
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3,2,1 and stop! An innovative, straightforward and clean route for the flash synthesis of metallacarboranes†
Ines
Bennour
,
Ana M.
Cioran
,
Francesc
Teixidor
 and
Clara
Viñas
*
and
Clara
Viñas
*
Institut de Ciència de Materials de Barcelona (ICMAB-CSIC). Campus UAB, 08193 Bellaterra, Barcelona, Spain. E-mail: clara@icmab.es
First published on 20th March 2019
Abstract
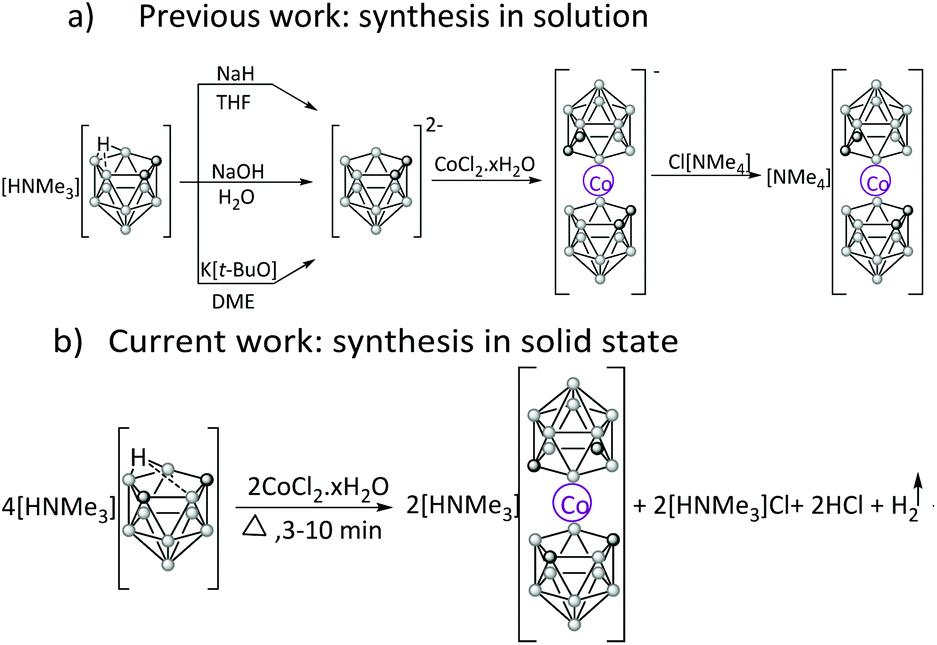
[Co(C2B9H11)2]−, analogs to metallocene, can be readily synthesised in high yield by a fast and clean solvent-free reaction of [HNMe3][C2B9H12] with CoCl2·xH2O. This innovative approach, applied to both ortho and meta isomers, yields the desired structures by simply heating the solid compounds to high temperature for a very short time.
Metallacarboranes are 3D boron hydride clusters that incorporate metal atoms or units in their polyhedral skeletons.1 They find many applications in materials, sensors/biosensors and medicine, among others.2 Metallacarboranes [M(C2B9H11)2]− (M = Fe3+, Co3+), which were reported in 1965
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 are the most studied. [M(C2B9H11)2]− were synthesised from the nido [C2B9H12]− cluster in two steps: (i) first, deprotonation of [C2B9H12]− produces nido [C2B9H11]2− which then (ii) reacts with metal-containing reagents yielding stable anionic metallacarboranes.3 The insertion of metal into the nido [C2B9H12]− ion is extremely versatile for two reasons: (i) the nido [C2B9H12]− ion withstands harsh reaction conditions (e.g. strong bases or reflux temperatures) and (ii) completion of the icosahedron is, in general, strongly thermodynamic and kinetic favoured. The synthesis of [M(C2B9H11)2]− has been reported in both non-aqueous and aqueous media by using NaH in dry THF or a freshly prepared hot 40% NaOH aqueous solution, respectively, to deprotonate the Hbridge of the nido-cluster.4 The complexation reaction with MCl2 required the reaction mixture to be stirred at reflux temperature under nitrogen for 3 hours. In both methods the work-up to isolate the pure [M(C2B9H11)2]− is tedious.4 A one-pot reaction method in anhydrous DME that uses K[t-BuO] as a base and anhydrous MCl2 was reported to produce [M(C2B9H11)2]− derivatives in high yield requiring much less work-up (Scheme 1a).5 This route is, however, time consuming and implies dealing with DME, a solvent with a high boiling point and hence, difficult to remove from the reaction flask.
