
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient epoxidation of vegetable oils catalyzed by a manganese complex with hydrogen peroxide and acetic acid
Jianming
Chen
,
Marc
de Liedekerke Beaufort
,
Lucas
Gyurik
,
Joren
Dorresteijn
,
Matthias
Otte†
 and
Robertus J. M.
Klein Gebbink
*
and
Robertus J. M.
Klein Gebbink
*
Organic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: r.j.m.kleingebbink@uu.nl
First published on 1st April 2019
Abstract
Epoxidized vegetable oils (EVOs) are versatile building blocks for lubricants, plasticizers, polyvinyl chloride (PVC) stabilizers, and surface coating formulations. In this paper, a catalytic protocol for the efficient epoxidation of vegetable oils is presented that is based on a combination of a manganese catalyst, H2O2 as an oxidant, and acetic acid as an additive. This protocol relies on the use of a homogeneous catalyst based on the non-noble metal manganese in combination with a racemic mixture of the N,N′-bis(2-picolyl)-2,2′-bispyrrolidine ligand (rac-BPBP). The optimized reaction conditions entail only 0.03 mol% of the manganese catalyst with respect to the number of double bonds (ca. 0.1 wt% with respect to the oil) and ambient temperature. This epoxidation protocol is highly efficient, since it allows most of the investigated vegetable oils, including cheap waste cooking oil, to be fully epoxidized to EVOs in more than 90% yield with excellent epoxide selectivities (>90%) within 2 h of reaction time. In addition, the protocol takes place in a biphasic reaction medium constituted by the vegetable oil itself and an aqueous acetic acid phase, from which the epoxidized product can be easily separated via simple extraction. In terms of efficiency and reaction conditions, the current epoxidation protocol outperforms previously reported catalytic protocols for plant oil epoxidation, representing a promising alternative method for EVO production.
Introduction
Nowadays, the decreasing supply of fossil resources and increasing environmental problems have triggered the search for durable alternative raw materials for chemical production. Renewable biomass has been identified world-wide as a prime candidate to replace fossil resources as the feedstock for the chemical industry.1 Among various biomass resources, vegetable oils (VOs) represent one of the most promising candidates due to their wide availability, biodegradable properties, and low costs.2–4 Common VOs are mixtures of triglycerides, which are composed of three fatty acid moieties connected by a glycerol backbone (Fig. 1). These fatty acids, either saturated or unsaturated, normally have 14 to 22 carbon atoms in each hydrocarbon chain, resulting is a relatively high overall carbon content.5 More importantly, the fatty acids in VOs are mostly unsaturated with 0 to 3 double bonds per carbon chain. Modifications of these C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds can produce new value-added chemicals or monomers for polymers, which has attracted research interest for many years.6,7 Functionalizing the double bonds via epoxidation is one of the most common approaches to produce epoxidized vegetable oils (EVOs), which are versatile building blocks for lubricants,8–10 plasticizers,11–13 polyvinyl chloride (PVC) stabilizers,14–16 and surface coating formulations.17–20
C bonds can produce new value-added chemicals or monomers for polymers, which has attracted research interest for many years.6,7 Functionalizing the double bonds via epoxidation is one of the most common approaches to produce epoxidized vegetable oils (EVOs), which are versatile building blocks for lubricants,8–10 plasticizers,11–13 polyvinyl chloride (PVC) stabilizers,14–16 and surface coating formulations.17–20
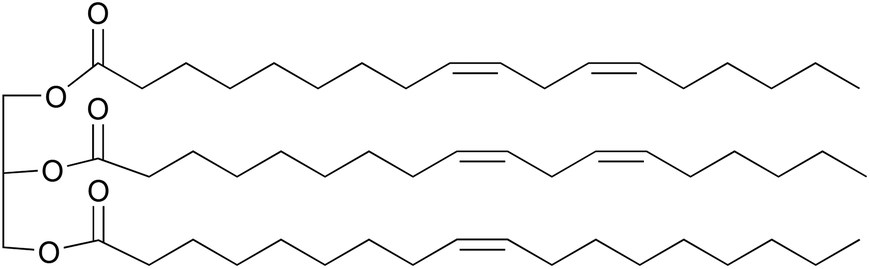 | ||
| Fig. 1 A typical triglyceride in sunflower oil, derived from the fatty acids linoleic acid (C18:2) and oleic acid (C18:1). | ||
The Prilezhaev process is currently adopted in industry for the production of EVOs. In this process double bonds are converted with percarboxylic acids formed in situ from a carboxylic acid (e.g. acetic acid) and hydrogen peroxide in the presence of a mineral acid such as H2SO4 or HCl (Scheme 1). This process has several drawbacks, such as low epoxide selectivity due to oxirane ring opening and corrosion issues, which are both caused by the strongly acidic reaction conditions. In the past few decades, tremendous efforts have accordingly been made for developing new catalytic systems, both homogeneous and heterogeneous, to form EVOs in a more selective and efficient manner.
 | ||
| Scheme 1 Conventional vegetable oil epoxidation process (Prilezhaev reaction). | ||
Some selected catalytic systems reported since 2000 for the epoxidation of VOs are listed in Table 1. Gerbase and co-workers reported a homogeneous CH3ReO3/H2O2/CH2Cl2 catalytic system, in which a very high yield of the epoxide (95%) was obtained under mild reaction conditions (entry 1).21 However, the use of an expensive noble metal (Re, 1 mol%) limits its application in a large-scale process. A cheaper metal (Mo) has been used by Farias et al. for the epoxidation of soybean oil.22 However, a relatively high reaction temperature of 110 °C was needed, to give only a moderate yield (54%) of the epoxide (entry 2). Lipases have also been used in the chemoenzymatic epoxidation of VOs. They have shown very high chemo-, regio-, and stereoselectivity without the formation of undesired ring-opening side-products.23 Vlček and Petrović used lipase Candida antarctica (Novozyme 435) to epoxidize soybean oil with H2O2 in high yield (entry 3).24 The protocol is sensitive to the reaction temperature; on one hand, a higher temperature is beneficial for double bond conversion; on the other hand this leads to deactivation of the lipase.4 Some other drawbacks of the use of lipases are their high cost and their relatively low reactivity because of limited interactions between the catalytic center and the large triglyceride substrates due to steric hindrance.4
| Entry | Oil | Oxidant | Catalyst (loading) | Solvent | Reaction conditions | Epoxide yield (%) |
|---|---|---|---|---|---|---|
| a TBHP = tert-butyl hydroperoxide. | ||||||
| 121 | Soybean oil | H2O2 | CH3ReO3 (1 mol%) | CH2Cl2 | 30 °C, 2 h | 95 |
| 222 | Soybean oil | TBHPa | [MoO2(acac)2] (1 mol%) | Toluene | 110 °C, 2 h | 54 |
| 324 | Soybean oil | H2O2 | Novozym 435 (4.0 wt%) | Toluene | 50 °C, 4 h | 90 |
| 425 | Soybean oil | H2O2 | [π-C5H5N(CH2)15CH3]3[PW4O16] (5.0 wt%) | — | 60 °C, 4 h | 90 |
| 526 | Soybean oil | H2O2 | [MeN(n-C8H17)3]{PO4[WO(O2)2]4} (31 wt%) | — | 40 °C, 3 h | 99 |
| 627 | Soybean oil | H2O2 | [MeN(n-C8H17)3]{PO4[WO(O2)2]4} supported on palygorskite (33 wt%) | — | 50 °C, 2 h | 79 |
| 730 | Soybean oil | H2O2 | Amorphous Ti/SiO2 (2.5 wt%) | tert-Butanol | 90 °C, >54 h | 88 |
| 831 | Soybean oil | TBHPa | Meso-Ti-HMS (2.5 wt%) | EtOAc | 60 °C, 24 h | 22 |
| 932 | Soybean oil | H2O2 | Nb2O5–SiO2 (12 wt%) | Et2O | 80 °C, 5 h | 10 |
| 1033 | Castor oil | H2O2 | Amberlite IR-120 (15 wt%) | Benzene | 50 °C, 10 h | 78 |
| 1134 | Soybean oil | TBHPa | MoO3/Al2O3 (Mo/CC = 1%) |
Toluene | 80 °C, 4 h | 16 |
| 1235 | Soybean oil | H2O2 | γ-Al2O3 (12 wt%) | Et2O | 80 °C, 10 h | 48 |
Entries 4–6 in Table 1 show examples of utilizing polyoxometalates as catalysts to epoxidize soybean oil with H2O2 as the oxidant. These epoxidation processes were carried out in a solvent-free medium, i.e., using a mixture of aqueous H2O2 and the plant oil. A somewhat elevated reaction temperature (40–60 °C) and a high catalyst loading (5–33 wt%) were required in these examples. For instance, Cheng et al. used 5 wt% of [π-C5H5N(CH2)15CH3]3[PW4O16] to obtain 90% of epoxidized soybean oil at a 60 °C reaction temperature.25 Immobilization of polyoxometalates onto an inorganic solid support is mostly used in order to increase their stability and reusability;4 nevertheless, this normally results in a lower catalytic activity. For example, Jiang et al. reported the use of the peroxophosphotungstate [MeN(n-C8H17)3]{PO4[WO(O2)2]4} for the catalytic epoxidation of soybean oil, which provided 99% yield at a very high catalyst loading (31 wt%, entry 5).26 Supporting this catalyst on modified halloysite nanotubes resulted in a diminished yield of 12%.26 Similarly, Chen et al. supported this complex on acid-activated palygorskite, giving rise to an epoxide yield of 79% (entry 6).27 The loading of this catalyst can be optimized to 0.1 mol% in the epoxidation of FAMEs, with 96% selectivity and 95% conversion at 60 °C. However, the epoxidation of VOs was not investigated in this study.28 Moores et al. developed a dipyridinium peroxophosphotungstate catalyst, which is able to fully epoxidize methyl oleate with a turnover number (TON) of 1868 using 0.22 mol% of the catalyst after 5 cycles at 60 °C.29 Also in this study the epoxidation of VOs was not reported.
