 5 μm NH2 100 Å, 250 × 21.2 mm AXIA packed, Phenomenex, Torrance, CA, USA) using HILIC conditions. Samples were loaded during isocratic conditions of 5% H2O in ACN, and eluted during gradient flow, with an increasing concentration of H2O, up to 60%. Elution fractions were collected in 8 ml polypropylene fraction collection tubes. The column was washed with 90% ACN:H2O before re-equilibration with three column volumes under the starting conditions. The flow rate was constant at 20 ml min−1, and the column temperature was maintained at 40 °C.
The fractions collected from the preparative scale purification were thoroughly analysed by MALDI-ToF MS, MS/MS and analytical HPLC, as described above. The concentrations in aliquots of purified M1EMA and M2EMA were determined by nuclear magnetic resonance (NMR) spectroscopy with an external reference, by using an optimised tuning and matching approach for comparison between two samples. A sample with a known concentration of HEMA was used as the external standard. The quantified samples were further used to construct external standard curves, allowing for quantification of MnEMAs via HPLC analysis.
Fractions containing the major products, M1EMA and M2EMA, were pooled and supplemented with 6 μg of HQ as an inhibitor. The respective product was concentrated in the SpeedVac maintaining temperatures <40 °C. M1EMA was concentrated to a visually clear, slightly viscous solution <50 μl, and M2EMA was concentrated to a brownish syrup <20 μl. The two samples were then resuspended in 500–550 μl D2O (99.8%) and centrifuged at 15000g for 10 min at room temperature to remove any particulates, and the supernatants were transferred to new tubes prior to NMR analyses.
Analysis by NMR spectroscopy
The purified fractions of M1EMA and M2EMA, and eventually their corresponding individual polymers, were used for structural analysis by NMR spectroscopy. 1H and 13C spectra were recorded at 10 and 45 °C on a Bruker Avance III spectrometer (Bruker, Billerica, MA, USA) at 500.17 MHz for 1H and at 125.77 MHz for 13C. Chemical shift assignment was made using two dimensional NMR experiments: 1H correlation spectroscopy (COSY), total correlation spectroscopy (TOCSY), heteronuclear multiple-bond correlation (HMBC), heteronuclear single quantum coherence (HSQC) NMR and 2D nuclear Overhauser effect spectroscopy (NOESY) with an 800 ms mixing time. Chemical shifts for the proton spectra (δH) were given in ppm relative to the residual internal solvent peak taking into account the temperature dependence25 (HDO, δH 4.94 ppm at 10 °C and δH 4.55 for 45 °C). The 13C spectrum chemical shift scale was obtained using the Bruker software Topspin 3.2 pl6 for calculating the unified scale according to the International Union of Pure and Applied Chemistry (IUPAC).26 The chosen temperatures for spectrum acquisition were different from room temperature in order to induce a shift of the residual internal solvent (HDO) to reveal or improve the resolution of the anomeric signals arising from the mannose units. Additionally, coupled 13C spectra were obtained at 125.77 MHz at 10 °C in D2O with a gated decoupling technique to study the direct coupling constant between C-1 and H-1 (1JC-1,H-1).Polymerisation
Conventional radical polymerisations of the M1EMA and M2EMA monomers were carried out using K2S2O5 and APS as the initiator system in a threaded cap NMR tube at a molar monomer to K2S2O5 to APS ratio of 100:4:17, and at a monomer concentration of 30 mM in D2O. The reaction solution was purged by bubbling nitrogen through a syringe for 1 h at room temperature. The polymerisation reaction was then carried out at 60 °C for 24 h under a nitrogen atmosphere. Subsequently, the tube was left to cool to room temperature, followed by immediate acquisition of 1H and 13C NMR spectra to confirm the polymerisation. Once the polymerisation was confirmed, the samples were further analysed by size exclusion chromatography (SEC). A sample volume of 20 μl (1 mg ml−1) was injected on a TSKgel® G4000PWXL column (TOSOH Bioscience GmbH, Griescheim, Germany) connected to a chromatography system (Waters 600E System Controller, Waters, Milford, MA, USA), using RI (Waters 2414 Differential Refractometer) and UV detection (Waters 486 Tunable Absorbance Detector) at 234 nm. De-ionized water served as the eluent at a flow rate of 0.5 ml min−1. The column was calibrated with dextran standards of 50, 150, 270, 410, and 1400 kDa (Fluka Chemie AG, Buchs, Swizerland). The remaining volume of the samples was diluted with D2O followed by acquisition of 1H and 13C NMR spectra at 10 and 45 °C.
13C NMR (D20, 10 °C) δC in ppm, the chemical shift scale from the unified scale according to IUPAC:26 169.73 (C-c), 135.76 (C-d), 126.99 (C-e), 99.92 (C-1), 76.31 (C-5), 72.91 (C-3), 70.45 (C-2), 67.31 (C-a), 66.75 (C-4), 64.29 (C-b), 62.49 (Impurity), 61.02 (C-6), 38.65 (Impurity), 17.43 (C-f).
13C NMR (D20, 10 °C) δ in ppm, the chemical shift scale from the unified scale according to IUPAC:26 169.73 (C-c), 135.75 (C-d), 127.01 (C-e), 100.22 (C-1′), 99.94 (C-1), 76.76 (C-4), 76.43 (C-5′), 74.88 (C-5), 72.72 (C-3′), 71.61 (C-3), 70.50 (C-2′), 69.84 (C-2), 67.38 (C-a), 66.68 (C-4′), 64.31 (C-b), 61 (C-6), 60.52 (C-6′), 38.65 (Impurity), 17.44 (C-f).
Results and discussion
Reactions with mannotetraose (M4) as the donor substrate and HEMA as the acceptor
TrMan5A has previously been shown to have synthetic capacity via transglycosylation when incubated with M4 as the glycosyl donor substrate and alcohols such as methanol, butanol and hexanol as glycosyl acceptors.21,22AnMan5B, however, does not use methanol or butanol as acceptors in reactions with M4.21 On the other hand, AnMan5B displays exceptionally high transglycosylation capacity with saccharide acceptors and even seems to prefer saccharides over water.21 To investigate whether TrMan5A and AnMan5B can yield glycoconjugates by using HEMA as the acceptor (Scheme 1), initial reactions were performed with M4 as the donor substrate. Reactions with allyl alcohol as the acceptor were included for comparison (Scheme 1C).In a first screening, samples from the enzymatic reactions with M4, HEMA or allyl alcohol were analysed using MALDI-ToF mass spectrometry. For TrMan5A, peaks corresponding to masses of molecules generated by a conjugation between manno-oligosaccharides and the allyl alcohol (Fig. 1A) or manno-oligosaccharides and HEMA (Fig. 1D) were detected, further referred to as “conjugates”. Also for AnMan5B, peaks corresponding to masses of conjugates with both the allyl alcohol and HEMA were detected (Fig. 1B and C) but at a lower peak intensity compared to TrMan5A. In addition, AnMan5B also generated significant transglycosylation products with saccharides as acceptors, in agreement with what has previously been reported.21 In the reactions with AnMan5B and the allyl alcohol, the only detected conjugate peak was allyl-mannobioside (M2-allyl) (Fig. 1B), showing an intensity that was significantly lower compared to the M2-allyl peak detected in the TrMan5A reactions. In reactions with AnMan5B and HEMA, low intensity peaks corresponding to masses of conjugates with up to six mannosyl units were detected (Fig. 1C).