3 are the most studied. [M(C2B9H11)2]− were synthesised from the nido [C2B9H12]− cluster in two steps: (i) first, deprotonation of [C2B9H12]− produces nido [C2B9H11]2− which then (ii) reacts with metal-containing reagents yielding stable anionic metallacarboranes.3 The insertion of metal into the nido [C2B9H12]− ion is extremely versatile for two reasons: (i) the nido [C2B9H12]− ion withstands harsh reaction conditions (e.g. strong bases or reflux temperatures) and (ii) completion of the icosahedron is, in general, strongly thermodynamic and kinetic favoured. The synthesis of [M(C2B9H11)2]− has been reported in both non-aqueous and aqueous media by using NaH in dry THF or a freshly prepared hot 40% NaOH aqueous solution, respectively, to deprotonate the Hbridge of the nido-cluster.4 The complexation reaction with MCl2 required the reaction mixture to be stirred at reflux temperature under nitrogen for 3 hours. In both methods the work-up to isolate the pure [M(C2B9H11)2]− is tedious.4 A one-pot reaction method in anhydrous DME that uses K[t-BuO] as a base and anhydrous MCl2 was reported to produce [M(C2B9H11)2]− derivatives in high yield requiring much less work-up (Scheme 1a).5 This route is, however, time consuming and implies dealing with DME, a solvent with a high boiling point and hence, difficult to remove from the reaction flask.
 | ||
| Scheme 1 | ||
Solvent-free transformations are an essential approach to the sustainability of organic and organometallic synthesis6 through the 21st century due to its efficiency in minimizing waste.7,8 In this work, the new expeditive syntheses of [M(C2B9H11)2]− (M = Co3+) and their Cc-substituted derivative compounds have been established by exploring the reactivity of nido [HNMe3][C2B9H12] with CoCl2·xH2O under solvent-free conditions in the absence of bases at high temperature during a very short time (minutes). The main objective was to design an easy, one-pot route to synthesise the pristine [Co(C2B9H11)2]− and their Cc-substituted derivatives in a high yield in a rapid and efficient reaction.
The solvent-free reaction between nido [HNMe3][7,8-C2B9H12] and CoCl2·xH2O (x = 0, 6) was performed by heating the two solid reagents at 350 °C (also at 250 °C) into a Pyrex tube. As displayed in Table 1, several parameters such as temperature, reaction time, nature of the CoCl2 reagent (anhydrous or hydrated) as well as the CoCl2/[NHMe3][C2B9H12] molar ratio were studied.
| Cluster | Entry | Conditions | Molar ratio CoCl2/[HNMe3][nido-C2B9H12] | Yield % | |
|---|---|---|---|---|---|
| Time (min) | T (°C) | ||||
| a Yield related to the cobaltabis(dicarbollide) mixture. | |||||

|
1 | 3 | 350 | 1.5 CoCl2 anh. | 83 |
| 2 | 5 | 350 | 1.5 CoCl2 anh. | 85 | |
| 3 | 7 | 350 | 1.5 CoCl2 anh. | 88 | |
| 4 | 7 | 350 | 1.5 CoCl2·6H2O | 90 | |
| 5 | 8 | 350 | 1.5 CoCl2·6H2O | 88 | |
|
6 | 2 | 350 | 1.5 CoCl2 anh. | 42 |
| 7 | 3 | 350 | 1.5 CoCl2 anh. | 46 | |
| 8 | 3 | 470 | 1.5 CoCl2 anh. | 68 | |
| 9 | 5 | 350 | 1.5 CoCl2 anh. | 75 | |
| 10 | 10 | 350 | 1.5 CoCl2 anh. | 90 | |
| 11 | 2 + 6 | 350 + 470 | 1.5 CoCl2·6H2O | 87 | |

|
12 | 2 + 6 | 350 + 470 | 1.5 CoCl2·6H2O | 88 |
| 13 | 2 + 6 | 350 + 470 | 2.5 CoCl2·6H2O | 94 | |
| 14 | 2 + 6 | 350 + 470 | 2.5 CoCl2 anh. | 60 | |
| 15 | 10 | 350 | 5.5 CoCl2 anh. | 83 | |

|
16 | 2 | 350 | 2.5 CoCl2 anh.. | 82a |
| 17 | 8 | 250 | 2.5 CoCl2 anh. | 68a | |
| 18 | 8 | 250 | 2.5 CoCl2·6H2O | 78a | |

|
