
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of pharmaceutical drugs from cardanol derived from cashew nut shell liquid†‡
Yiping
Shi
 ,
Paul C. J.
Kamer§
and
David J.
Cole-Hamilton
*
,
Paul C. J.
Kamer§
and
David J.
Cole-Hamilton
*
EaStChem, School of Chemistry, University of St. Andrews, Fife, KY16 9ST, Scotland, UK. E-mail: djc@st-and.ac.uk
First published on 13th February 2019
Abstract
Cardanol from cashew nut shell liquid extracted from cashew nut shells was successfully converted into various useful pharmaceutical drugs, such as norfenefrine, rac-phenylephrine, etilefrine and fenoprofene. 3-Vinylphenol, the key intermediate for the synthesis of these drugs, was synthesised from cardanol by ethenolysis to 3-non-8-enylphenol followed by isomerising ethenolysis. The metathesis reaction worked very well using DCM, but the greener solvent, 2-methyl tetrahydrofuran, also gave very similar results. Hydroxyamination of 3-vinylphenol with an iron porphyrin catalyst afforded norfenefrine in over 70% yield. Methylation and ethylation of norfenefrine afforded rac-phenylephrine and etilefrine respectively. A sequence of C–O coupling, isomerising metathesis and selective methoxycarbonylation afforded fenoprofene in good yield. A comparison of the routes described in this paper with some standard literature syntheses of 3-vinylphenol and of the drug molecules shows significant environmental advantages in terms of precursors, yields, number of steps, conditions and the use of catalysts. The Atom Economy of our processes is generally similar or significantly superior to those of the literature processes mainly because the side products produced during synthesis of 3-vinylphenol (1-octeme, 1,4-cyclohexadiene and propene) are easily separable and of commercial value, especially as they are bio-derived. The E Factor for the production of 2-vinylphenol by our process is also very low compared with those of previously reported syntheses.
Introduction