Many heterogeneous catalytic systems have also been reported to be used in the epoxidation of VOs. Several representative examples are listed in Table 1, entries 7–12. As can be seen from these examples, all reactions were performed at high temperatures (mostly >80 °C), and in most of the cases relatively low reactivities were obtained (yields mostly <70%). For instance, reaction temperatures between 60 and 80 °C were required in the reactions using meso-Ti-HMS, Nb2O5–SiO2, MoO3/Al2O3, or γ-Al2O3 as a catalyst, giving rise to 10–48% yields of epoxidized soybean oil (entries 8, 9, 11, and 12). Fierro and co-workers have developed an amorphous Ti/SiO2/H2O2/t-BuOH catalytic protocol for the epoxidation of soybean oil with a relatively low catalyst loading (2.5 wt%), achieving a relatively high epoxide yield (88%, entry 7).30 However, a high reaction temperature (90 °C) and long reaction time (>54 h) were used. Using Amberlite IR-120 as catalyst, 78% epoxidized castor oil can be obtained under relatively mild reaction conditions (50 °C, entry 10).33 However, the toxic solvent benzene and a large amount of the catalyst (15 wt%) were used in this case.
To sum up, the conventional Prilezhaev process and previously reported catalytic systems for the epoxidation of VOs generally entail at least one of the following disadvantages: low selectivity, low catalyst efficiency, usage of high-cost catalysts, harsh reaction conditions, long reaction time, or the use of a harmful organic solvent. In order to meet the increasing demand for the production of EVOs on a large-scale, the development of more efficient, practical catalytic systems for the selective epoxidation of VOs and their derivatives under mild reaction conditions is desirable. Ideally, such catalytic systems would conform to the principles of green chemistry in modern chemistry.4
Spannring et al. have reported on a one-pot oxidative cleavage protocol of unsaturated fatty acids (UFAs) and fatty acid methyl esters (FAMEs) to form aldehydes as primary products, with the catalytic epoxidation of double bonds as the first and key step.36 This epoxidation step was carried out using the abundant, environmentally benign first-row transition metal iron, supported by a bis-alkylamine-bis-pyridine (N2Py2) ligand, in the presence of H2O2 and acetic acid (AcOH). The actual oxidizing species in this system is considered to be a highly electrophilic FeVO intermediate, which is generated through the lysis of the O–O bond of an FeIII–OOH species formed upon exposure of the starting Fe(II) complex to H2O2.37 Even though this catalytic system is relatively efficient, i.e., full conversion of the substrate can be achieved with 0.5 mol% of the catalyst per double bond, a further reduction of the catalyst loading seems necessary for the large-scale application of these protocols. Furthermore, these catalytic protocols make use of relatively toxic acetonitrile (MeCN) as the reaction solvent.
As a first-row transition metal, manganese has similar advantages to iron in terms of cost, availability, and low toxicity. In addition, several studies have reported that Mn(N2Py2) complexes generally show higher conversions and yields as compared to their Fe(N2Py2) analogs in both aliphatic C–H oxidation and alkene epoxidation.38–41 The generally accepted mechanism through which these Mn-catalysts operate starts with the oxidation of the starting MnII-complex with H2O2 to form a MnIII-hydroperoxo species.41 Subsequently, this species binds a carboxylic acid, which aids in the formation of a MnV-oxo-carboxylato intermediate and water through a cyclic, hydrogen-bonded transition state. It is this MnV-oxo intermediate that converts the alkene substrate to the epoxide product. Based on these findings, the current study aimed to explore the use of Mn(N2Py2) type complexes in combination with H2O2 and AcOH for the catalytic epoxidation of VOs and their derivatives. This study has resulted in the development of a highly efficient, Mn-based protocol that can be conducted at room temperature in only 2 h of reaction time, generally providing EVOs in more than 90% yield and with over 90% epoxy-group selectivity. In addition, the use of the MeCN solvent could be strongly limited and even avoided through further optimization of the reaction conditions. Details on the use of this Mn-based epoxidation protocol for a series of different VOs will also be discussed.