19 | 2 | 350 | 2.5 CoCl2 anh. | 68a |
| 20 | 8 | 250 | 2.5 CoCl2 anh. | 62a | |
| 21 | 8 | 250 | 2.5 CoCl2·6H2O | 70a | |

|
22 | 8 | 470 | 2.5 CoCl2 anh. | 50 |

|
23 | 5 | 350 | 2.5 CoCl2 anh. | 40 |
| 24 | 10 | 350 | 2.5 CoCl2 anh. | 47 | |
| 25 | 15 | 350 | 2 CoCl2 anh. | 52 | |
| 26 | 15 | 350 | 2.5 CoCl2 anh. | 74 | |
Upon completion of the reaction, water was added to the crude product before extracting with ethyl ether. Surprisingly, the formation of cobalt metal was not observed. This was typical in the wet methods as Co(II) dismutates to Co(III) and Co.4 The orange organic phase was separated from the pink aqueous phase, the solvent removed in vacuum, and the residue taken up in water. This solution was treated with an aqueous solution of Cl[NMe4] to give the less soluble [NMe4][3,3′-Co(1,2-C2B9H11)2] complex, which was confirmed by 11B- and 1H NMR spectra. The yield provided in Table 1 is related to the [NMe4]+ cation in all experiments.
As demonstrated in entries 3 and 4, no significant effect on final yield was observed when using anhydrous CoCl2 with respect to the CoCl2·6H2O.
Once the synthesis of the pristine [3,3′-Co(1,2-C2B9H11)2]− complex was achieved, we tested if the synthesis of Ccluster-substituted cobaltabis(dicarbollide) complexes could be accomplished. Reaction was run by using the same method and conditions but starting with the monosubstituted [HNMe3][8-R-7,8-C2B9H10] (R = Me, Ph) ligands.
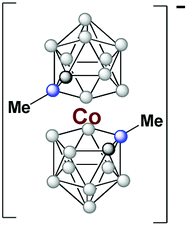
This new reaction led to the formation of [NMe4][3,3′-Co(1-Me-1,2-C2B9H10)2] and [NMe4][3,3′-Co(1-Ph-1,2-C2B9H10)2] sandwich with yields equal to 90% after 10 min and 94% after 8 min, respectively (entries 10 and 13). It was noticed that the presence of one methyl or aryl group linked to the Ccluster decreases the reaction's yield; however, very good yields were still obtained in only 10 minutes of heating in comparison with the traditional method that takes around 1440 minutes to give rise to the same compound, but with a yield difference of approx. 5%. Time of reaction also has a positive impact on the final yield. As illustrated in Table 1, entries 7 and 9, the yield rises significantly with increase in reaction time, namely from 3 to 5 minutes. Furthermore, if temperature is increased by 120 °C (entries 7 and 8), a consequently boost in yield of about 22% is observed. Reactions with CoCl2·xH2O (x = 0, 6) have also been conducted at 250 °C just extending the reaction time 6 extra minutes with comparable yields to these in the table.
Based on these utmost positive results, the complexation reactions of disubstituted nido units ([HNMe3][7-R-8-R′-7,8-C2B9H10] (R = R′ = Et; R = Me, R′ = Et and R = Ph, R′ = Et)) were investigated. Our approach was shown to work with both [HNMe3][7-Me-8-Et-7,8-C2B9H10] and [HNMe3][7,8-Et2-7,8-C2B9H10], leading to [Co(C2B9H11)2]− derivatives with yields above 70%. However, the ethyl units were lost in part (vide infra). On the other hand, the linkage of the aryl substituent to the C2B3 face in the presence of the ethyl group hinders the formation of the complex. Therefore, our next step was to study the effect of the lone pair of electrons on the complexation reactions. Starting from closo 1-R-2-R′-1,2-C2B10H10 (R = R′ = SEt; R = Me, R′ = SEt and R = Ph, R′ = SEt), via a partial deboronation reaction, nido ligands [HNMe3][7-R-8-R′-7,8-C2B9H10] (R = R′ = SEt; R = Me, R′ = SEt and R = Ph, R′ = SEt)9 were synthesised (see ESI†).