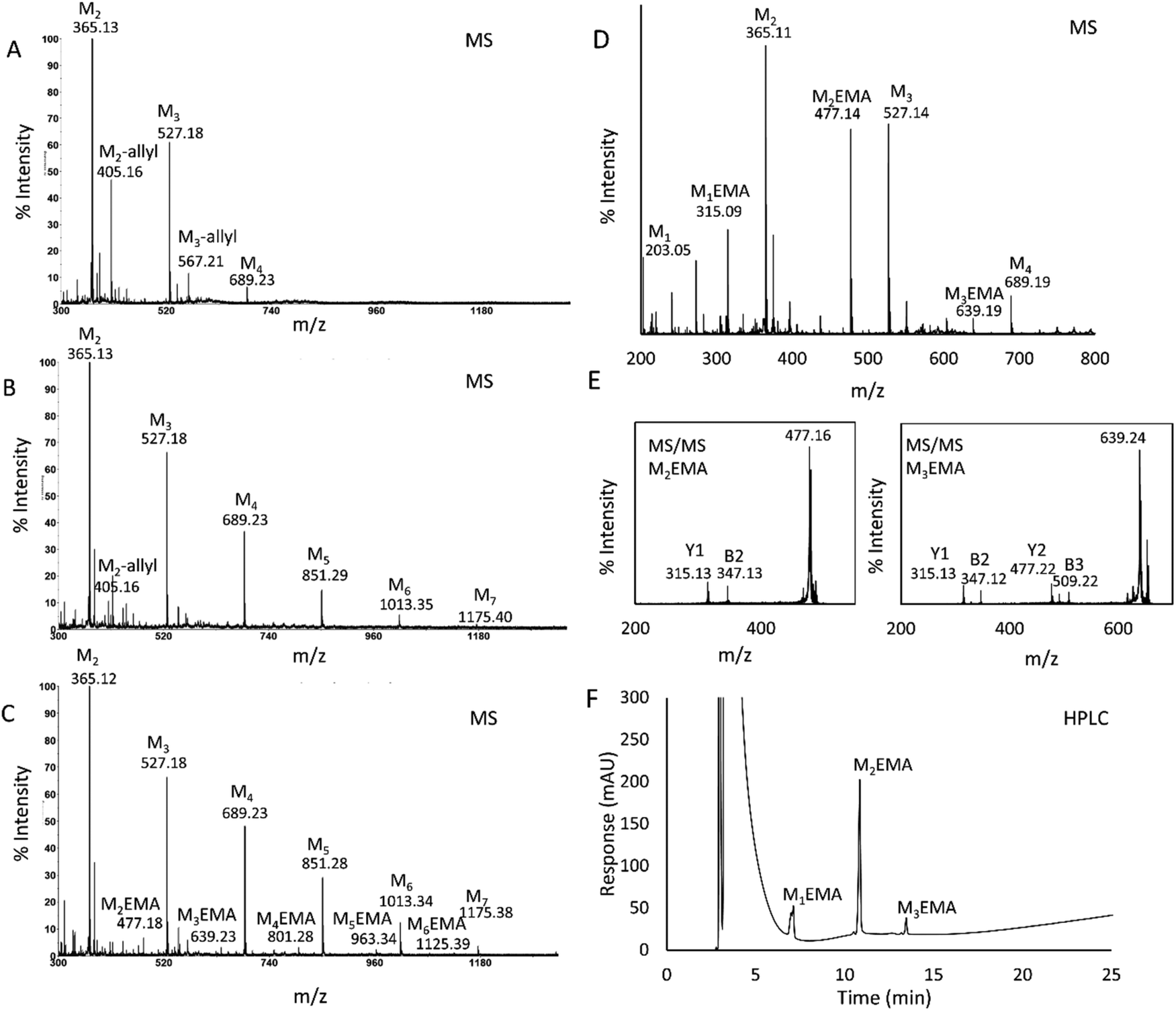 | ||
| Fig. 1 Analysis of 1 h reactions with TrMan5A (2 μM) or AnMan5B (2 μM) using M4 (5 mM) as the donor substrate and HEMA (25 vol%) or allyl alcohol (25 vol%) as the acceptor. In MALDI-ToF MS (A–E), peaks were detected as monoisotopic masses of single charge sodium adducts. The theoretical m/z for the peaks corresponding to oligosaccharides and conjugates, marked in the figure, are: M2 (m/z: 365.11), M3 (m/z: 527.16), M4 (m/z: 689.21), M5 (m/z: 851.27), M6 (m/z: 1013.32), M7 (m/z: 1175.37), M2-allyl (m/z: 405.14), M3-allyl (m/z: 567.19), M1EMA (m/z: 315.11), M2EMA (m/z: 477.16), M3EMA (m/z: 639.21), M4EMA (m/z: 801.26), M5EMA (m/z: 963.32) and M6EMA (m/z: 1125.37). (A) MS spectra of the reaction with TrMan5A, M4 and the allyl alcohol, showing the donor substrate M4, oligosaccharide products M2 and M3 and peaks of conjugations between the allyl alcohol and M2 and M3; M2–3-allyl. (B) MS spectra of the reaction with AnMan5B, M4 and the allyl alcohol, showing the donor substrate M4, oligosaccharide products M2 and M3, transglycosylation products M5, M6, and M7 and a small peak of M2-allyl. (C) MS spectra of the reaction with AnMan5B, M4 and HEMA, showing the donor substrate M4, oligosaccharide products M2 and M3, transglycosylation products M5, M6, and M7 and small peaks of conjugates between HEMA and M2–M6: M2–6EMA. (D) MS spectra of the reaction with TrMan5A, M4 and HEMA, showing the donor substrate M4, oligosaccharide products M1, M2 and M3, and peaks of conjugates between HEMA and M2 and M3: M2–3EMA. (E) MS/MS fragmentation of M2EMA and M3EMA, showing Y1 and B2 ions for M2EMA and Y1, Y2, B2 and B3 ions for M3EMA, which were of expected m/z for molecules fragmenting in the glycosidic bond (theoretical m/z for Y1: 315.11, B2: 347.10, Y2: 477.16, B3: 509.12). (F) HPLC chromatogram showing separation of M1EMA, M2EMA and M3EMA. HPLC analysis was carried out with an analytical NH2 column run under HILIC conditions. 10 μl sample (2× diluted) was injected. Three separate peaks were detected after the void: rf 1: 7.0 min, 2: 10.8 min and 3: 14.1 min. Fractions were collected and the peaks were individually identified as M1EMA, M2EMA and M3EMA respectively, by MALDI-ToF MS. | ||
Based on the complexity and heterogeneity of the product profile generated by AnMan5B, together with the lower peak intensity for the conjugates compared to the TrMan5A reactions, the enzyme chosen for further studies was TrMan5A. HEMA was selected as the acceptor for further study, as the polymerisation of saccharide acrylates can be demonstrated without the need for co-monomers.4–6
In the MS spectra of reactions with TrMan5A, M4 and HEMA (Fig. 1D), peaks corresponding to masses of manno-oligosaccharides and of conjugates between mannose and HEMA; 2-(β-mannosyloxy) ethyl methacrylate (M1EMA), mannobiose and HEMA; 2-(β-mannobiosyloxy) ethyl methacrylate (M2EMA) and mannotriose and HEMA; and 2-(β-mannotriosyloxy) ethyl methacrylate (M3EMA) were detected (Fig. 1D). MALDI-ToF-ToF MS/MS analysis of the peaks corresponding to M2EMA and M3EMA further supported the fact that they originated from mannosyls conjugated with HEMA (Fig. 1E). The MS/MS spectra showed Y and B ions that could be matched to M2EMA and M3EMA fragmenting in the glycosidic bond (Fig. 1E).27,28
A TLC analysis of reactions with TrMan5A, M4 and HEMA also showed that the enzymatic reaction generated, in addition to oligosaccharides, products that migrated further on the plate compared to oligosaccharide standards (Fig. S1†), i.e. products that were less hydrophilic than oligosaccharides. These products were assumed to be conjugates with HEMA. None of the controls (substrate, enzyme and HEMA control) showed spots at the same level on the plate. A previously reported TLC analysis of reactions with M4 and TrMan5A indicated no spots migrating further than mannose.21 The combined results from the MALDI-ToF MS and TLC analyses showed that the TrMan5A β-mannanase can use HEMA as the acceptor in reactions with M4 as the donor substrate, to generate 2-(β-manno(oligo)syloxy) ethyl methacrylates (MnEMAs), and that the MnEMAs can be separated from manno-oligosaccharides based on differences in hydrophilicity.
HPLC analysis of the reaction with TrMan5A, M4 and HEMA, separated on an NH2 column run under HILIC conditions, resulted in the separation of three clear peaks (Fig. 1F). Fractions were collected and elutes were identified by MALDI-ToF MS as the conjugates M1EMA, M2EMA and M3EMA. The MS analysis verified that the peaks in the HPLC chromatogram each contained one conjugate and no other conjugate or saccharide.
Assessment of reactions with polymeric mannans and HEMA
The experiments with M4 gave valuable insights into both enzymatic performance and the setup of analytical procedures. The interesting finding that TrMan5A can yield MnEMAs when incubated with M4 as the donor substrate, and that these can be separated on a HPLC column, provided promising results when proceeding to set up reactions using polymeric mannans, LBG and AcGGM, as donor substrates. To our knowledge these abundant and naturally occurring mannans have previously not been evaluated as donor substrates in enzymatic synthesis reactions.In MALDI-ToF MS analysis of reactions with LBG and HEMA, conjugates between HEMA and hexoses up to a degree of polymerisation (DP) of 3 were detected (Fig. S2B†), together with non-conjugated oligosaccharide products of DP2–4, also present in the reactions without HEMA (Fig. S2A†). In addition, samples from reactions with LBG and HEMA were analysed using HPLC, and peaks with the same retention times as previously determined for M1–M3EMA were detected. This led to the conclusion that TrMan5A can synthesise MnEMAs also when using LBG as the donor substrate.