Results and discussion
A series of non-heme Mn-complexes bearing N2Py2 ligands have been synthesized to study the epoxidation of UFAs, FAMEs, and VOs (Fig. 2). These N2Py2 ligands, including the well-known BPMEN,42 BPMCN,43 and BPBP44 (BPMEN = N,N′-dimethyl-N,N‘-bis(2-picolyl)ethylenediamine, BPMCN = N,N′-dimethyl-N,N′-bis(2-picolyl)-cyclohexane-trans-1,2-diamine, and BPBP = N,N′-bis(2-picolyl)-2,2′-bispyrrolidine), can be readily converted to the corresponding Mn complexes [Mn(OTf)2(BPMEN)]45 (1), [Mn(OTf)2(R,R-BPMCN)]45 (2) and [Mn(OTf)2(BPBP)]46 (3), respectively, upon treatment with Mn(OTf)2 in THF (Fig. 2; see the Experimental section for details). Among the BPBP-based complexes, Mn-complexes derived from different BPBP stereoisomers,47i.e., S,S-, R,S-, rac-, and mix-BPBP, were synthesized and tested for their epoxidation activity. In contrast to the enantiomerically pure versions of the ligand, less costly non-enantiomerically pure versions of the BPBP ligand (such as rac- and mix-BPBP) would be preferably used in the epoxidation of VOs, in which enantioselectivity issues are not at stake from a product application point of view. The development and application of mix-BPBP in Fe-catalyzed epoxidation reactions was earlier reported by Spannring et al.36Mix-BPBP constitutes a mixture of R,R-BPBP, S,S-BPBP, and (meso) R,S-BPBP. In the current study, rac-BPBP was also used, which constitutes a mixture of R,R- and S,S-BPBP (see the Experimental section for details). Another N2Py2 ligand used in this study is BPBI (N,N′-bis(2-picolyl)-2,2′-bis-isoindoline), which was previously developed in our group for the synthesis of the Fe(BPBI) complex employed in aliphatic C–H oxidation reactions.48 Recently, this ligand and its derivatives have also been reported for the synthesis of the corresponding Mn-complexes, for which good reactivities have been observed in alkene epoxidation.49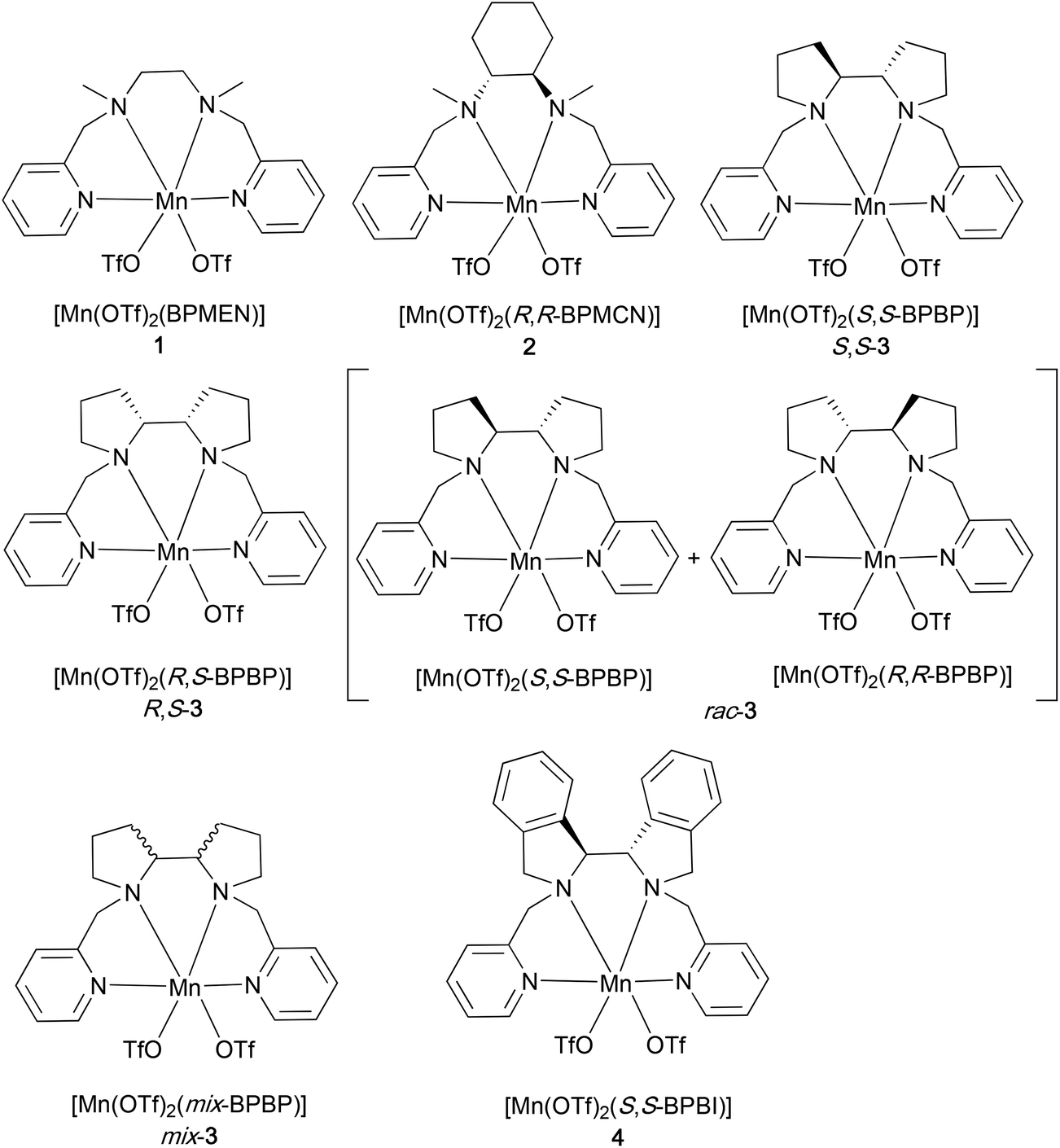 | ||
| Fig. 2 Non-heme Mn(II) complexes with N2Py2 ligands used in this study. | ||
Epoxidation of UFAs and FAMEs
Oleic acid (C18:1) was chosen as the model substrate to optimize the reaction conditions for the epoxidation of UFAs by the Mn-complexes in combination with H2O2 as the oxidant and AcOH as an additive (Table 2). Initial experiments were carried out in the absence of the Mn-complex to evaluate the crucial role of manganese in catalysis. On adding only H2O2 (2 equiv.)/AcOH, no substrate conversion was observed at all after 1 h (Table 2, entry 1). In contrast, 2 equiv. of m-chloroperoxybenzoic acid (mCPBA) were able to fully epoxidize the substrate, yielding the epoxide quantitatively (entry 2). This result is in line with a previous report on the high efficiency of mCPBA in the epoxidation of UFAs and VOs.50 When Mn(OTf)2, the Mn salt used for the synthesis of the Mn complexes, together with H2O2 (2 equiv.) and AcOH (9 equiv., undiluted) was used, no conversion of oleic acid was detected (entry 3). Subsequently, using similar reaction conditions to the reported Fe(mix-BPBP)/H2O2/AcOH catalytic protocol for the epoxidation of UFAs,36 catalytic experiments were performed with several Mn-complexes as catalysts (0.5 mol%). Complex 1 showed 40% conversion and yielded 32% of epoxidized oleic acid (entry 4). Similar to 1, 32% of the epoxide was found in the reaction with chiral complex 2, albeit with a somewhat higher conversion (48%; entry 5). The epoxide yield was found to increase to 46% with S,S-3, even though substrate conversion did not increase further (48%, entry 6). The generally increased catalytic performance from 1 to 2 to S,S-3 seems related to the enhanced stability of the manganese complexes, as a result of the increased rigidity of their ligand backbone.44,51 Using complex 4, a significant drop was found in both the conversion and yield (28% and 28%, respectively; entry 7).|
|
||||
|---|---|---|---|---|
| Entry | Cat. (mol%) | H2O2 equiv. (addition time) | Conv.b (%) | Yieldb (%) |
| a General reaction conditions: Oleic acid (0.5 mmol), catalyst, and AcOH (9 equiv.) were mixed and stirred in MeCN (2 mL) at room temperate (RT), and subsequently H2O2 (1 M solution in MeCN) was added, with a 1 h total reaction time. b Determined by NMR analysis. c 2 equiv. of mCPBA were used as the oxidant in the absence of AcOH. d [Fe(OTf)2(S,S-BPBP)] was used as the catalyst. e 1 equiv. of AcOH was used. f 5 equiv. of AcOH were used. g 18 equiv. of AcOH were used, 1.25 h total reaction time. | ||||
| 1 | — | 2 (at once) | 0 | 0 |
| 2c | — | — | >99 | 99 |
| 3 | Mn(OTf)2 (0.5) | 2 (at once) | 0 | 0 |
| 4 | 1 (0.5) | 2 (at once) | 40 | 32 |
| 5 | 2 (0.5) | 2 (at once) | 48 | 32 |
| 6 | S ,S-3 (0.5) | 2 (at once) | 48 | 46 |
| 7 | 4 (0.5) | 2 (at once) | 28 | 28 |
| 8 | S ,S-3 (0.5) | 1.5 (30 min) | >99 | 99 |
| 9 | S ,S-3 (0.1) | 1.5 (30 min) | >99 | 99 |
| 10d | S,S-3-Fe (0.1) | 1.5 (30 min) | 70 | 70 |
| 11 | R ,S-3 (0.1) | 1.5 (30 min) | 10 | 10 |
| 12 | mix -3 (0.1) | 1.5 (30 min) | >99 | 99 |
| 13 | rac-3 (0.1) | 1.5 (30 min) | >99 | 99 |
| 14 | rac-3 (0.05) | 1.5 (30 min) | >99 | 99 |
| 15e | rac-3 (0.05) | 1.5 (30 min) | 10 | 7 |
| 16f | rac-3 (0.05) | 1.5 (30 min) | 52 | 50 |
| 17 | rac-3 (0.01) | 1.5 (30 min) | 80 | 80 |
| 18g | rac-3 (0.02) | 1.5 (45 min) | >99 | 99 |

Since S,S-3 outperforms the other Mn-catalysts in terms of epoxide yield, reaction condition optimization was carried out using BPBP-based Mn-complexes as catalysts. As reported previously,44,52 adding the oxidant slowly improves the substrate conversion in aliphatic C–H oxidation reactions. A slow addition protocol, i.e., dropwise addition of H2O2 over 30 min using a syringe pump, was therefore tested in oleic acid epoxidation. Considering that S,S-3 shows very high epoxide selectivities and that N2Py2-based iron complexes were shown to decompose under the oxidizing conditions,47,52 the oxidant loading was lowered to 1.5 equiv. On using these conditions, substrate conversion and product yield both significantly increased to 99% (entry 8). Further lowering of the catalyst loading to 0.1 mol% still showed quantitative conversion of oleic acid into its epoxide product (entry 9). In comparison, using the same amount of S,S-3-Fe ([Fe(OTf)2(S,S-BPBP)]), only 70% of conversion and yield were obtained (entry 10). This observation is consistent with previously reported results by Bryliakov and Talsi, which show that the Mn(N2Py2) complexes exhibit higher reactivities than the corresponding Fe(N2Py2) complexes in epoxidation reactions.41,53Meso-complex R,S-3 turned out to be almost inactive in the epoxidation reaction, with only 