Out of these three compounds, only [HNMe3][7-Me-8-SEt-7,8-C2B9H10], yielded 50% of the target complex, which confirms the inhibition of the solid state reaction when hindrance and/or electronic alterations exist (entry 22). Nevertheless, the yield of the [HNMe3][3,3′-Co(1-Me-2-SEt-C2B9H9)2] was low compared to the previously obtained complexes. The yield for this particular [HNMe3][7-Me-8-SEt-7,8-C2B9H10] ligand using traditional methods was of 79% with K[t-BuO] and 35% when NaH was employed.5 Having obtained such good results and satisfactory yields by using this new solid state complexation reaction on ortho–nido species, the behaviour of the meta–nido isomer, [HNMe3][7,9-C2B9H12], was our target.
The [NMe4][2,2′-Co(1,7-C2B9H11)2] complex was obtained in 74% yield after heating at 350 °C for 15 min. The meta-isomer needed more time to produce a comparable good yield to the ortho-isomer. Table 1 summarises the different guidelines used to find the optimal parameters for all obtained sandwich complexes. The direct isolation of the complexes having cobalt in the formal +3 oxidation state was not accompanied by the formation of cobalt metal. This observation suggests that the initially Co2+ is rapidly oxidized to form the more stable d6 metallabis(dicarbollide) complex 18 e− count avoiding reduction from Co2+ to Co0. This is proof that the free-solvent synthesis of [3,3′-Co(1,2-C2B9H11)2]− takes place via another mechanism than the previously reported synthesis in solution.
To discern on the reaction pathway, several studies were run. The solvent-free reaction's progress was controlled by the colour change of the mixture as well as liberation of gas. To determine the identity of the evolved gas, a water solution monitored by a pH meter was prepared. As the pH of this aqueous solution did not change, the generated gas was not NMe3, but probably hydrogen. The next study was to measure the pH of the aqueous phase of the extraction process, which provided a value of 6.33 suggesting that an acid had been generated. Both experiments along with the observation that no Co0 was produced and the 11B NMR evidence that [3,3′-Co(1,2-C2B9H11)2]− was obtained, agree with the balanced equation displayed at Scheme 1(b). The solvent-free synthetic reaction takes place by an oxidation–reduction reaction that involves the two redox couples Co3+/Co2+ and H2O/OH−. Moreover, X-ray diffraction of [HNMe3][2,2′-Co(1,7-C2B9H11)2] fully supports that the cation is [HNMe3]+ (Fig. 1).10
 | ||
Fig. 1 Perspective view of the [HNMe3][2,2′-Co(1,7-C2B9H11)2] unit with 50% ellipsoids. Selected bond lengths (Å): C1–B3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C7–B3 = 1.727(7) Å, C1–B6C7–B11 = 1.683(7) Å, B11–B6 = 1.706(7) Å. C7–B3 = 1.727(7) Å, C1–B6C7–B11 = 1.683(7) Å, B11–B6 = 1.706(7) Å. | ||
The MS spectra of [NMe4][Co(1-Me-2-Et-1,2-C2B9H9)2] and [NMe4][Co(1,2-Et2-1,2-C2B9H9)2] are clear in showing that some of the ethyl groups are lost/won during the formation of the cobaltabis(dicarbollide) cluster from the nido components and CoCl2(xH2O), x = 0, 6. We do not know at what stage of the complex building the transfer of ethyl units occurs, but it is clear that at 250 °C some of the ethyl units are lost or won. Our interpretation is that at some stage of the building process β-hydride elimination occurs, (ESI†) that is consistent with NMR spectra indicating co-existence of some [Co(C2B9H11)2]− derivatives.