Cashew nut shell liquid (CNSL) is a natural oil and a byproduct of cashew nut manufacture. Around 30–35% of the shell weight is CNSL,1 and about ∼1 M tonnes of CNSL is produced annually worldwide.2 Presently, in the cashew nut industry, cashew nut shells are disposed of as furnace materials, and are burnt in a semi-open pit for thermal energy generation for roasting the nuts. They are also burnt for cooking applications, which is a quick and cheap way for disposal. However, these applications have very low combustion efficiency.3CNSL contains a mixture of products including anacardic acid, 1, cardanol, 2, cardol, 3, and 2-methylcardol, 4, which contain different functional groups, the aromatic ring, the hydroxyl group, the carboxylic acid and the double bonds in the alkyl chain (Fig. 1). For this reason, cashew nut shell liquid has versatile applications, and it can be used for a wide range of chemical modifications to produce useful monomer precursors. CNSL derived material can be used as chemical intermediates, additives,4,5 stabilizers,6 lubricants,7 diesel engine fuel alternatives,8 anti-oxidants,9 anticorrosive paints, sodium cardanol sulfate detergent,10 coating, brake linings11 and resins.12 Although there is a large number of applications of cashew nut shell liquid (CNSL) and its components, anacardic acid and cardanol, there are very few examples of the transformation of cashew nut shell liquid and its components into small value-added molecules. The examples predominantly focused on the metathesis of the double bonds in anacardic acid and cardanols using either homogeneous or heterogeneous catalysis (the latter with Ru catalysts supported on mesoporous molecular sieve SBA-15).13
 | ||
| Fig. 1 Components of CNSL. | ||
Metathesis reactions of anacardic acid and cardanol for the synthesis of fine chemicals and new hybrid functional materials have been reviewed.14 Cardanol porphyrins, cardanol phthalocyanines and cardanol fullerenes can all be produced by metathesis of cardanol derivatives.14 These interesting studies enabled the synthesis of challenging molecules. However, it would be more interesting to synthesise products with practical use. It has previously been reported in our group that the tsetse fly attractants, 3-ethyl phenol and 3-propyl phenol can both be produced selectively in good yields (>80%) from cardanol.15
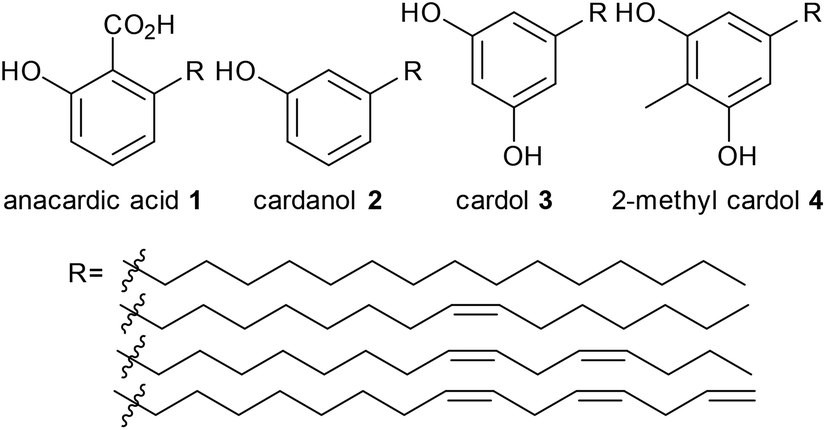