As a first trial towards using a renewable resource from the forest industry as the donor substrate, AcGGM extracted from spruce17 was also evaluated in enzymatic reactions with HEMA. MALDI-ToF MS analysis of these reactions gave more complex spectra due to the higher complexity of the AcGGM substrate compared to that of LBG (Fig. S2C and D†). Spectra from reactions, both with and without HEMA, showed peaks corresponding to hexoses with DP1–7 (H1–H7) (Fig. S2C and D†). Since the AcGGM substrate is partly acetylated,17 oligosaccharides of hexoses with one, two or three acetyl groups were present (Fig. S2C and D†). In the reactions where HEMA was included, peaks that correspond to masses of conjugates between HEMA and hexoses up to DP4 were detected (Fig. S2D†). Both non-acetylated and acetylated conjugates with HEMA were detected in the spectra (Fig. S2D†). Although the results from reactions with AcGGM provide novel and promising results, the commercially available donor substrate LBG was chosen for further reactions in this study, due to the limited amount of available AcGGM and in order to limit the product heterogeneity.
Enzyme stability
Analysis of the stability of TrMan5A in various HEMA concentrations was performed to refine the reaction conditions for the synthesis of MnEMAs. The results from the stability analysis of TrMan5A are presented in Fig. 2. TrMan5A is stable at 37 °C for 24 h in the absence of HEMA. Also, with 5 and 10 vol% HEMA the enzyme maintained its activity after incubation for 5 h, and after 24 h at these concentrations of HEMA the remaining activity was 85–90%. With 15 and 20 vol% HEMA the enzyme was stable for 1 h and the activity then decreased to 85% and 65%, respectively, after 5 h, and to 60% and 35%, respectively, after 24 h. With 25 vol% HEMA the enzyme maintained around 85% of its activity for 1 h. Then the activity decreased to 40% after 5 h, and became negligible after 24 h. With concentrations of up to 20 vol% HEMA, TrMan5A is operational for at least 24 h at 37 °C and therefore this concentration of HEMA and this temperature were chosen for further reactions. A prolonged incubation under these conditions (20% HEMA, 37 °C) showed that TrMan5A had lost all activity after 48 h.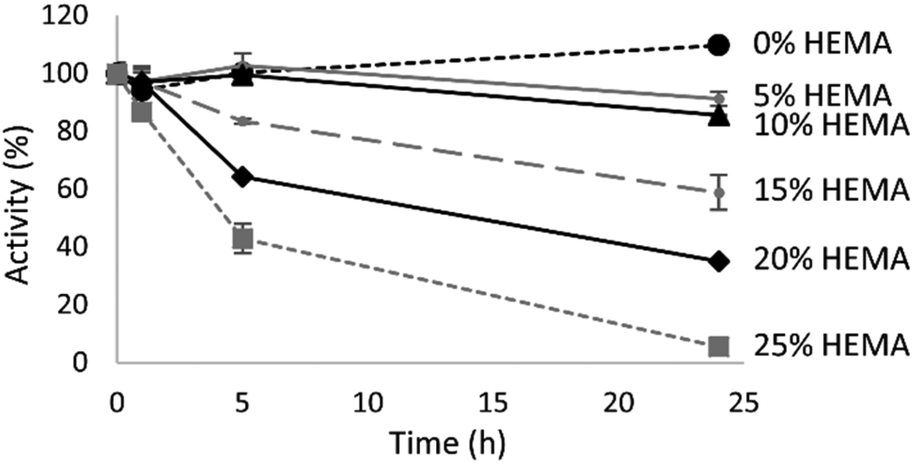 | ||
| Fig. 2 Residual activity of TrMan5A in 0–25 vol% HEMA. Enzyme activity was analysed with an azo-carob galactomannan assay after 0–24 h. | ||
Reaction parameters
In order to obtain amounts of MnEMAs that allowed further characterisation, the effect of varying different reaction parameters on the synthesis of MnEMAs was investigated. Time-course studies over 0–48 h were performed, altering donor substrate and acceptor concentrations and enzyme load, respectively. Based on the detectable peak areas from the HPLC analysis of the conjugates, various conditions were evaluated (Fig. 3). Using the same amount of the enzyme (2 μM TrMan5A), while increasing the donor substrate concentration (0.25–3% (w/v) LBG) and acceptor concentration (10 vol% vs. 20 vol% HEMA), resulted in a higher HPLC peak area of the MnEMA products, where M2EMA showed the largest peaks (Fig. 3A). The highest product formation was achieved at 3% LBG and 20% HEMA (25% less M2EMA was detected using 10% HEMA). The M2EMA detection using 1% or 0.25% LBG instead of 3% LBG decreased by 39% and 87%, respectively. The reaction progression over time using 3% LBG and 20% HEMA showed a decrease of M2EMA after prolonged incubation (34% less detected at 24 h compared to 2 h) (Fig. 3B). The peak response of M1EMA, however, increased steadily over the time course (Fig. 3B). The decrease in M2EMA levels could potentially be caused by secondary hydrolysis.13 Such hydrolysis of the terminal mannosyl unit would also explain the increasing levels of M1EMA. With a lower enzyme load (0.2 μM rather than 2 μM) secondary hydrolysis appeared to be limited and the maximum amounts of conjugates, based on HPLC peak areas, were obtained after a 48 h incubation of 0.2 μM TrMan5A with 3% (w/v) LBG and 20 vol% HEMA.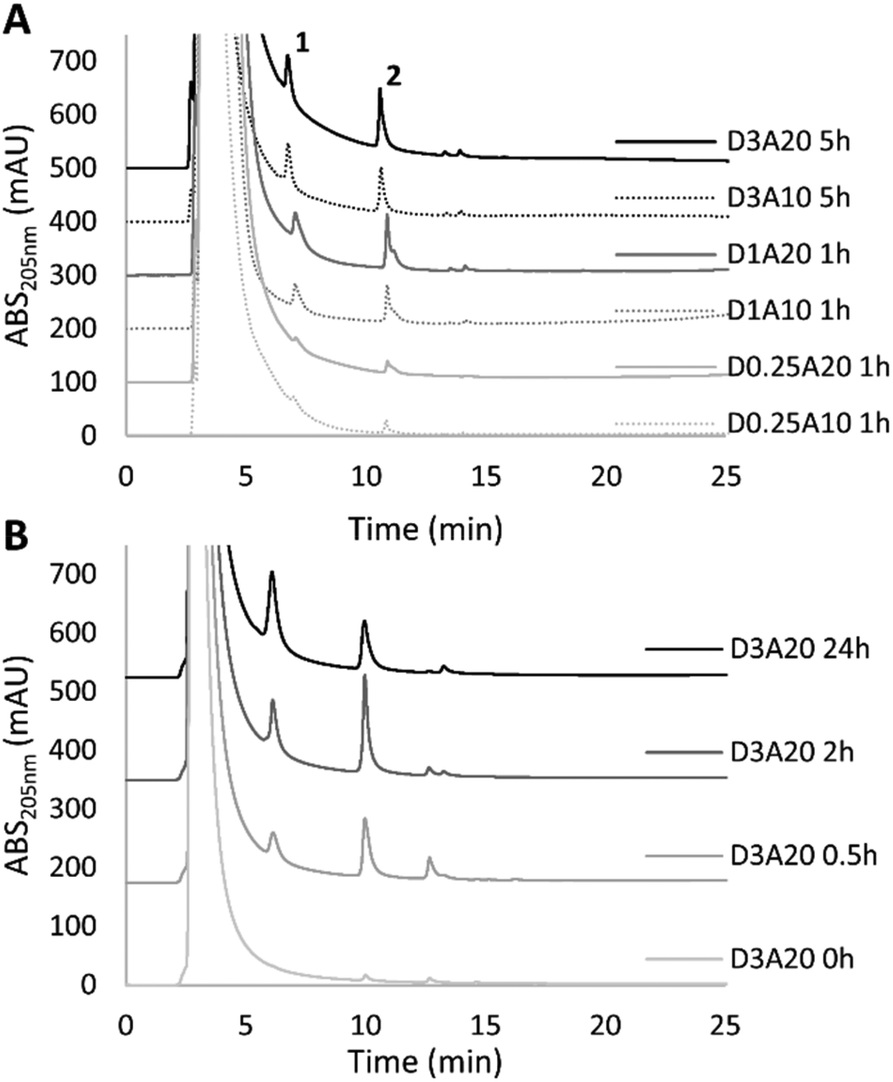 | ||
| Fig. 3 (A) Study on the impact of reaction parameters with 2 μM TrMan5A, LBG (0.25, 1 or 3% (w/v)) and HEMA (10 or 20 vol%). Presented HPLC chromatograms represent the incubation time required to obtain the maximum peak areas of M1EMA (peak 1) and M2EMA (peak 2) under the respective conditions. D = donor (LBG) concentration in % (w/v), A = acceptor (HEMA) concentration (vol%). Samples of 3% (w/v) LBG reactions were diluted 1:3 with ACN, while all others were diluted 1:1 with ACN. (B) Time course study on the reaction with 2 μM TrMan5A, LBG (3% (w/v)) and HEMA (20 vol%), from 0 to 24 h. Analysis after 3× 1:1 HEMA extraction with diethyl ether (see the Methods section). | ||
In order for the observed secondary hydrolysis to occur, the M2EMA molecule needs to be able to bind in the active site-cleft of TrMan5A through subsites −1, +1 and +2 with the two mannoses in subsites −1 and +1, to expose the target terminal glycosidic bond for the catalytic amino acids.29 This would also mean that the EMA moiety can be accommodated in the +2 subsite which is an important site for glycan interactions both in hydrolysis and transglycosylation.20 Furthermore, for the synthesis of MnEMAs shown in the current paper to occur, HEMA needs to be accommodated in subsite +1 to be positioned for conjugation with the mannosyl in subsite −1. The picture emerges that the EMA unit can replace mannosyl units in this type of enzyme–ligand interaction within distal subsites (i.e. not −1). This view is also supported by the interesting observation that mannanase AnMan5B uses HEMA as the acceptor (Fig. 1C), but not shorter alcohols.21 Interaction or accommodation of EMA units in this type of enzyme potentially opens up for further reactions involving this chemical group and enzyme catalysis.