10% substrate being converted and product being formed (entry 11), which is in line with the catalytic behavior of the corresponding iron complex [Fe(OTf)2(R,S-BPBP)] in both alkene epoxidation and aliphatic C–H oxidation.47 Furthermore, using 0.1 mol% of mix-3 provided an identical reaction outcome to that of the reaction with S,S-3 (quantitative yield, entry 12). This observation corroborates the notion that S,S-3 and R,R-3 are the catalytically active components in mix-3 and that R,S-3 does not contribute to the activity of mix-3. In accordance with this notion, the use of 0.1 mol% of rac-3 in the reaction provided a quantitative yield of the epoxide as well (entry 13). Since the rac-BPBP ligand mixture can be readily isolated from the mix-BPBP mixture via flash column chromatography and no ligand resolution is needed, rac-3 represents a much cheaper catalyst than an enantiopure Mn(BPBP) complex. In addition, the use of rac-3 as opposed to mix-3 could be advantageous for practical applications, since rac-3 is devoid of inactive metal-containing components which could facilitate regulatory registration and since a lower amount of the catalyst could be used because rac-3 contains more active catalyst per gram of the catalyst material.
No drop in catalytic conversion and yield was found when further reducing the amount of the catalyst by 50% (0.05 mol% rac-3, entry 14). Decreasing the loading of AcOH to either 1 or 5 equiv. led to incomplete substrate conversions (10% or 52%, respectively, entries 15 and 16). Costas et al. have earlier shown the beneficial effect of the addition of larger amounts of acetic acid in epoxidation reactions catalyzed by S,S-3.54 The use of increased amounts of AcOH does furthermore improve the solubility of fatty acid substrates in the AcOH/CH3CN medium, as was noted earlier in Fe-catalyzed oxidative cleavage reactions.36 Further lowering of the catalyst loading to 0.01 mol% rac-3 using 9 equiv. of AcOH resulted in 80% of substrate conversion and product yield within the standard reaction time of 1 h, providing a boundary of catalyst activity per time unit (entry 17). Variation of the reaction parameters based on these combined observations finally led to a protocol that uses 0.02 mol% of rac-3 catalyst loading, 45 min of H2O2 (1.5 equiv.) addition time, and an AcOH loading of 18 equiv., respectively; these reaction settings lead to full oleic acid conversion and quantitative formation of its epoxide (entry 18).
Using the catalytic protocol optimized for the epoxidation of oleic acid, the protocol was tested for the catalytic epoxidation of a series of other UFAs and FAMEs (Table 3). Elaidic acid, the trans-isomer of oleic acid, undergoes epoxidation quantitatively under the protocol conditions (entry 2). The same catalytic outcome was obtained when changing the carboxyl group in oleic acid to a carbomethoxy group as in methyl oleate (entry 3). Notably, a very high TON of 4950 was obtained in this case, which largely outperforms a previously reported benchmark in the epoxidation of methyl oleate (TON = 1868).29 Erucic acid (C22:1), on the other hand, seems more difficult to epoxidize using the current reaction conditions: only 36% of the substrate was converted in this case, yielding 36% yield of the epoxide (entry 4). Erucic acid is a solid at room temperature (melting point = 34 °C) and not well soluble in MeCN. The resulting biphasic solid–liquid reaction medium is likely to limit the catalytic activity, leading to poor catalytic results. Upon increase of the reaction temperature to 36 °C, the resulting biphasic liquid–liquid reaction medium allowed for more effective catalysis to occur and, accordingly, erucic acid was fully converted to give 90% of the epoxide product under these conditions (entry 5). In turn, methyl erucate undergoes the epoxidation process smoothly using the standard protocol, forming the epoxide in quantitative yield (entry 6). This catalytic system can also be perfectly applied to UFAs with more double bonds, as shown in the cases of linoleic acid with two double bonds and linolenic acid with three double bonds (both with quantitative yields, entries 7 and 8). For the small set of UFAs and FAMEs tested, excellent epoxide yields and high turnover numbers (TON >4500) were obtained, meaning that the rac-3/H2O2/AcOH catalytic system is more efficient than previous examples28,29 and is promising to be widely applied in the epoxidation of a wide range of UFAs and FAMEs.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | x, y, z | Lipid number | R | Conv.b (%) | Yieldb (%) | TONc |
|
a Unless stated otherwise, reaction conditions are substrate (0.5 mmol CC, 1 equiv.), rac-3 (0.02 mol%, added as a 10 mM solution in MeCN), H2O2 (1.5 equiv., 1 M solution in MeCN) and AcOH (18 equiv.) in CH3CN at room temperature; the oxidant was added over 45 min, and the reaction mixture was stirred for an additional 30 min. All reagent loadings are with respect to (w.r.t.) the amount of CC bonds.
b Determined by NMR analysis.
c TON = [mol epoxide]/[mol rac-3].
d Reaction temperature was 36 °C.
|
|||||||
| 1 | Oleic acid | 6, 1, 5 | C18:1 cis-9 | H | >99 | 99 | 4950 |
| 2 | Elaidic acid | 6, 1, 5 | C18:1 trans-9 | H | >99 | 99 | 4950 |
| 3 | Methyl oleate | 6, 1, 5 | C18:1 cis-9 | Me | >99 | 99 | 4950 |
| 4 | Erucic acid | 6, 1, 9 | C22:1 cis-13 | H | 36 | 36 | 1800 |
| 5d | Erucic acid | 6, 1, 9 | C22:1 cis-13 | H | >99 | 90 | 4500 |
| 6 | Methyl erucate | 6, 1, 9 | C22:1 cis-13 | Me | >99 | 99 | 4950 |
| 7 | Linoleic acid | 3, 2, 5 | C18:2 cis-9,12 | H | >99 | 99 | 4950 |
| 8 | Linolenic acid | 0, 3, 5 | C18:3 cis-9,12,15 | H | >99 | 99 | 4950 |

Epoxidation of VOs
Next, the Mn-catalyzed epoxidation of VOs was explored using the rac-3/H2O2/AcOH catalytic protocol (Table 4). In order to obtain the optimal reaction conditions, sunflower oil was chosen as the benchmark oil. The Wijs method55 was used to determine the iodine value (I.V.) of sunflower oil, providing information on its CC bond content (I.V. = 130). All reagent loadings listed in Table 4 are w.r.t. the amount of CC bonds in the oils. Iodine values were determined again after the epoxidation reaction in order to calculate the conversion of CC bonds (see the Experimental section). The amount of epoxy-groups formed in the reaction is described as the percentage of oxirane oxygen in the resulting oils, which was determined according to AOCS Official Method Cd 9–57.56 The formal yields were calculated from the measured oxirane oxygen content and the theoretical maximum oxirane oxygen content (see the Experimental section).