We have also used homogeneous metathesis,16–18 isomerising metathesis,15 methoxycarbonylation18 and isomerisation17,19 to make higher added value chemicals such as 3-nonylphenol,16,17 1-octene,16,17 1,3-cyclohexadiene,16,17 which allows ethenolysis using simple first generation metathesis catalysts,16,20 potential monomers containing hydroxy and carboxyl groups,18 fused bicyclic compounds19 and large ring macrocyles.18 To the best of our knowledge, the only report of converting CNSL, anacardic acid or cardanol into pharmaceutical drugs is the synthesis of ginkgolic acid, a tyrosinase inhibitor, from anacardic acid, which appeared while this manuscript was under review.21 although we have reported the synthesis of a substituted isochromen-1-one which has some structural similarities to massoia lactone from anarcardic acid.19 Pharmaceutical drugs are mainly synthesised from fossil fuels by stoichiometric reactions, and in some cases toxic reagents such as cyanide are involved, which renders the synthesis environmentally unfriendly. Our target was to synthesise pharmaceutical drugs from renewable feedstocks which do not compete with food. An inedible by-product from food processing, in this case cashew nut shell liquid, is therefore a very good starting material. The aromatic and olefin functional groups make cardanol suitable for this purpose. After searching through the drug database, five drugs (fenoprofen, 5, norfenefrine, 6, phenylephrine, 7, etilefrine, 8, and metaraminol, 9) were found to be of potential interest. The importance and the current synthesis of these drugs will be discussed in detail later. Their general structures, containing a meta-hydroxyl group, are similar to the ones in cardanol, 2, or anacardic acid, 1, from CNSL (highlighted in red, Scheme 1). Only a few steps of modification are needed to convert food by-products into high value and important medicines, which will be discussed in detail later.
 | ||
| Scheme 1 Retrosynthesis of target drugs, with the relationship to cardanol, 2. | ||
Results and discussion
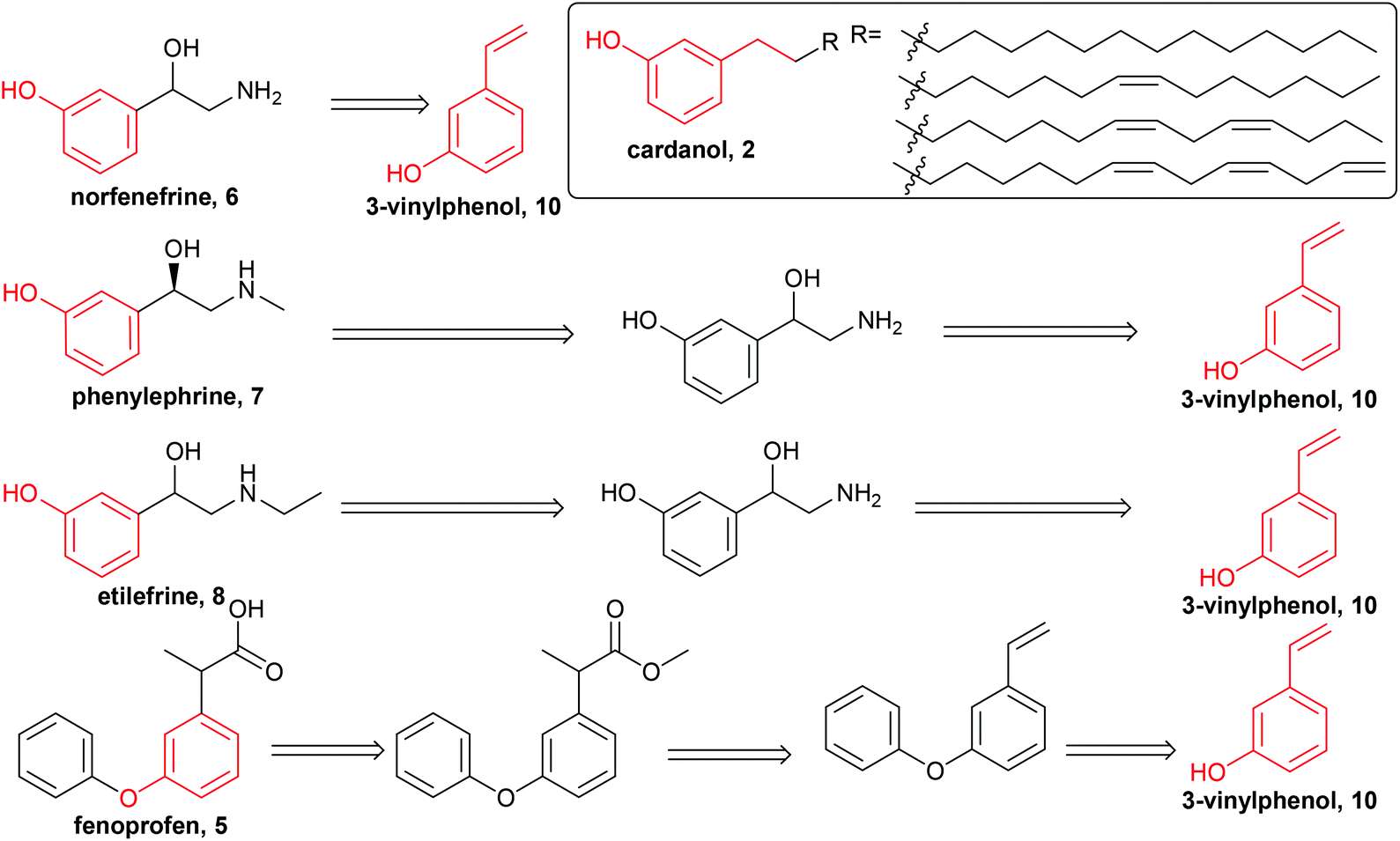
The initial retrosyntheses of the target drugs are summarised in Scheme 1, from which it can be concluded that 3-vinylphenol, 10, is the key intermediate for the synthesis of the relevant drugs. Therefore, the initial study was focused on converting CNSL and derivates (e.g. anacardic acid and cardanol) into 3-vinylphenol, 10.3-vinylphenol
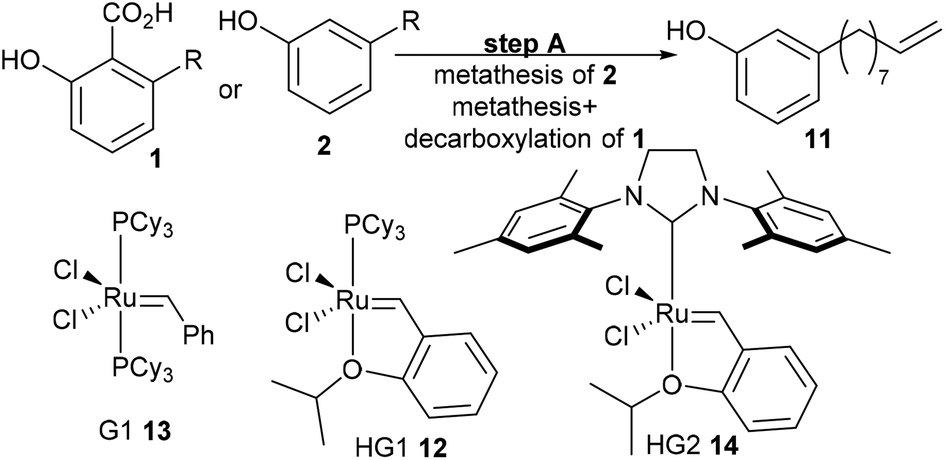