Preparative production of MnEMA
Based on the reaction conditions that gave the highest amount of MnEMA products, reactions were performed in a 50 ml scale. These conditions were deduced from the peak area on analytical HPLC and resulted in the use of 0.2 μM TrMan5A, 3% (w/v) LBG and 20 vol% HEMA, incubated for 48 h. Separation of MnEMA products was performed using preparative HPLC by collecting and further analysing the fractions (Fig. 4). To determine the purity of the fractions collected from the preparative HPLC run, each fraction was analysed by MALDI-ToF MS (Fig. 4); the fractions were determined to be free from sugars and only containing one type of MnEMA. The MnEMA peaks were further studied by MALDI-ToF-ToF MS/MS, revealing a fragmentation pattern corroborating the identity of each peak (data not shown).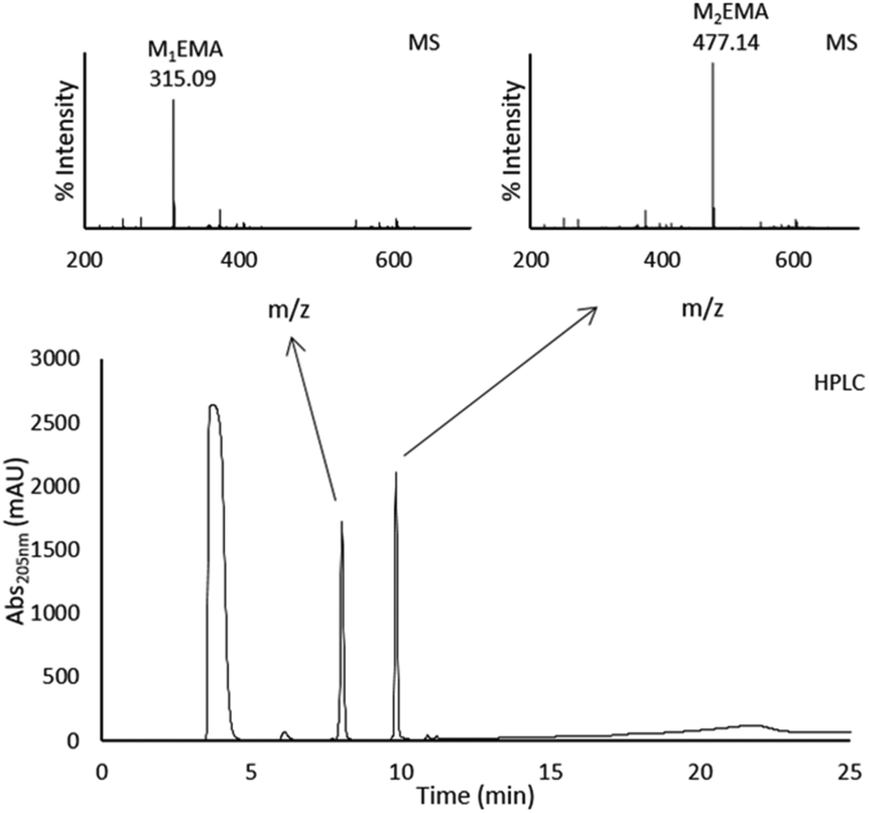 | ||
| Fig. 4 Chromatogram of preparative HPLC purification carried out with a preparative NH2 column, run under HILIC conditions. 8 ml of sample was injected during an isocratic flow of 95% ACN and 5% H2O, followed by a gradient from 95 to 40% ACN over 13 min. Two major peaks were detected after the void: rf 1: 8.0 min, 2: 9.9 min. Fractions were collected and the peaks were individually identified as M1EMA and M2EMA by MALDI-ToF MS. Theoretical m/z for M1EMA: 315.11 and M2EMA: 477.16. | ||
The sample concentrations, analysed using NMR, were determined to be 11.5 g L−1 (in 500 μl) for M1EMA and 20.2 g L−1 (in 540 μl) for M2EMA. These quantified samples were then used as external standards in HPLC analysis for the quantification of MnEMAs in different reactions.
Reaction yields
The concentrations of MnEMAs in the preparative scale (50 ml) reactions were quantified as 0.306 g L−1 (±0.008) M1EMA and 0.953 g L−1 (±0.009) M2EMA, corresponding to a yield of 3.63% (±0.026) (Table 1). The yield was calculated based on the mannosyl units in the conjugates (M1EMA and M2EMA) vs. the mannosyl units in the donor substrate (LBG, with the theoretical amount of galactose subtracted). It can be noted that when, in a smaller scale, using lower concentrations of LBG as the donor substrate (0.25–1% (w/v)) (Fig. 3A), the yield was higher (6.52% (±0.22) and 7.42% (±0.64) respectively), but the reactions resulted in lower concentrations of conjugates. The lower yields from reactions with higher LBG concentrations could potentially be due to the increased viscosity of the donor substrate, which may cause diffusion limitations. These reactions were also incubated longer, which might have led to partial loss of enzyme activity during the reaction time. Using M4 as the donor substrate (Fig. 1C) resulted in a higher yield (13.8% (±1.0)) compared to that when using the more complex LBG as the donor substrate (Table 1). This yield is comparable with the yield obtained from reactions where cellobiose was used a donor substrate in the enzymatic synthesis of glucosyl-methacrylamide monomers (12–18%),16 although higher concentrations of both the donor substrate and acceptor were used in that study. A strong point in our approach is that unmodified natural glycans can be used as donors, rather than activated (para-nitrophenyl or nucleotide) sugars which likely results in higher rates and yields.16| M1EMA (g L−1) | M2EMA (g L−1) | Yield (%) | Enzyme concentration | Incubation time | |
|---|---|---|---|---|---|
| M4 5 mM (0.33% (w/v)) | 0.106 (±0.003) | 0.544 (±0.043) | 13.78 (±1.0) | 2 μM | 1 h |
| LBG 0.25% (w/v) | 0.036 (±0.005) | 0.150 (±0.002) | 6.52 (±0.22) | 2 μM | 1 h |
| LBG 1% (w/v) | 0.202 (±0.029) | 0.654 (±0.081) | 7.42 (±0.54) | 2 μM | 1 h |
| LBG 3% (w/v) | 0.306 (±0.008) | 0.953 (±0.009) | 3.63 (±0.026) | 0.2 μM | 48 h |
Interestingly, the obtained yield for the synthesis of M2EMA using M4 as the donor substrate was about 12 times higher compared to that for the synthesis of hexyl mannobioside, also using TrMan5A and similar concentrations and conditions.22 Although, the limited water solubility of hexanol likely plays a role, the potentially favourable accommodation or interaction of HEMA in the TrMan5A active site cleft (as discussed above) can be an additional positive factor.
The drastic difference in yields between the more complex LBG and the oligosaccharide M4 indicates that pre-hydrolysis of the target substrate may result in higher reaction efficacy. Pre-hydrolysis would generate shorter saccharides and also decrease the viscosity of the donor substrate, which could favour the enzymatic synthesis. The approach with pre-hydrolysis has previously been used and applied for enzymatic synthesis of alkyl-glycosides, using insoluble ivory nut mannan as the starting material.22 Additional strategies to improve the yield from reactions with mannans could be to remove side groups. Galactose substitutions along the main backbone of the galactomannan substrate may cause steric hindrance for the β-mannanase and limit productive binding.30 Thus, a synergistic hydrolysis of galactose residues, by the action of an α-galactosidase,31 could enable further action on the substrate by the β-mannanase30,32 and improve the yield.