| Entry | MeCN | Catalyst loading (mol%) | H2O2 addition time (min) | AcOH (eq.) | Overall reaction time (h) | Conv. (%) | Epoxide yield (%) | Epoxide selectivity (%) |
|---|---|---|---|---|---|---|---|---|
| a General reaction conditions: Sunflower oil (1 g), rac-3 (0.02–0.04 mol%, w.r.t. double bonds, added as a 10 mM solution in MeCN, ca. 100–200 μL) and AcOH (2.6 mL for 9 equiv., 5.3 mL for 18 equiv.) were mixed in the absence or presence of MeCN (2 mL), and stirred vigorously (1000 rpm) at room temperature; H2O2 (1.5 equiv., w.r.t. double bonds, 35% aqueous solution) was added over 45–120 min, and the reaction mixture was stirred for an additional 30–60 min. b 100 mg of sunflower oil was used, H2O2 was added as a 1 M solution in MeCN (0.8 mL). c Stirring rate = 500 rpm. | ||||||||
| 1b | 2 mL | 0.02 | 45 | 9 | 1.75 | >99 | 95 | 95 |
| 2 | 2 mL | 0.02 | 45 | 9 | 1.75 | 83 | 75 | 90 |
| 3 | — | 0.02 | 45 | 9 | 1.75 | 45 | 25 | 56 |
| 4 | — | 0.04 | 45 | 9 | 1.75 | 80 | 45 | 56 |
| 5 | — | 0.02 | 45 | 18 | 1.75 | 82 | 53 | 65 |
| 6 | — | 0.02 | 90 | 18 | 2 | 84 | 73 | 87 |
| 7 | — | 0.02 | 120 | 18 | 3 | 89 | 65 | 73 |
| 8 | — | 0.03 | 90 | 18 | 2 | >99 | 90 | 90 |
| 9 | — | 0.03 | 90 | 9 | 2 | 80 | 70 | 88 |
| 10c | — | 0.03 | 90 | 18 | 2 | 83 | 75 | 90 |
Using similar reaction conditions to the optimized ones for epoxidation of UFAs and FAMEs, except for half the amount of AcOH used, the epoxidation of sunflower oil (100 mg) was performed with 0.02 mol% of rac-3 (added as a 10 mM solution in MeCN), 1.5 equiv. of H2O2 (added as a 1 M solution in MeCN over 45 min), and 9 equiv. of AcOH in MeCN (2 mL) under vigorous stirring (1000 rpm) at ambient temperature. Full conversion of double bonds in the oil was achieved, with 95% of epoxy-group yield (Table 4, entry 1). To make this epoxidation more practical, the reaction scale was increased to 1 g of sunflower oil in the same amount of MeCN (i.e., 2 mL, entry 2). Both the conversion and yield dropped to 83% and 75%, respectively, albeit with a similar epoxide selectivity (95% for entry 1 and 90% for entry 2). For 1 g scale reactions like this, H2O2 was added as a commercial 35% aqueous solution. With the aim of making the epoxidation protocol more environmentally friendly, the reaction conditions were further optimized not to use MeCN as the organic solvent. It is noteworthy that in all the reactions in Table 4, the catalyst was added as a 10 mM stock solution in MeCN (ca. 100–200 μL), for the purpose of easy operation. As shown in entry 3, the epoxidation process proceeds very poorly in the absence of the MeCN solvent: only 45% of the double bonds were converted, forming 25% of the epoxide with an epoxide selectivity of 56%. Doubling the loading of the Mn-catalyst to 0.04 mol% under these conditions increases both conversion (80%) and yield (45%) at the same epoxidation selectivity of 56% (entry 4).
In separate catalyst tests the Mn-catalyst was found to be insoluble in sunflower oil but slightly soluble in AcOH, whereas the oil is insoluble in AcOH. This results in a biphasic reaction medium (oil![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH = 1:2.4, v/v; 9 equiv. AcOH), with the Mn-catalyst residing in the AcOH layer. Addition of more AcOH (18 equiv., oil:AcOH = 1:4.8, v/v) was considered to increase the total liquid–liquid interface under vigorous stirring. Entry 5 clearly shows that the reaction benefited significantly from the additional amount of AcOH. Compared to entry 3, considerably more double bonds were converted (82% over 45%) and a moderate yield of the epoxide was obtained (53%). Yet, the epoxide selectivity under these conditions is still relatively low (65%). In order to suppress the formation of side-products, H2O2 was delivered to the reaction mixture more slowly over a period of 90 min. This resulted in a significant improvement in epoxide selectivity to 87%, at almost the same conversion (84%, entry 6). A further increase in the H2O2 delivery time (120 min) and an extension of the overall reaction time to 3 h led to a drop in epoxide selectivity (to 73%), even though a slightly increased conversion was observed (89%, entry 7). 1H NMR analysis of the resulting mixture obtained from extraction with diethyl ether and subsequent condensation showed the presence of remarkable amounts of diol compounds (∼20%). Apparently, the longer overall reaction time (3 h) allows for a more pronounced ring-opening of the initially formed epoxides under the acidic conditions.
:AcOH = 1:2.4, v/v; 9 equiv. AcOH), with the Mn-catalyst residing in the AcOH layer. Addition of more AcOH (18 equiv., oil:AcOH = 1:4.8, v/v) was considered to increase the total liquid–liquid interface under vigorous stirring. Entry 5 clearly shows that the reaction benefited significantly from the additional amount of AcOH. Compared to entry 3, considerably more double bonds were converted (82% over 45%) and a moderate yield of the epoxide was obtained (53%). Yet, the epoxide selectivity under these conditions is still relatively low (65%). In order to suppress the formation of side-products, H2O2 was delivered to the reaction mixture more slowly over a period of 90 min. This resulted in a significant improvement in epoxide selectivity to 87%, at almost the same conversion (84%, entry 6). A further increase in the H2O2 delivery time (120 min) and an extension of the overall reaction time to 3 h led to a drop in epoxide selectivity (to 73%), even though a slightly increased conversion was observed (89%, entry 7). 1H NMR analysis of the resulting mixture obtained from extraction with diethyl ether and subsequent condensation showed the presence of remarkable amounts of diol compounds (∼20%). Apparently, the longer overall reaction time (3 h) allows for a more pronounced ring-opening of the initially formed epoxides under the acidic conditions.
To maximize the epoxide yield, the catalyst loading was slightly increased to 0.03 mol% and the delivery time of H2O2 was brought back to 90 min. In this case, the double bonds were fully converted and an excellent epoxide yield (90%) was found (entry 8). Considering the potential epoxide ring-opening side reaction under acidic conditions, using these altered conditions but at a lower AcOH loading (9 equiv.) led to a decrease of both the conversion and yield (80% and 70%, respectively, entry 9). Finally, the impact of vigorous stirring was examined by bringing down the stirring rate to 500 rpm, which resulted in a decrease of the conversion from 99% to 83% (entry 10). The necessity of using a larger amount of AcOH and a high stirring speed indicates that the epoxidation reaction is enhanced with more pronounced mixing, which suggests that the catalytic reaction takes place at the biphasic liquid–liquid interface.
Overall, the reaction conditions described in entry 8 provide an optimized reaction parameter set for the epoxidation of sunflower oil in the absence of MeCN and lead to full conversion of all double bonds in the oil and 90% of epoxide formation with the corresponding diols as likely byproducts. These conditions even provide a better reaction outcome than the original conditions using acetonitrile as a solvent (compare entries 2 and 8 for 1 g scale reactions).