3-Vinylphenol, 10, could be obtained by an ethenolysis of cardanol acid to give 3-(non-8-enyl)phenol, 11, as the intermediate, which was followed by a one-pot isomerising ethenolysis.15 Following a similar reaction procedure previously reported in our group,15 3-(non-8-enyl)phenol, 11, was obtained in 96% yield after ethenolysis of cardanol with HG1 catalyst, 12 (Table 1, entry 1). The yield was slightly lower with G1 catalyst, 13 (85%, Table 1, entry 2), and only 11% yield of 3-(non-8-enyl)phenol, 11, was obtained when cardanol was metathesized with HG2 catalyst, 14 (Table 1, entry 3). Very similar results were obtained when the greener solvent, 2-methyl THF, was used (Table 1, entry 4). However, when anacardic acid, 1, was used as substrate, very poor conversions were observed using either HG1, 12, or HG2 catalysts, 14 (Table 1, entries 5 and 6). Reproduction of the isomerizing metathesis of 11 was carried out using ruthenium (M1, 15) and palladium dimer, 16, the desired 3-vinylphenol, 10, was obtained in 78% yield (Scheme 2), which is consistent with the literature results.15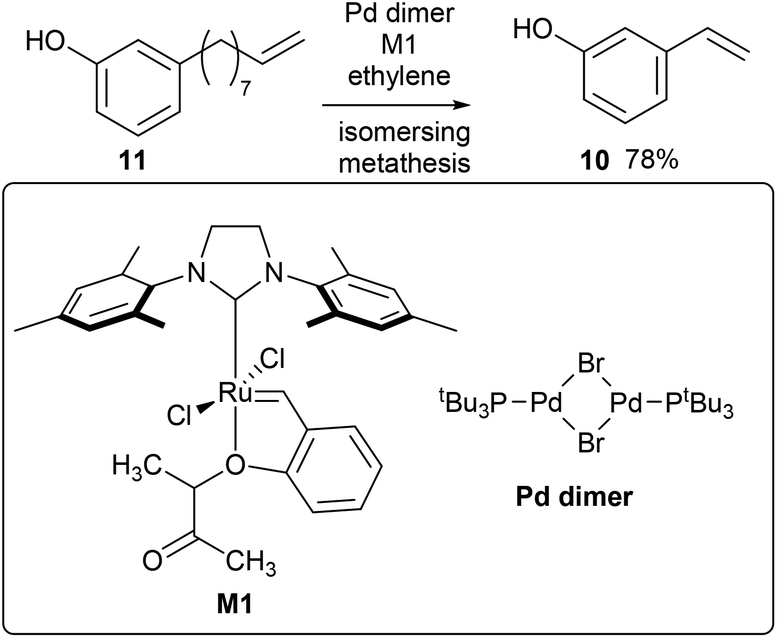 | ||
| Scheme 2 Synthesis of 3-vinyl phenol by isomerising metathesis using a palladium dimer (isomerisation) and M1 (metathesis) catalyst. | ||
|
|
|||
|---|---|---|---|
| Entry | Substrate | Catalyst | Yield (%) |
| a Reagents and conditions: Substrate (3 mmol), catalyst (0.3 mol%), anhydrous DCM (7.5 mL), ethene (10 bar), 24 h, r.t. b 2-Methyl THF (7.5 mL) instead of DCM. | |||
| 1 | Cardanol | HG1 | 96 |
| 2 | Cardanol | G1 | 85 |
| 3 | Cardanol | HG2 | 11 |
| 4b | Cardanol | HG1 | 93 |
| 5 | Anacardic acid | HG2 | <1 |
| 6 | Anacardic acid | HG1 | 1 |
The synthesis of 3-vinylphenol, 10, could also, in principle, be carried out by a two-step synthesis: isomerisation of cardanol to the benzylic cardanol followed by ethenolysis. Previous studies in our group showed that the [Pd2(dba)3]/DTBPMB/MSA system, which is active for the isomerising methoxycarbonylation of methyl oleate,22 and is a good isomerisation catalyst in the absence of CO, is effective for isomerising cardanol, although the yield of benzylic alkene was only 40%. This is the thermodynamic maximum. Although the styrene will be the most thermodynamically favoured isomer, the conjugation energy is insufficient to overcome the entropic advantage of having the double bond in other positions in the chain completely. Therefore, the cardanol was shortened by ethenolysis to produce 3-(non-8-enyl)phenol, 11, with the method previously mentioned (96% yield, Table 1, entry 1). 