Structural analysis of the synthesised conjugates M1EMA and M2EMA
The TrMan5A β-mannanase uses the retaining double-displacement mechanism and is thus unequivocally expected to produce β-anomeric transglycosylation products.12,33 With this knowledge and the MALDI-ToF MS mass determination of purified compounds we can predict their chemical structures, as 2-(β-mannosyloxy) ethyl methacrylate (M1EMA) and 2-(β-mannobiosyloxy) ethyl methacrylate (M2EMA) (Fig. 5).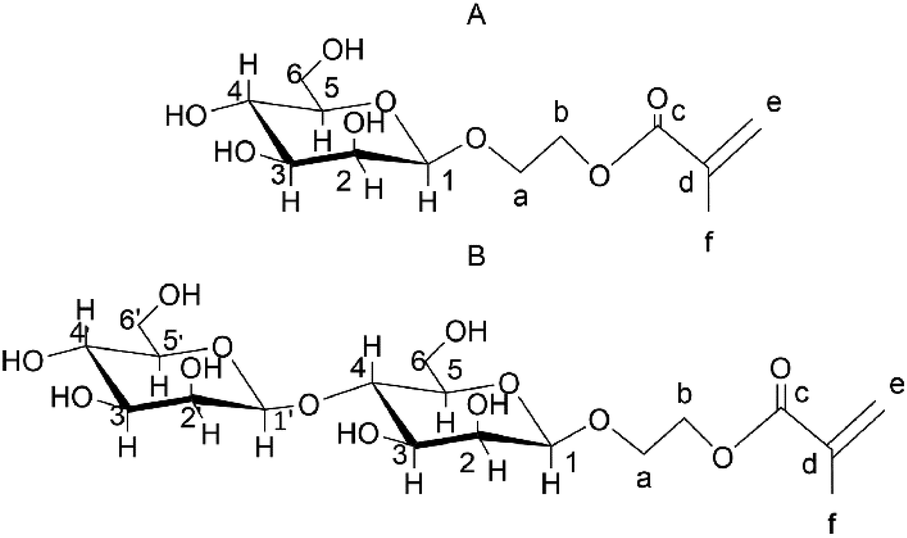 | ||
| Fig. 5 The expected β-configured structures of the synthesised mannosyl acrylates. (A) M1EMA and (B) M2EMA. Number 1 is assigned to the anomeric position and then following the clockwise direction. For the acrylate part, carbon atoms have been labelled a–f from the glycosidic bond oxygen. | ||
The structure of M2EMA was fully resolved by NMR spectroscopy. The 1H and 13C spectra for M2EMA can be seen in Fig. 6 and Fig. S3,† respectively. Wholly resolved 1H and 13C spectra of M2EMA, showing the crowded “sugar region”, can be found in ESI Fig. S4 and S5.† The structural determination was carried out by first comparing the spectra with published data, and then the assignment of signals was made unequivocally based on empirical rules for chemical shifts, coupling constants, multiplicity analysis and integration coupled with 2D NMR spectral analysis.34 The use of 2D NMR techniques enables full assignment without relying on analogy with reference data.35
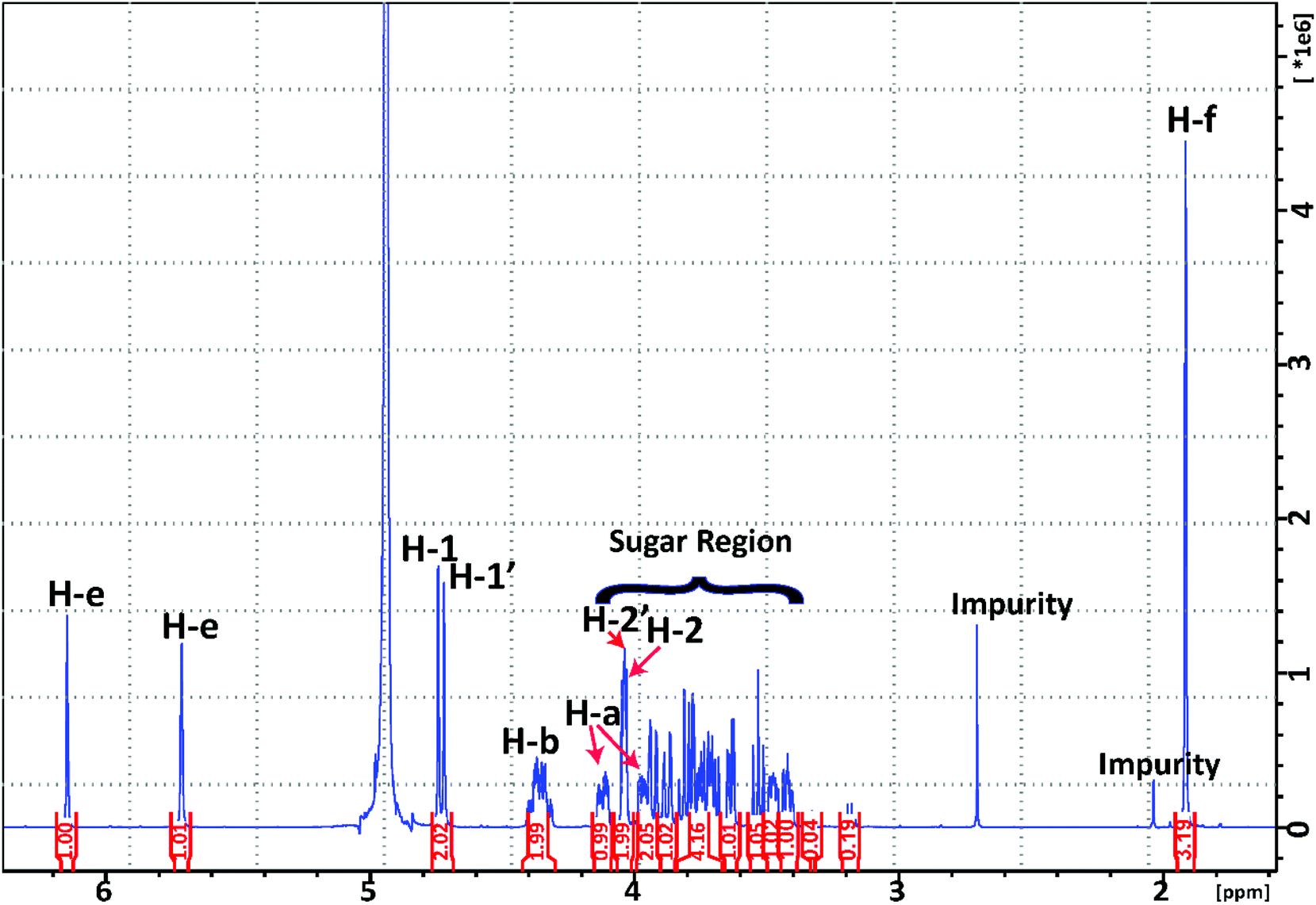 | ||
| Fig. 6 1H NMR spectrum of the synthesised M2EMA collected at 10 °C. | ||
When analysing the M2EMA sample, two different signals were identified in the anomeric region (δH 4.4–5.52 ppm) corresponding to the anomeric protons of the two mannoside units (H-1 and H-1′ in Fig. 5B). The chemical shifts (δ in ppm) were observed at 4.74 and 4.72 ppm, respectively (Fig. 6). These values are in very good agreement with previously analysed internal and terminal non-reducing β-mannosyls of β-manno-oligosaccharides (4.72–4.75 ppm),33,36 while non-reducing β-mannosyls or β-galactosyls of oligosaccharides show higher values (5.0–5.2 ppm).36,37 Apart from these well separated signals arising from the anomeric protons, Fig. 6 shows a multitude of signals assigned to the ‘sugar region’ in δH ∼ 4.06–3.38 ppm and are in agreement with previously reported ranges for mannose based mono- and disaccharides, including methylated ones.37
In the next step the signals associated with the acrylate unit, in particular the signals corresponding to the acrylate double bond (H-e in Fig. 5), were observed at δH 6.14 and 5.71 ppm and those associated with the –CH3 protons (H-f in Fig. 5) at δH 1.92 ppm. These are in agreement with previously reported values at 5.70–6.14 ppm for H-e and at 1.87–1.91 ppm for H-f, respectively.38 They were also shown to be in agreement with the signals observed in the spectra recorded from 2-hydroxyethyl methacrylate used for the synthesis (D2O, 25 °C). The empirical assignment was in agreement with the initial assignment. Fig. 7 shows the overlapping of HMBC and HSQC spectra of M2EMA, identifying relevant cross peaks that confirm the synthesis of the compound against the proposed structure (Fig. 5B). These data reveal the formation of the glycosidic linkage between the acrylate and the mannose unit at the anomeric carbon position (C-a to C-1 to C-2), and the linkage between the two mannose units (C-4 to C-1′ to C-2′). As no other chemical shifts showed such correlations in the overlapped 2D spectra, we confirm the formation of these linkages. We further confirmed that no signal could be found that would correspond to the presence of (1 → 6)-linked α-D-galactopyranosyl units.