Conclusions
A new, fast and environmentally-friendly solid state reaction for the syntheses of cobaltabis(dicarbollides) is presented. Our approach is a significant improvement on the traditional syntheses in solution in both speed of reaction and generated yield. We demonstrate that the [Co(C2B9H11)2]− building reaction works well with starting plain, single or double Ccluster-substituted nido clusters. However, care has to be taken when β-hydride elimination may take place because in this case the resulting [Co(C2B9H11)2]− derivative may be different to the expected one based on the precursor nido cluster due to the β-hydride elimination, as commonly occurs in organometallic chemistry. The presence of substituents bearing a free pair of electrons also influences the complex formation. Meta-isomers also give rise to the corresponding cobaltabis(dicarbollide), but in this case, the reaction time needs to be increased. A suggested mechanism of the complexation reaction is proposed, based on identifying the chemical nature of the evolved gas, the pH of the mixture, the crystal structure of the target complex and the absence of Co2+ dismutation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by MINECO (CTQ2016-75150-R), Generalitat de Catalunya (2017 SGR 1720).Notes and references
- R. N. Grimes, Carboranes, Elsevier Inc., New York, 3rd edn, 2016 Search PubMed; C. E. Housecroft, Cluster Molecules of the p-Block Elements, Oxford University Press Inc., New York, 1994 Search PubMed.
- N. S. Hosmane and R. Eagling, Handbook of Boron Science with applications in Organometallics, Catalysis and Medicine (4 Vols.), World Scientific Publishing Europe Ltd., London, 2018 CrossRef PubMed; R. Ziólkowski, A. B. Olejniczak, L. Górski, J. Janusik, Z. J. Leśnikowski and E. Malinowska, Bioelectrochemistry, 2012, 87, 78 CrossRef PubMed; T. García-Mendiola, V. Bayon-Pizarro, A. Zaulet, I. Fuentes, F. Pariente, F. Teixidor, C. Viñas and E. Lorenzo, Chem. Sci., 2016, 7, 5786 RSC; E. Hey-Hawkins and C. Viñas, Boron-Based Compounds: Potential and Emerging Applications in Medicine, John Wiley & Sons Ltd., 2018 Search PubMed.
- M. F. Hawthorne, D. C. Young and P. A. Wegner, J. Am. Chem. Soc., 1965, 87, 1818 CrossRef CAS; M. F. Hawthorne and T. D. Andrews, J. Chem. Soc., Chem. Commun., 1965, 443 RSC.
- M. F. Hawthorne, D. C. Young, T. D. Andrews, D. V. Howe, R. L. Pilling, A. D. Pitts, M. Reintjes, L. F. Warren Jr. and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 879 CrossRef CAS.
- C. Viñas, J. Pedrajas, J. Bertran, F. Teixidor, R. Kivekäs and R. Sillanpää, Inorg. Chem., 1997, 36, 2482 CrossRef.
- G. Kaupp, Top. Curr. Chem., 2005, 254, 95 CrossRef CAS; K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef PubMed; G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159 RSC; G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701 CrossRef PubMed; V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 1606 RSC; V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, J. Am. Chem. Soc., 2002, 124, 6244 CrossRef PubMed.
- R. Noyori, Chem. Commun., 2005, 1807 RSC; M. Avalos, R. Babiano, P. Cintas, J. L. Jiménez and J. C. Palacios, Angew. Chem., Int. Ed., 2006, 45, 3904 CrossRef CAS PubMed; M. Jansen and J. C. Schön, Angew. Chem., Int. Ed., 2006, 45, 3406 CrossRef PubMed.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792 CrossRef PubMed.
- M. F. Hawthorne, D. C. Young, P. M. Garrett, D. A. Owen, S. G. Schwerin, F. N. Tebbe and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 862 CrossRef CAS; C. Viñas, J. Pedrajas, J. Bertran, F. Teixidor, R. Kivekäs and R. Sillanpää, Inorg. Chem., 1997, 36, 2482 CrossRef; C. Viñas, J. Pedrajas, F. Teixidor, R. Kivekäs, R. Sillanpää and A. J. Welch, Inorg. Chem., 1997, 36, 2988 CrossRef.
- Crystal data for C7H32B18CoN: monoclinic, space group C2/m, a = 9.130(11) Å, b = 10.446(10) Å, c = 11.101(9) Å, V = 1014.44 Å3, Z = 2, T = 100(2) K, measured reflections 24871, independent reflections 856, Rint = 0.0325, R1 = 0.0677, wR2 = 0.1743. CCDC 1882757.†.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis characterization of the cobaltabis(dicarbollide) complexes and X-ray studies of [HNMe3][2,2′-Co(1,7-C2B9H12)2]. CCDC 1882757. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8gc03943g |
| This journal is © The Royal Society of Chemistry 2019 |