3-(Non-8-enyl)phenol, 11, was isomerized with [Pd2(dba)3]/DTBPMB/MSA in toluene at 80 °C, after 64 hours, the benzylic product, 3-(non-1-en-1-yl)phenol, 17, was obtained in 68% selectivity over all the other isomers (Table 2, entry 1). Heating at 80 °C for 96 hours led to 71% selectivity of 17 (Table 2, entry 2). Isomerisation of 3-(non-8-enyl)phenol with Pd catalyst can also be carried out in other solvents. Very similar results were obtained in DCM, THF or in a greener solvent, 2-MeTHF (Table 2, entries 3–8). [Rh(acac)(CO)2] together with DTBPMB ligand is known to be effective in isomerising hydroformylation of cardanol,23 therefore, it was interesting to find out whether [Rh(acac)(CO)2]/DTBPMB is effective in the isomerization reaction in the absence of CO/H2 gas. However, no benzylic alkene was observed after 64 hours, and only 28% of the terminal alkene was converted to other internal alkenes (Table 2, entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat prec. | solvent | T (°C) | t (h) | Sel. (benzylic alkene/others)b (%) |
| a Reagents and conditions: Substrate (50 mg, 0.2 mmol, 1 equiv.), catalyst (5 mol%) DTBPMB (0.5 equiv.), MSA (0.7 equiv.), solvent (1 mL). b Selectivity calculated by quantitative 1H NMR. | |||||
| 1 | [Pd2(dba)3] | Toluene | 80 | 64 | 68 |
| 2 | [Pd2(dba)3] | Toluene | 80 | 96 | 71 |
| 3 | [Pd2(dba)3] | DCM | 40 | 64 | 68 |
| 4 | [Pd2(dba)3] | DCM | 40 | 96 | 70 |
| 5 | [Pd2(dba)3] | THF | 70 | 64 | 62 |
| 6 | [Pd2(dba)3] | THF | 70 | 96 | 62 |
| 7 | [Pd2(dba)3] | 2-MeTHF | 80 | 64 | 63 |
| 8 | [Pd2(dba)3] | 2-MeTHF | 80 | 96 | 63 |
| 9 | [Rh(acac)(CO)2] | Toluene | 80 | 64 | 0 |
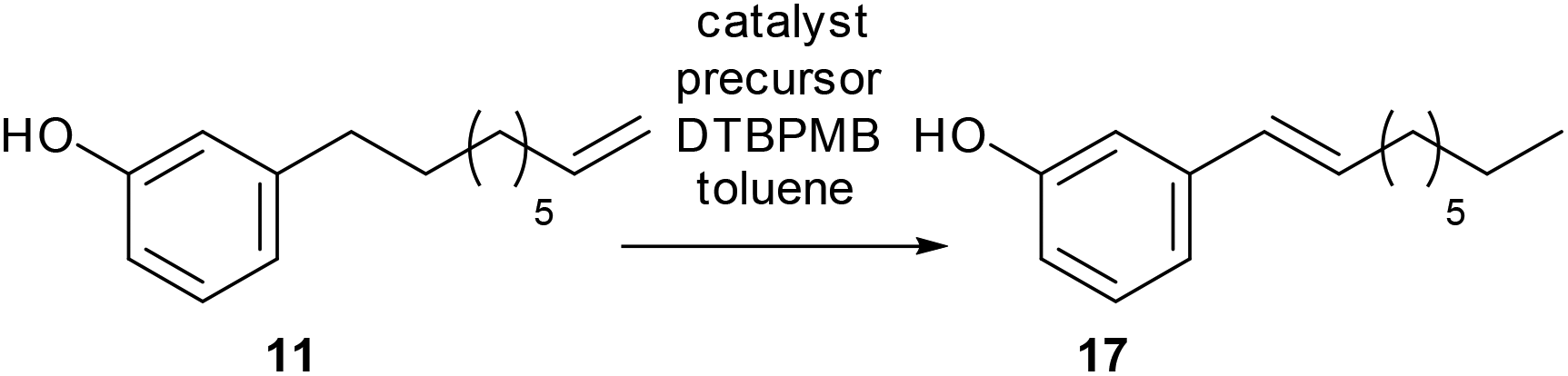
After isomerization of 3-(non-8-enyl)phenol, 11, the metathesis reaction was then carried out on the crude product from the isomerisation step (71% of 17, Table 2, entry 2). The crude product was metathesized using M1 catalyst, 15, and 3-vinylphenol, 10, was obtained in 65% yield by GC-FID together with alkenyl phenols with longer chain lengths (Scheme 3).