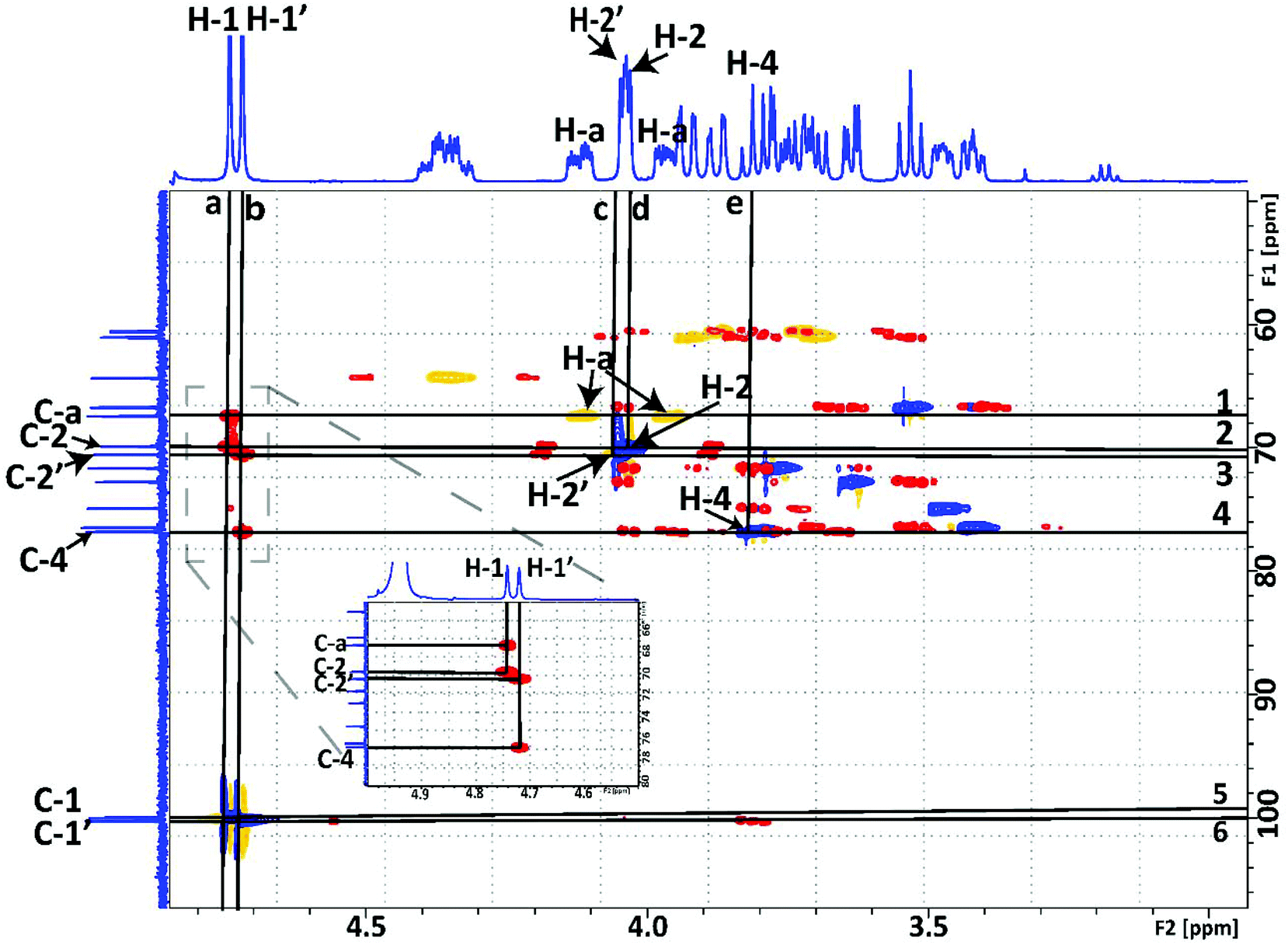 | ||
| Fig. 7 Overlay of 1H–13C HMBC (red, positive signals) and HSQC (blue, positive signals; yellow, negative signals) NMR spectra of the synthesised M2EMA. Chemical shifts in ppm (δ). The horizontal lines are drawn at (from top to bottom): (1) δC 67.38 of C-a, along that line direct bond J-coupling to H-a protons from the crosspeak in HSQC at δH 4.12 and at δH 3.97; (2) δC 69.83 of C-2, along that line direct bond J-coupling to H-2 protons from the crosspeak in HSQC at δH 4.03; (3) δC 70.5 of C-2′, along that line direct bond J-coupling to H-2′ protons from the crosspeak in HSQC at δH 4.04; (4) δC 76.8 of C-4, along that line direct bond J-coupling to H-4 protons from the crosspeak in HSQC at δH 3.8; (5) δC 99.97 of C-1 with the cross peak in HSQC with the H-1 protons at δH 4.74 ppm indicating a direct bond J-coupling between C-1 and H-1; and (6) δC 100.21 of C-1′ with the cross peak in HSQC with H-1′ at δH 4.72 indicating a direct bond J-coupling between C-1′ and H-1′. The vertical lines are drawn at (a) δH 4.74 of H-1. Inset: along this vertical line two long-range J-couplings to C-2 at δC 69.83 and to C-a of the acrylate at δC 67.38; and (b) δH 4.72 of H-1′ of the second mannose unit are observed. Inset: along this vertical line two long-range J-couplings to C-4 at δC 70.5 and to C-4′ at δC 76.8 are observed; vertical lines c, d and e drawn at crossings described in horizontal lines 3, 2 and 4, respectively. All spectra were collected at 10 °C in D2O. | ||
We also confirmed that the synthesised M1EMA corresponded to the proposed structure (Fig. 5A). Fig. S8† reveals the formation of the glycosidic linkage between the acrylate and the mannose unit at the anomeric carbon (C-a to C-1 to C-2), in analogy to the analysis shown in Fig. 7. Fully resolved 1H and 13C spectra for this compound can be found in the ESI (Fig. S9 and S10†).
Configuration at the anomeric carbon
The direct coupling constant 1JC-1,H-1 is the most reliable indication of an anomeric configuration.34 It has been reported that this coupling constant in pyranose derivatives of carbohydrates is approximately 10 Hz lower for an axial (β) H-1 compared to an equatorial H-1 (α), 160 Hz against 170 Hz, respectively. This difference is found even when there are differences in the magnitudes related to the electronegativity of the substituent at C-1.39 In both our samples this direct coupling constant, 1JC-1,H-1, was determined to be ∼160 Hz, which is in agreement with that observed in pyranose derivatives for an axial (β) H-1 (Fig. S6†). As an additional confirmation of the β-configuration, we observed a strong NOE between H-1 (anomeric proton) and H2, H3 and H5 in M1EMA, which has been observed for β-D-mannose35 (Fig. S7†). We hereby conclude that the resolved structure of enzymatically synthesised M2EMA corresponds to the hypothesised molecular structure (Fig. 5B).Polymerisation of the enzymatically produced monomers