To further investigate the substrate tolerance and validation of this epoxidation strategy, the epoxidation of various VOs was examined using this optimized rac-3/H2O2/AcOH catalytic system (Table 4, entry 8). All reactions were performed without using MeCN as the solvent. It is noteworthy that in the cases of 1-gram scale reactions, the catalyst was added as a 10 mM stock solution in MeCN for the purpose of easy operation. In addition, a fixed amount of AcOH (5.3 mL) was used for all 1-gram scale reactions, regardless of the different iodine values of the oils. As shown in entries 1–7 in Table 5, most of the VOs can be epoxidized using these standard reaction conditions with full conversions and excellent yields (up to 99%). For olive oil a somewhat lower reactivity was observed (90% conversion and 85% yield, entry 6). Notably, among these VOs rapeseed oil, linseed oil, and soybean oil are promising feedstocks to produce epoxidized VOs in industry, because of their wide availability and low prices.4 The present epoxidation protocol provides an excellent tool to produce EVOs from these three VOs, with nearly quantitative epoxide yields in all cases (98%, 99% and 99%, respectively; entries 4, 5 and 7).
| Entry | Oil | Iodine value | Catalyst loading (mol%) | Conversion (%) | Epoxide yield (%) | Epoxide selectivity (%) | TONb |
|---|---|---|---|---|---|---|---|
| a Unless stated otherwise, reaction conditions are: Oil (1 g), rac-3 (0.03 mol% w.r.t. double bonds, added as a 10 mM solution in MeCN, ca. 100–180 μL), and AcOH (5.3 mL) at room temperature; H2O2 (1.5 equiv. w.r.t. double bonds) was added over 90 min, and the reaction mixture was stirred for an additional 30 min, stirring rate = 1000 rpm. b TON = [mol epoxide]/[mol rac-3]. c 5 g of oil was used, and 26.5 mL of AcOH was added. d Solid catalyst was added. | |||||||
| 1 | Sunflower oil | 130 | 0.03 | >99 | 90 | 90 | 3000 |
| 2 | Walnut oil | 144 | 0.03 | >99 | 96 | 96 | 3200 |
| 3 | Peanut oil | 91 | 0.03 | >99 | 90 | 90 | 3000 |
| 4 | Rapeseed oil | 121 | 0.03 | >99 | 98 | 98 | 3267 |
| 5 | Linseed oil | 165 | 0.03 | >99 | 99 | 99 | 3300 |
| 6 | Olive oil | 88 | 0.03 | 90 | 85 | 94 | 2833 |
| 7 | Soybean oil | 128 | 0.03 | >99 | 99 | 99 | 3300 |
| 8 | Rice oil | 105 | 0.04 | >99 | 92 | 92 | 2300 |
| 9 | Sesame oil | 115 | 0.04 | 80 | 75 | 94 | 1875 |
| 10 | Cooked sunflower oil | 118 | 0.03 | >99 | 98 | 98 | 3267 |
| 11c,d | Sunflower oil | 130 | 0.03 | >99 | 90 | 90 | 3000 |
Full epoxidation of the double bonds in rice oil could also be achieved using a small increase in catalyst loading to 0.04 mol% (entry 8). With 0.03 mol% of the catalyst, only 81% conversion was found for rice oil. However, under these conditions some 20% of double bonds remain in sesame oil, with 75% epoxides being formed (entry 9). Overall, entries 1–9 show that the performance of this catalytic system is independent of the iodine values of the starting VOs. Not only vegetable oils with a low iodine number (such as peanut oil, I.V. = 91), but also the ones with a high iodine number (such as linseed oil, I.V. = 165) show full conversion and very high epoxide yields (90% for peanut oil and 99% for linseed oil).
From a cost point of view, waste cooking oils are potentially promising feedstocks for the production of EVOs since they are generally 30–60% cheaper than regular vegetable oils.6 Applying the current epoxidation protocol to cooked sunflower oil led to full conversion of double bonds and 98% epoxide yield (entry 10). Next, a scale-up experiment with 5 grams of sunflower oil was carried out (entry 11). No drop in conversion and yield was observed in comparison with the 1-gram scale experiment (entry 1 vs. entry 11). Notably, the Mn-catalyst was added as a solid in this case, meaning that the reaction can be performed with the same efficiency in the complete absence of MeCN. Furthermore, separation of the resulting epoxidized sunflower oil was conducted straightforwardly by simple extraction of the reaction mixture with Et2O and follow-up removal of the organic solvents. The obtained epoxidized sunflower oil showed high purity according to 1H NMR analysis.
It can be concluded from Table 5 that the Mn-based catalytic protocol is capable of epoxidizing a variety of VOs, mostly leading to epoxide yields of 90% or higher (only sesame oils showed a 75% yield) with low catalyst loadings (0.03–0.04 mol%). As a result, high TON values were obtained (1875–3300), which are much higher than those of the previously reported homogeneous systems21,22 summarized in Table 1 (54–95, entries 1 and 2). In addition, the system provides very high selectivities for the epoxide products in the range of 90–99% for all oils tested in this study.
One-pot oxidative cleavage of UFAs and FAMEs
As mentioned earlier, epoxidized UFAs, their ester derivatives, and EVOs can be further transformed into other industrially valuable compounds.4 One example is through the oxidative cleavage of the C–C bond in the oxirane ring to yield a monofunctional aldehyde and an α,ω-aldehyde fatty acid (or methyl ester) as the primary products (Scheme 2).5 The primary aldehyde product, e.g. nonanal in the case of oleic acid, can be used as a direct, yet renewable replacement for petrochemically derived nonanal in plasticizer production.5 On the other hand, the aldehyde moiety in an α,ω-aldehyde fatty acid (ester) can readily be converted to an acid, hydroxy, or amino group. These bifunctional fatty acids (or esters) can be used for the production of nylons and polyesters, both large volume and high value products.5 Spannring et al. have previously developed the Fe-catalyzed one-pot oxidative cleavage of UFAs and FAMEs into aldehydes with H2O2 and sodium periodate.36 This protocol comprises consecutive reaction steps involving double bond epoxidation catalyzed by the [Fe(OTf)2(mix-BPBP)]/H2O2/AcOH catalytic system (in which 0.5 mol% of the Fe-catalyst was employed), hydrolysis of the epoxide in the presence of H2SO4 to form a vicinal diol, and cleavage of the diol to give the two aldehydes with NaIO4 as the oxidant (Scheme 2).36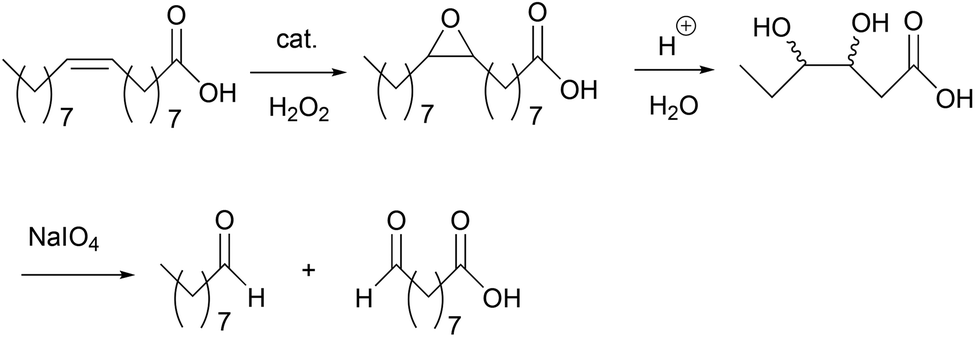 | ||
| Scheme 2 Oxidative cleavage of oleic acid into aldehyde products.36 | ||
Similarly, the Mn-based epoxidation protocol developed in this paper was used in the one-pot cleavage of a number of UFAs and FAMEs. The cleavage reaction followed the same procedure as before: (1) epoxidation of the double bond using the optimized reaction conditions with the rac-3/H2O2/AcOH (0.02 mol%, 1.5 equiv. and 18 equiv., respectively) catalytic system as shown in Table 3; (2) hydrolysis of the oxirane ring by addition of 0.5 equiv. of H2SO4 (1 equiv. of H+); and (3) cleavage of the diol with 1 equiv. of NaHCO3 and 1 equiv. of NaIO4. GC analyses were conducted after the one-pot procedure to determine the amount of nonanal and detect the formation of nonanoic acid (over-oxidized product from nonanal). It is noteworthy that only the formation of nonanal was quantified by GC, and that the formation of the α,ω-aldehyde fatty acid (ester) was considered to have taken place in equal amounts.