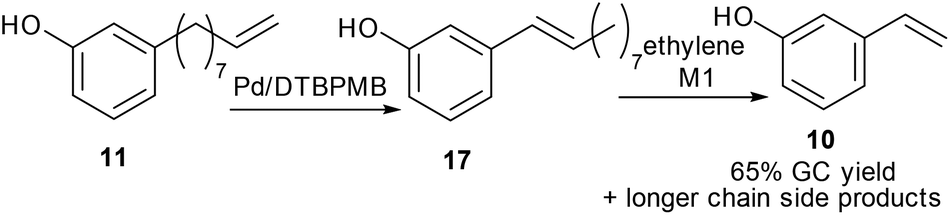 | ||
| Scheme 3 Synthesis of 3-vinylphenol in two steps from 3-non-8-enyl phenol. | ||
Synthesis of norfenefrine
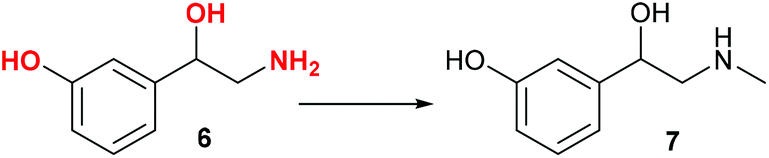
Norfenefrine, 6, is an adrenergic agent used as a sympathomimetic drug, which is pharmaceutically active in its racemic form. Norfenefrine, 6, plays a role as a minor neurotransmitter in the brain and regulates blood pressure in acute hypotensive states.25As presented in Scheme 1, 3-vinylphenol, 10, which was successfully obtained from cardanol by ethenolysis followed by isomerising metathesis, could possibly be used as an intermediate for the synthesis of norfenefrine, 6, by a hydroxyamination reaction on the double bond. Morandi reported a reaction of this kind which converts 1-methoxy-3-vinylbenzene, 18, to 2-amino-1-(3-methoxyphenyl)ethan-1-ol, 19, in 70% yield using an iron catalyst and O-pivaloylhydroxylammonium triflate (Scheme 4, Reaction A).24 We employed a similar method using 3-vinylphenol, 10, as substrate, and the desired norfenefrine, 6, which is also known as 3-(2-amino-1-hydroxyethyl)phenol, was obtained in 71% isolated yield (Scheme 4, Reaction B).
 | ||
| Scheme 4 Hydroxyamination of styrene type substrate. (A) Literature hydroxyamination of 1-methoxy-3-vinylbenzene, 18.24 (B) Experimental synthesis of norfenefrine, 6, from 3-vinylphenol, 10. | ||
Synthesis of metaraminol
Metaraminol, 9, which is used in the prevention and treatment of hypotension,26 has a very similar structure as norfenefrine, 6. It is active in its enantiopure form. It is interesting to investigate if the hydroxyamination reaction used for the synthesis of norfenefrine, 6, would work when using 3-(prop-1-en-1-yl)phenol, 20, as substrate. According to literature data, hydroxyamination of styrene, 21, occurs in 74% yield, while using β-methyl styrene, 22, as substrate gave around 20% yield (Scheme 5, Reaction A).24 The extra methyl group on the side chain led to a dramatic decrease of the yield. A reaction using the same hydroxyamination conditions as for 3-vinylphenol was carried out on 3-(prop-1-en-1-yl)phenol, 20. The desired racemic metaraminol, 9, was obtained in 24% NMR yield in this case (Scheme 4, Reaction B). This is in line with the results from the literature. | ||
| Scheme 5 Synthesis of metaraminol, 9, and related literature results.24 | ||
Synthesis of racemic phenylephrine
Phenylephrine which has a similar structure to that of norfenefrine, is a sympathomimetic, vasoconstrictor, mydriatic and cardiotonic agent. It can be used as decongestant, pupil dilator, vasopressor and in the treatment of hemorrhoids and priapism, to dilate the pupil or to increase the blood pressure, it is active as the L-isomer.25 Selective methylation of norfenefrine, 6, could result in the formation of racemic phenylephrine, 7. However, there are three active sites for the direct alkylation, both of the hydroxy groups and the amine groups are readily available for the reaction.When reacting norfenefrine, 6, with methyl iodide in the presence of a base, a rather complicated mixture of products was obtained, which might be because of methylation on alcohol, mono- and dimethylated products, and selective methylation was not achieved by this method (Table 3, entries 1 and 2). It has been reported that 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) is a good solvent for the selective alkylation of amines, producing a high proportion of mono-alkylated products over di-alkylated products.22 Methylation of norfenefrine with methyl iodide in HFIP in the absence of base gave very poor conversion, and low yield of the desired phenylephrine (Table 3, entry 3). By changing the methylation agent from methyl iodide to methyl triflate, much higher conversion was observed, and the desired phenylephrine, 7, was obtained in 79% yield (Table 3, entry 4).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | MX | Solvent | T (°C) | t (h) | Conv.b (%) | Yieldb (%) |
| a Conditions: Norfenefrine (1 mmol), MeX (1.5 mmol), HFIP (1 mL). b Conversions and yields were calculated by 1H NMR spectroscopy. c KOH (2 equiv.). | ||||||
| 1c | MeI | Acetone | 60 | 2 | 100 | n.d. |
| 2c | MeI | DMSO | 100 | 16 | 100 | n.d |
| 3 | MeI | HFIP | 25 | 24 | 4 | 2 |
| 4 | MeOTf | HFIP | 25 | 1 | 84 | 79 |
Synthesis of etilefrine
Another drug with a similar structure to that of norfenefrine, 6, is etilefrine, 8, which is a cardiac stimulant, and is used as an antihypotensive. Etilefrine is pharmaceutically active in its racemic form. Etilefrine can increase cardiac output, stroke volume, venous return and blood pressure by intravenous infusion.27 It is also an analeptic and sympathomimetic agent.25Reductive amination was used for the selective alkylation of nitrogen over the oxygen. When reacting norfenefrine, 6, with acetaldehyde in the presence of sodium borohydride, both the desired etilefrine, 8, and the side diethylated product 23 were obtained, and they were easily separated by flash column chromatography. The expected etilefrine, 8, was obtained in 54% yield, and the diethylated product was obtained in 35% yield (Scheme 6).