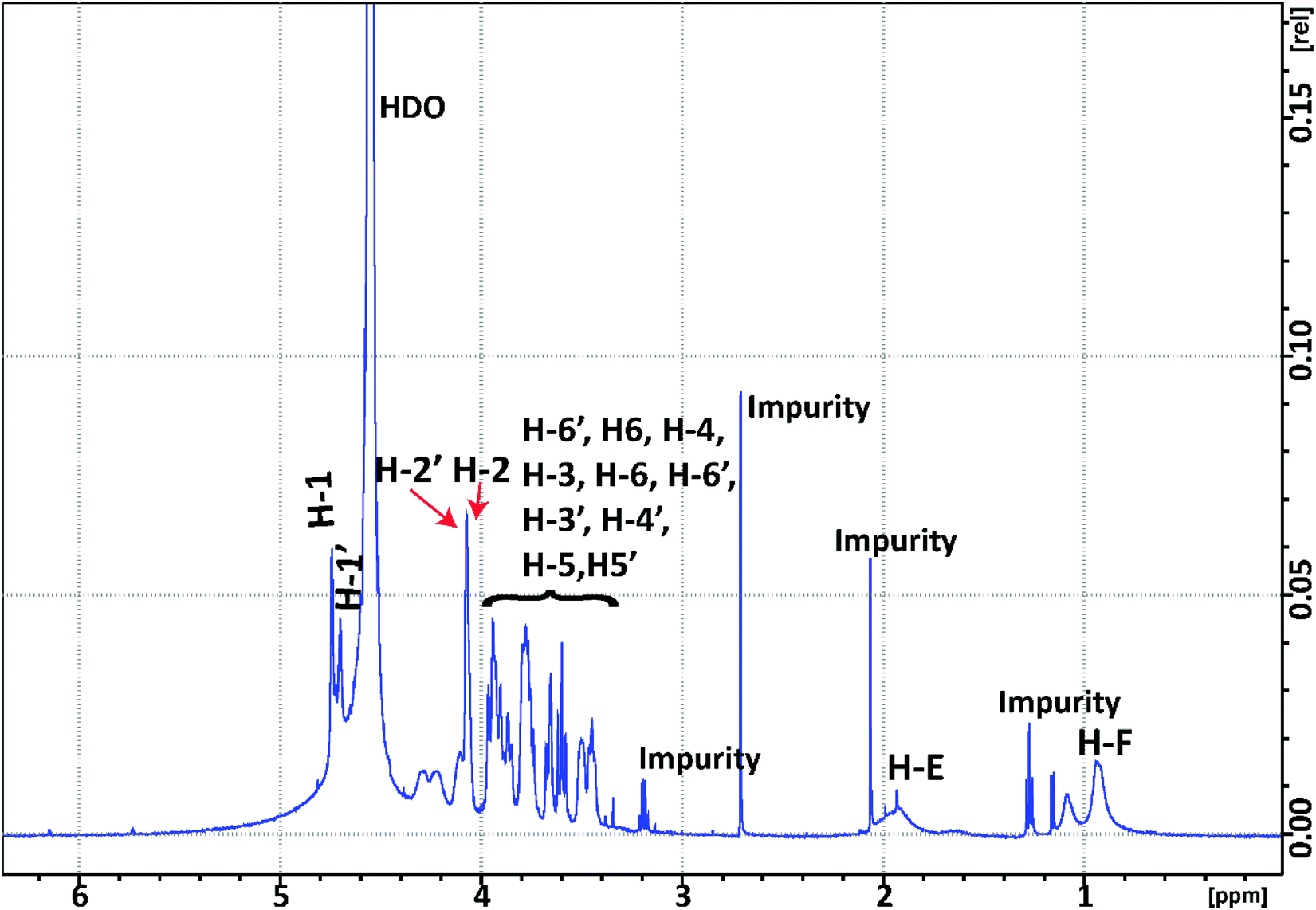 | ||
| Fig. 8 1H NMR spectrum of poly(M2EMA) collected at 45 °C, D2O. HDO – residual water peak. | ||
The polymerisation solutions were also analysed by SEC (Fig. 9). The main peak of poly(M2EMA) eluted with the void volume (5.2 ml). The main peak of poly(M1EMA) eluted at 6 ml, which corresponds to an apparent molecular weight larger than that of the highest dextran standard included (dextran, 1400 kDa, elution volume: 6.7 ml). It should be noted that both poly(M2EMA) and poly(M1EMA) showed additional minor peaks in the SEC chromatogram. For poly(M2EMA), a peak eluting at 7.4 ml (close to the elution volume for dextran 410 kDa) was observed. For poly(M1EMA) a minor peak eluted at 9.5 ml, which is a larger volume than that for the smallest dextran standard used (dextran 50 kDa eluted at 8.8 ml). In comparison, a standard of polyethylene glycol (PEG) of 400 Da eluted at 11.5 ml. Based on the present data, it is not possible to conclude whether the synthesised polymers form aggregates, which could potentially be the reason why multiple peaks were present in the SEC analysis. However, the SEC data further confirmed that the enzymatic synthesis of polymerisable monomers was successful, resulting in novel glycopolymers. Further study of these glycopolymers is an interesting future line of research but it is out of the scope of this study.
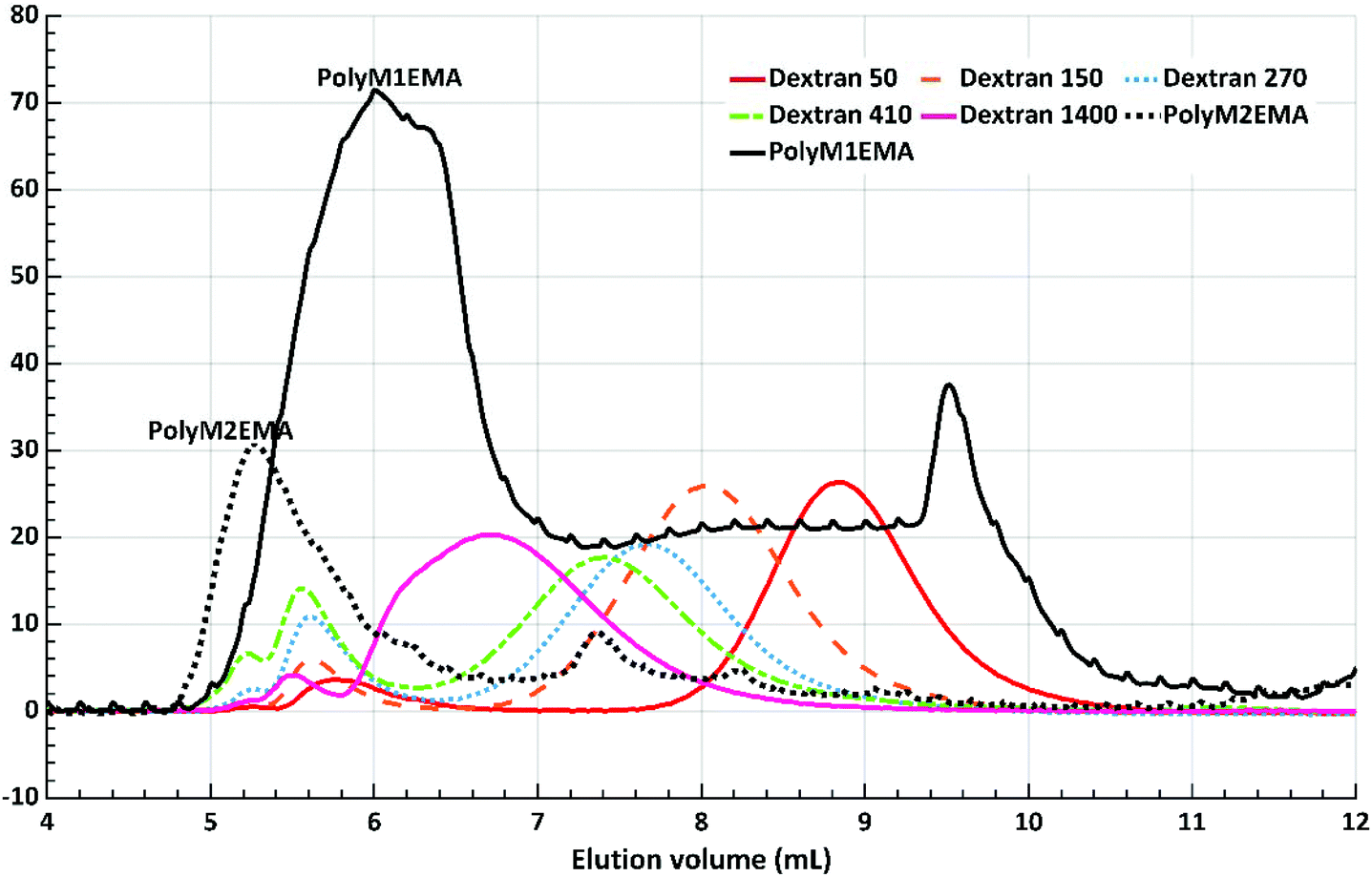 | ||
| Fig. 9 SEC chromatogram of poly(M2EMA) and poly(M1EMA), including dextran standards (Mw 50, 150, 270, 410 and 1400 kDa). Poly(M2EMA) eluted with the void volume (5.2 ml). | ||
Glycopolymers such as the ones prepared in our study are of great interest as they can be produced from renewable abundant carbohydrate sources. Glycopolymers based on acrylate monomers have received particular attention due to their multiple potential applications.2,40–42 Acrylates and methacrylates are extensively used in industry because they are highly versatile and can be polymerised or copolymerised with numerous monomers to tune material properties. They can be used to produce functional materials that range from soft rubbers to non-film forming materials.43 By attaching a glycan structure to the acrylate functionality, yielding our glycomonomers, we expect to contribute to further functional versatility. Through different polymerisation techniques we may increase the possibilities for new applications. For example, we envision synthesising a range of novel polymers with different properties via copolymerisation with different comonomers. The glycan part will then likely modulate the solubility and aggregation properties, and also potentially contribute with specific affinity for biomaterial surfaces and tissues, further widening the applications in biomaterial technology and biomedicine.