Table 6 shows that all reactions resulted in full substrate conversion with 0.02 mol% of rac-3 in a total reaction time of 5.75–7.25 h. In the cases of oleic acid, methyl oleate, and methyl erucate, very high nonanal yields were achieved (98%, 94% and 91%, respectively, entries 1, 3, and 5). A moderate nonanal yield of 70% was observed in the oxidative cleavage of elaidic acid (entry 2). 1H-NMR analysis of the resulting reaction mixture showed that in this case there was still ca. 20% of the intermediate epoxide remaining, which is likely due to the fact that a trans-epoxide, formed from epoxidation of the trans double bond in elaidic acid, is harder to hydrolyze than a cis-epoxide.57 Oxidative cleavage of erucic acid yielded only 55% of nonanal (entry 4), whereas neither over-oxidized nonanoic acid nor unreacted intermediates (epoxide and diol) were detected by GC or NMR analyses. In all these reactions, no formation of nonanoic acid was observed, indicating that this one-pot cleavage methodology is highly selective for aldehyde products. In general, this Mn-initiated protocol for one-pot oxidative cleavage of UFAs and FAMEs outperforms the reported Fe-based catalytic system in terms of catalytic efficiency. In the later protocol, 0.5 mol% of [Fe(OTf)2(mix-BPBP)] was used to yield 69–96% of nonanal in a general reaction time of 20–72 h.36
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | n | R | Conv.b (%) | Nonanal yieldc (%) |
| a General reaction conditions at ambient temperature: step 1: substrate (0.5 mmol), rac-3 (0.02 mol%), AcOH (9 mmol), and H2O2 (0.75 mmol, added in 45 min) in CH3CN (2 mL), 1.25 h reaction time; step 2: 1 mL of CH3CN and 1 mL of H2SO4 (0.25 M) in water were added, 3 h reaction time; and step 3: 0.5 mmol of NaHCO3 and 0.5 mmol of NaIO4 were added, 1.5 h reaction time. b Determined by NMR analysis. c Determined by GC analysis. d The reaction time for step 3 was 3 h. e Same as (d), and in addition the reaction in step 1 was heated to 36 °C. | |||||
| 1 | Oleic acid | 7 | H | >99 | 98 |
| 2d | Elaidic acid | 7 | H | >99 | 70 |
| 3d | Methyl oleate | 7 | Me | >99 | 94 |
| 4e | Erucic acid | 11 | H | >99 | 55 |
| 5d | Methyl erucate | 11 | Me | >99 | 91 |
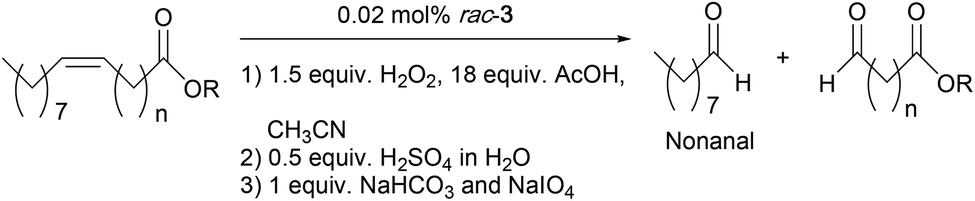
Conclusions
A highly efficient catalytic protocol has been developed for the epoxidation of UFAs, FAMEs, and VOs based on the use of the abundant first-row transition metal manganese in combination with aqueous H2O2 as the oxidant. Using [Mn(OTf)2(rac-BPBP)] (rac-3) as the catalyst, the double bonds in most of the vegetable oils tested can be fully converted with a very low catalyst loading: 0.03 mol% w.r.t. double bonds or ca. 0.1 wt% w.r.t. oil. In all the VOs tested, this catalytic system is highly selective for epoxides (>90%), and most of the EVOs were obtained in more than 90% yield within 2 h of reaction time at ambient temperature. Therefore, this protocol has a wide applicability for a variety of VOs, with a range of iodine values from 88 to 165. In the cases of walnut oil, linseed oil, rapeseed oil, cooked sunflower oil, and soybean oil, (nearly) quantitative epoxide yields can be achieved under standard reaction conditions. By increasing the addition time of the H2O2 oxidant to 90 min and the volume ratio of oil:AcOH to ca. 1:4.8, it is possible to perform the reaction with the use of MeCN only as a delivery medium of the catalyst solution rather than a solvent in 1-gram scale reactions. Notably, the high catalytic efficiency was retained in a scale-up experiment with 5 g of sunflower oil in which the use of MeCN could be completely omitted. The volume of AcOH and the stirring rate are essential as the reaction medium is biphasic due to the absence of MeCN. In addition, the resulting EVOs can be obtained straightforwardly from the reaction crude via simple extraction with Et2O. Further transformation of epoxidized UFAs and FAMEs has also been carried out via an oxidative cleavage procedure, providing nonanal and an α,ω-aldehyde fatty acid (ester) with moderate to excellent yields (55–98%). This process is performed in a one-pot manner, initiated by the rac-3/H2O2/AcOH catalytic system in the first epoxidation step.
Overall, the present homogeneous Mn-catalyzed epoxidation protocol provides a highly efficient and practical tool for the production of EVOs under very mild reaction conditions in short reaction times, and can be carried out using only minute amounts of MeCN as a solubilizing agent or even in the complete absence of MeCN, allowing for facile product isolation. This protocol is expected to represent a promising alternative to conventional epoxidation methods and outperforms previously reported catalytic protocols for EVO production in terms of efficiency and reaction conditions.
Experimental section
General
All the complexation reactions were performed under a nitrogen atmosphere using standard Schlenk techniques and all other reactions including catalytic reactions were conducted under ambient conditions. The solvents diethyl ether and MeCN were purified with an MBraun MB SPS-800 solvent purification system. Demineralized water and technical grade CH2Cl2 were used for reactions. Tetrahydrofuran for complexation reactions was dried with sodium, and was distilled under nitrogen prior to use. Vegetable oils were obtained from local supermarkets. All other reagents, substrates, and reaction products were obtained commercially and used without further purification. Column chromatography was performed using neutral alumina. 1H and 13C NMR spectra were recorded with a 400 MHz Varian spectrometer at 25 °C; chemical shifts (δ) are given in ppm referenced to the residual solvent peak. Gas chromatography (GC) was performed on a Perkin–Elmer Clarus 500 Gas Chromatograph equipped with an Agilent HP 5 column with a 5% phenyl–95% methylpolysiloxane ratio and a flame-ionization detector. ESI-MS measurements were performed with a Waters LCT Premier XE KE317. The ligands S,S-BPBPM,44R,S-BPBP,47mix-BPBP,47 and S,S-BPBI45 and Mn complexes 1,452,45 and S,S-346 were synthesized following reported procedures. Elemental microanalyses were carried out by the Mikroanalytisches Laboratorium Kolbe, Germany.Synthesis of ligands and Mn complexes
1058 found: 6221052. Elemental analysis calcd (%) for C30H26F6MnN4O6S2·2H2O: C 44.62, H 3.74, N 6.94, found C 46.12, H 3.91, N 6.87.
Catalysis
The conversion of the substrate was determined as follows:

The yield of epoxidized products was determined as follows:


Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. C. acknowledges the China Scholarship Council (CSC) for a doctoral scholarship.Notes and references
- T. E. Bull, Science, 1999, 285, 1209 CrossRef CAS.
- S. G. Tan and W. S. Chow, Polym.-Plast. Technol. Eng., 2010, 49, 1581–1590 CrossRef CAS.
- A. K. R. Somidi, R. V. Sharma and A. K. Dalai, Ind. Eng. Chem. Res., 2014, 53, 18668–18677 CrossRef CAS.
- S. M. Danov, O. A. Kazantsev, A. L. Esipovich, A. S. Belousov, A. E. Rogozhin and E. A. Kanakov, Catal. Sci. Technol., 2017, 7, 3659–3675 RSC.
- P. Spannring, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. Klein Gebbink, Catal. Sci. Technol., 2014, 4, 2182–2209 RSC.
- J. McNutt and Q. S. He, J. Ind. Eng. Chem., 2016, 36, 1–12 CrossRef CAS.