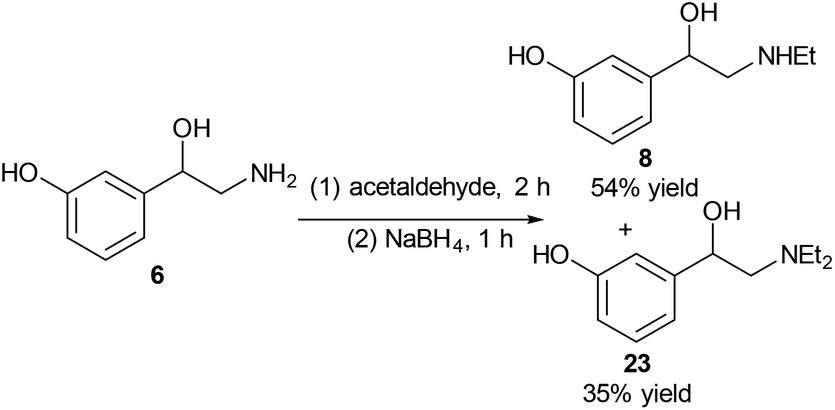 | ||
| Scheme 6 Synthesis of etilefrine, 8 from norfenefrine, 6. | ||
Due to the success of the methylation procedure for the selective mono-methylation of norfenefrine, we employed these conditions in the synthesis of etilefrine using ethyl triflate as the ethyl source. The ethylation of norfenefrine with ethyl triflate in HFIP led to a higher yield of the desired etilefrine (79%) compared to the reductive amination, together with 17% of the diethylated product after 1 hour at room temperature (Scheme 7).
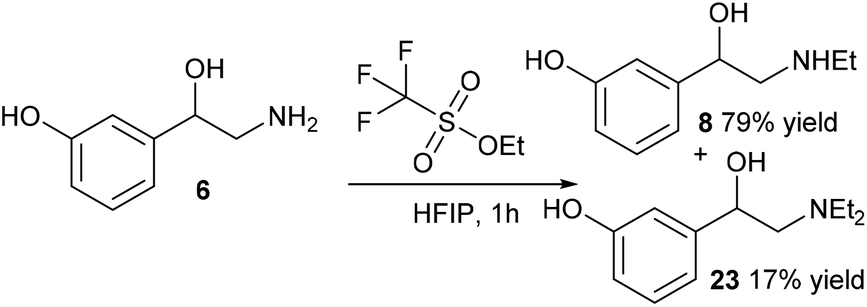 | ||
| Scheme 7 Ethylation of norfenefrine using ethyl triflate. | ||
Synthesis of fenoprofen
Another drug, which contains a meta-hydroxy phenyl group similar to CNSL, is fenoprofen, 5, which is a nonsteroidal anti-inflammatory drug (NSAID) and is marketed in the USA as Nalfon, which is also pharmaceutically active in its racemic form. The current cost for its oral capsules (400 mg) is around $256 for a supply of 90 capsules.28 It is effective for the treatment of fever, pain and swelling caused by inflammation.A few synthetic pathways have been tested (ESI, section S1‡). The best reaction pathway is to convert cardanol to the O-Ph cardanol, 24, firstly, which could avoid the complexity of the C–O coupling with unstable intermediates (e.g. styrene intermediate 10 in Scheme S1‡).