Conclusions
We have successfully demonstrated the enzyme catalysed synthesis of mannosyl acrylate monomers and their polymerisation into novel polyacrylates with one or two pendant mannosyl units. We show that the choice of the β-mannanase used for synthesis is important since different product profiles were obtained with the two tested enzymes. β-Mannanase AnMan5A generated longer saccharide conjugates compared to TrMan5A with M4 as the donor and either HEMA or an allyl alcohol as the acceptor. These product profiles are in good agreement with previous transglycosylation reactions using saccharides or simpler alcohols as acceptors.21 We furthermore demonstrate that abundant and renewable hemicellulosic β-mannans (locust bean galactomannan and softwood hemicellulose) can directly be used as donor glycans in the β-mannanase catalysed synthesis of these mannosyl acrylate monomers. Different conditions for reactions using β-mannanase TrMan5A as a catalyst, locust bean gum galactomannan as a donor and HEMA as an acceptor were investigated. A 50 ml scale reaction generated sufficient amounts of HILIC purified M1EMA and M2EMA to verify the predicted chemical structures of these two novel glycomonomers, using 1H and 13C NMR spectroscopy. M1EMA and M2EMA polymerised into novel water soluble acrylic polymers carrying pendant mannosyl or mannobiosyl units, respectively. These pendant glycan moieties are expected to be evenly distributed along the polyacrylate chain and we believe that varying the ratio between them individually, as well as using other acrylate monomers during polymerisation, will enable the design of future glycopolymers with differences in properties such as solubility, aggregation and surface/biomolecular interactions.The new monomers thus have the potential to open up for new applications, also taking advantage of the specific interaction of mannosyl units with biomolecules.2 A route forward to reach such applications could, upscaling aside, involve an increase of the yield during the enzymatic synthesis. An interesting approach there would be either pre-hydrolysis of the galactomannan22 or enzyme synergy by the inclusion of a sidegroup cleaving enzyme. This could increase the exposure of the β-mannan backbone to the β-mannanase, which has been shown for other enzyme systems.30 These two approaches would probably also aid when further investigating abundant forestry resources (AcGGM) as glycan donors, for which the first trials were performed here.
It is also worth mentioning that different β-mannanases clearly yield different glycan lengths during synthesis. Hence, an increased understanding of the catalysts would aid in future enzyme development approaches to govern product profiles,22 as well as limit secondary hydrolysis, further increasing the capability to synthesise defined acrylate glycomonomers. In the current paper, we also further establish mass spectrometry as a powerful tool for screening when conditions or enzyme variants are initially evaluated.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This study was financed by the Swedish Foundation for Strategic Research (SSF) through grant RBP14-0046 and Carl Tryggers Stiftelse. We thank Eimantas Šileikis for technical help related to preparative HPLC. We are also grateful for the help and assistance of Dr Christoffer Karlsson in the NMR analysis of the monomers.References
- H. V. Scheller and P. Ulvskov, in Annu. Rev. Plant Biol, ed. S. Merchant, W. R. Briggs and D. Ort, 2010, vol. 61, pp. 263–289 Search PubMed.
- S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392–1412 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- M. Obata, M. Shimizu, T. Ohta, A. Matsushige, K. Iwai, S. Hirohara and M. Tanihara, Polym. Chem., 2011, 2, 651–658 RSC.
- W. M. J. Kloosterman, D. Jovanovic, S. G. M. Brouwer and K. Loos, Green Chem., 2014, 16, 203–210 RSC.
- W. M. J. Kloosterman, S. Roest, S. R. Priatna, E. Stavila and K. Loos, Green Chem., 2014, 16, 1837–1846 RSC.
- S. Cheng, Y. Zhao and Y. Wu, Prog. Org. Coat., 2018, 118, 40–47 CrossRef CAS.
- M. Ambrosi, A. S. Batsanov, N. R. Cameron, B. G. Davis, J. A. K. Howard and R. Hunter, J. Chem. Soc., Perkin Trans. 1, 2002, 45–52 CAS.
- J. J. Gridley and H. M. I. Osborn, J. Chem. Soc., Perkin Trans. 1, 2000, 1471–1491 RSC.
- L. Wen, G. Edmunds, C. Gibbons, J. Zhang, M. R. Gadi, H. Zhu, J. Fang, X. Liu, Y. Kong and P. G. Wang, Chem. Rev., 2018, 118, 8151–8187 CrossRef CAS PubMed.
- T. J. Boltje, T. Buskas and G. J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed.
- M. L. Sinnott, Chem. Rev., 1990, 90, 1171–1202 CrossRef CAS.
- F. van Rantwijk, M. W. V. Oosterom and R. A. Sheldon, J. Mol. Catal. B: Enzym., 1999, 6, 511–532 CrossRef CAS.
- P. Bojarova and V. Kren, Trends Biotechnol., 2009, 27, 199–209 CrossRef CAS PubMed.
- M. Ochs, M. Muzard, R. Plantier-Royon, B. Estrine and C. Rémond, Green Chem., 2011, 13, 2380–2388 RSC.
- A. Adharis, D. Vesper, N. Koning and K. Loos, Green Chem., 2018, 20, 476–484 RSC.
- J. Lundqvist, A. Jacobs, M. Palm, G. Zacchi, O. Dahlman and H. Stalbrand, Carbohydr. Polym., 2003, 51, 203–211 CrossRef CAS.
- S. Willfor, K. Sundberg, M. Tenkanen and B. Holmbom, Carbohydr. Polym., 2008, 72, 197–210 CrossRef CAS.
- A. Andersson, T. Persson, G. Zacchi, H. Stalbrand and A. S. Jonsson, Appl. Biochem. Biotechnol., 2007, 137, 971–983 CrossRef PubMed.
- A. Rosengren, P. Hagglund, L. Anderson, P. Pavon-Orozco, R. Peterson-Wulff, W. Nerinckx and H. Stalbrand, Biocatal. Biotransform., 2012, 30, 338–352 CrossRef CAS.
- A. Rosengren, S. K. Reddy, J. S. Sjoberg, O. Aurelius, D. T. Logan, K. Kolenova and H. Stalbrand, Appl. Microbiol. Biotechnol., 2014, 98, 10091–10104 CrossRef CAS PubMed.
- J. Morrill, A. Manberger, A. Rosengren, P. Naidjonoka, P. von Freiesleben, K. Krogh, K. E. Bergquist, T. Nylander, E. N. Karlsson, P. Adlercreutz and H. Stalbrand, Appl. Microbiol. Biotechnol., 2018, 102, 5149–5163 CrossRef CAS PubMed.
- P. Hagglund, T. Eriksson, A. Collen, W. Nerinckx, M. Claeyssens and H. Stalbrand, J. Biotechnol., 2003, 101, 37–48 CrossRef CAS PubMed.
- M. Bounias, Anal. Biochem., 1980, 106, 291–295 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. De Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Magn. Reson. Chem., 2008, 46, 582–598 CrossRef CAS PubMed.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
- G. L. Sassaki and L. M. de Souza, in Tandem Mass-spectrometry - Molecular Characterisation, 2013, ch. 4, pp. 82–115 Search PubMed.
- E. Sabini, H. Schubert, G. Murshudov, K. S. Wilson, M. Siika-Aho and M. Penttila, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 3–13 CrossRef CAS.
- V. Bagenholm, S. K. Reddy, H. Bouraoui, J. Morrill, E. Kulcinskaja, C. M. Bahr, O. Aurelius, T. Rogers, Y. Xiao, D. T. Logan, E. C. Martens, N. M. Koropatkin and H. Stalbrand, J. Biol. Chem., 2017, 292, 229–243 CrossRef PubMed.
- S. K. Reddy, V. Bagenholm, N. A. Pudlo, H. Bouraoui, N. M. Koropatkin, E. C. Martens and H. Stalbrand, FEBS Lett., 2016, 590, 2106–2118 CrossRef CAS PubMed.
- S. Malgas, J. S. van Dyk and B. I. Pletschke, World J. Microbiol. Biotechnol., 2015, 31, 1167–1175 CrossRef CAS PubMed.
- V. Harjunpaa, A. Teleman, M. Siika-Aho and T. Drakenberg, Eur. J. Biochem., 1995, 234, 278–283 CrossRef CAS PubMed.
- W. A. Bubb, Concepts Magn. Reson., Part A, 2003, 19, 1–19 CrossRef.
- P. K. Agrawal, Phytochemistry, 1992, 31, 3307–3330 CrossRef CAS PubMed.
- M. Tenkanen, M. Makkonen, M. Perttula, L. Viikari and A. Teleman, J. Biotechnol., 1997, 57, 191–204 CrossRef CAS PubMed.
- F. Pereira, Carbohydr. Res., 2011, 346, 960–972 CrossRef CAS PubMed.
- J. Voepel, J. Sjöberg, M. Reif, A.-C. Albertsson, U.-K. Hultin and U. Gasslander, J. Appl. Polym. Sci., 2009, 112, 2401–2412 CrossRef CAS.
- K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293–297 RSC.
- S. Kitazawa, M. Okumura, K. Kinomura and T. Sakakibara, Chem. Lett., 1990, 19, 1733–1736 CrossRef.
- M. Al-Bagoury, K. Buchholz and E.-J. Yaacoub, Polym. Adv. Technol., 2007, 18, 313–322 CrossRef CAS.
- M. Obata, R. Otobuchi, T. Kuroyanagi, M. Takahashi and S. Hirohara, J. Polym. Sci., Part A-1: Polym. Chem., 2017, 55, 395–403 CrossRef CAS.
- S. Srivastava, Des. Monomers Polym., 2009, 12, 1–18 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc03947j |
| ‡ These authors contributed equally to the study. |
| This journal is © The Royal Society of Chemistry 2019 |