- Y. B. Huang, M. Y. Yao, P. P. Xin, M. C. Zhou, T. Yang and H. Pan, RSC Adv., 2015, 5, 74783–74789 RSC.
- A. Adhvaryu and S. Z. Erhan, Ind. Crops Prod., 2002, 15, 247–254 CrossRef CAS.
- A. Campanella, E. Rustoy, A. Baldessari and M. A. Baltanás, Bioresour. Technol., 2010, 101, 245–254 CrossRef CAS PubMed.
- L. Fantoni and C. Simoneau, Food Addit. Contam., 2003, 20, 1087–1096 CrossRef CAS PubMed.
- H. Schuster, L. A. Rios, P. P. Weckes and W. F. Hoelderich, Appl. Catal., A, 2008, 348, 266–270 CrossRef CAS.
- J. Salimon, N. Salih and E. Yousif, Eur. J. Lipid Sci. Technol., 2010, 112, 519–530 CAS.
- J. O. Metzger, Eur. J. Lipid Sci. Technol., 2009, 111, 865–876 CrossRef CAS.
- H.-S. Hwang, A. Adhvaryu and S. Z. Erhan, J. Am. Oil Chem. Soc., 2003, 80, 811–815 CrossRef CAS.
- H.-S. Hwang and S. Z. Erhan, J. Am. Oil Chem. Soc., 2001, 78, 1179–1184 CrossRef CAS.
- X. Wu, X. Zhang, S. Yang, H. Chen and D. Wang, J. Am. Oil Chem. Soc., 2000, 77, 561–563 CrossRef CAS.
- M. D. Soucek, A. H. Johnson and J. M. Wegner, Prog. Org. Coat., 2004, 51, 300–311 CrossRef CAS.
- W. D. Wan Rosli, R. N. Kumar, S. Mek Zah and M. M. Hilmi, Eur. Polym. J., 2003, 39, 593–600 CrossRef CAS.
- N. O. Shaker, E. M. Kandeel, E. E. Badr and M. M. El-Sawy, J. Dispersion Sci. Technol., 2008, 29, 421–425 CrossRef CAS.
- S. F. Thames and H. Yu, Surf. Coat. Technol., 1999, 115, 208–214 CrossRef CAS.
- A. E. Gerbase, J. R. Gregório, M. Martinelli, M. C. Brasil and A. N. F. Mendes, J. Am. Oil Chem. Soc., 2002, 79, 179–181 CrossRef CAS.
- M. Farias, M. Martinelli and D. P. Bottega, Appl. Catal., A, 2010, 384, 213–219 CrossRef CAS.
- J. O. Metzger and U. Bornscheuer, Appl. Microbiol. Biotechnol., 2006, 71, 13–22 CrossRef CAS PubMed.
- T. Vlček and Z. S. Petrović, J. Am. Oil Chem. Soc., 2014, 83, 247–252 CrossRef.
- W. Cheng, G. Liu, X. Wang, X. Liu and L. Jing, Eur. J. Lipid Sci. Technol., 2015, 117, 1185–1191 CrossRef CAS.
- J. Jiang, Y. Zhang, L. Yan and P. Jiang, Appl. Surf. Sci., 2012, 258, 6637–6642 CrossRef CAS.
- H. Zhang, H. Yang, H. Guo, J. Yang, L. Xiong, C. Huang, X. Chen, L. Ma and Y. Chen, Appl. Clay Sci., 2014, 90, 175–180 CrossRef CAS.
- T. B. Khlebnikova, Z. P. Pai, L. A. Fedoseeva and Y. V. Mattsat, React. Kinet. Catal. Lett., 2009, 98, 9–17 CrossRef CAS.
- L. C. de la Garza, K. D. O. Vigier, G. Chatel and A. Moores, Green Chem., 2017, 19, 2855–2862 RSC.
- A. Campanella, M. A. Baltanás, M. C. Capel-Sánchez, J. M. Campos-Martín and J. L. G. Fierro, Green Chem., 2004, 6, 330–334 RSC.
- X. Ye, P. Jiang, P. Zhang, Y. Dong, C. Jia, X. Zhang and H. Xu, Catal. Lett., 2010, 137, 88–93 CrossRef CAS.
- M. Di Serio, R. Turco, P. Pernice, A. Aronne, F. Sannino and E. Santacesaria, Catal. Today, 2012, 192, 112–116 CrossRef CAS.
- M. R. Janković, S. V. Sinadinović-Fišer and O. M. Govedarica, Ind. Eng. Chem. Res., 2014, 53, 9357–9364 CrossRef.
- Y. X. Miao and J. P. Liu, Adv. Mater. Res., 2014, 881–883, 140–143 Search PubMed.
- R. Turco, C. Pischetola, R. Tesser, S. Andini and M. Di Serio, RSC Adv., 2016, 6, 31647–31652 RSC.
- P. Spannring, V. Yazerski, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. Klein Gebbink, Chem. – Eur. J., 2013, 19, 15012–15018 CrossRef CAS PubMed.
- G. Olivo, O. Cussó and M. Costas, Chem. – Asian J., 2016, 11, 3148–3158 CrossRef CAS PubMed.
- R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Molecules, 2016, 21, 1454 CrossRef PubMed.
- D. Shen, C. Miao, D. Xu, C. Xia and W. Sun, Org. Lett., 2015, 17, 54–57 CrossRef CAS PubMed.
- R. V. Ottenbacher, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, Org. Lett., 2012, 14, 4310–4313 CrossRef CAS PubMed.
- O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196–1202 CrossRef CAS.
- K. Chen and L. Que Jr., Chem. Commun., 1999, 1375–1376 RSC.
- M. Costas, A. K. Tipton, K. Chen, D. H. Jo and L. Que Jr., J. Am. Chem. Soc., 2001, 123, 6722–6723 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed.
- A. Murphy, G. Dubois and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 5250–5251 CrossRef CAS PubMed.
- R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, Adv. Synth. Catal., 2011, 353, 885–889 CrossRef CAS.
- V. Yazerski, P. Spannring, D. Gatineau, C. H. M. Woerde, S. M. Wieclawska, M. Lutz, H. Kleijn and R. J. M. Klein Gebbink, Org. Biomol. Chem., 2014, 12, 2062–2070 RSC.
- J. Chen, M. Lutz, M. Milan, M. Costas, M. Otte and R. J. M. Klein Gebbink, Adv. Synth. Catal., 2017, 359, 2590–2595 CrossRef CAS.
- X. Chen, B. Gao, Y. Su and H. Huang, Adv. Synth. Catal., 2017, 359, 2535–2541 CrossRef CAS.
- H. A. J. Aerts and P. A. Jacobs, J. Am. Oil Chem. Soc., 2004, 81, 841–846 CrossRef CAS.
- M. Grau, A. Kyriacou, F. Cabedo Martinez, I. M. de Wispelaere, A. J. P. White and G. J. P. Britovsek, Dalt. Trans., 2014, 43, 17108–17119 RSC.
- N. A. Vermeulen, M. S. Chen and M. C. White, Tetrahedron, 2009, 65, 3078–3084 CrossRef CAS.
- R. V. Ottenbacher, D. G. Samsonenko, E. P. Talsi and K. P. Bryliakov, Org. Lett., 2012, 14, 4310–4313 CrossRef CAS PubMed.
- O. Cussó, I. Garcia-Bosch, D. Font, X. Ribas, J. Lloret-Fillol and M. Costas, Org. Lett., 2013, 15, 6158–6161 CrossRef PubMed.
- C. Paquot, Stand. Methods Anal. Oils, Fats Deriv., 1979, 66–70 Search PubMed.
- C. Paquot, Stand. Methods Anal. Oils, Fats Deriv., 1979, 92–93 Search PubMed.
- R. E. Parker and N. S. Isaacs, Chem. Rev., 1959, 59, 737–799 CrossRef CAS.
- S. E. Denmark, J. Fu and M. J. Lawler, Org. Synth., 2006, 83, 121–130 CrossRef CAS.
Footnote |
| † Current address: Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany. |
| This journal is © The Royal Society of Chemistry 2019 |