Cardanol, 3, was therefore reacted with bromobenzene first in the presence of anhydrous potassium tert-butoxide under literature conditions for the C–O coupling of the phenol (Step A, Scheme 8).29 Only about 30% of cardanol was converted to the O-Ph product, 24, at 45 °C. Increasing the temperature increased the conversion, and at 100 °C, the starting material was fully converted. Preparative scale gave similar results, and the O-Ph cardanol, 24, was successfully synthesised in 82% yield.
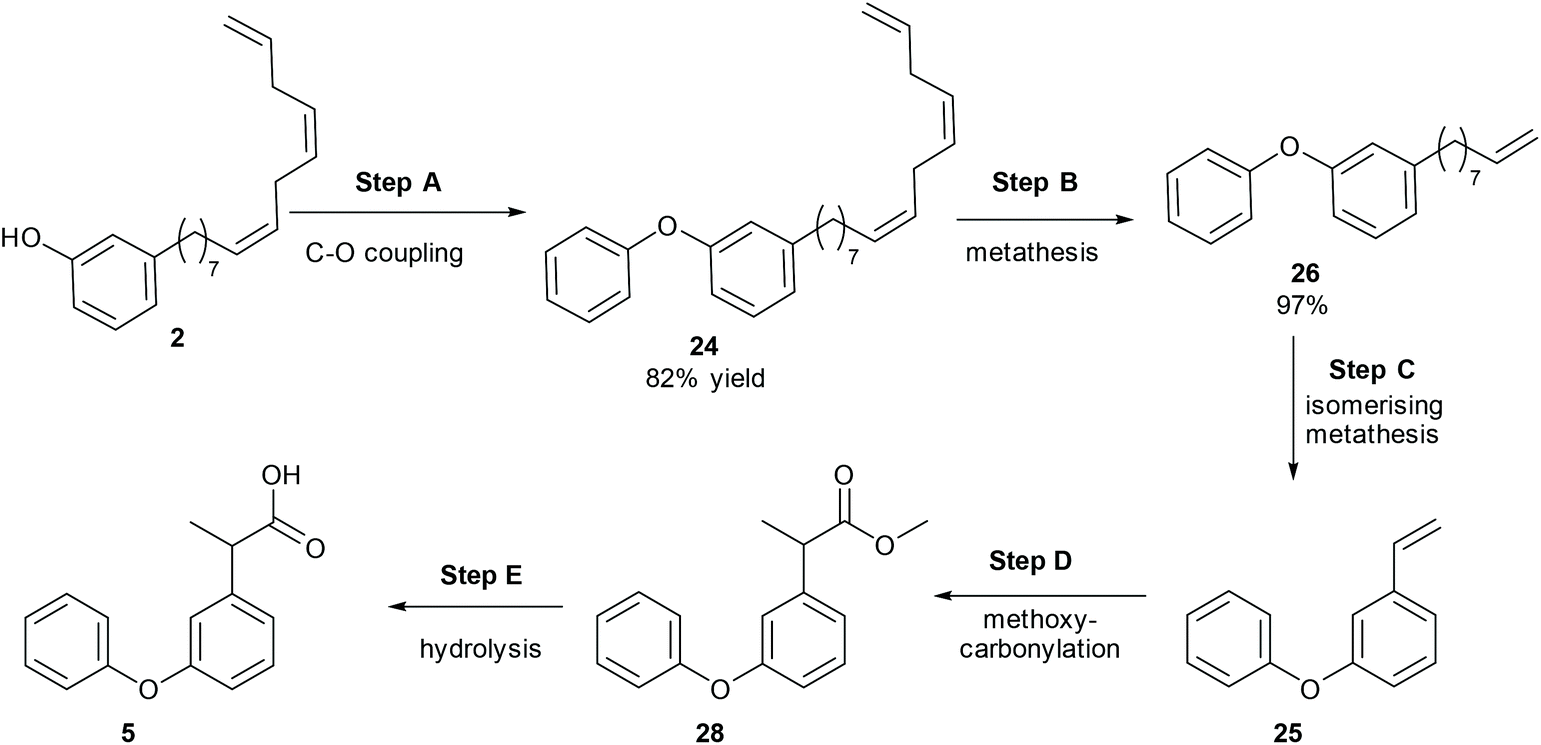 | ||
| Scheme 8 The synthesis of fenoprofen. | ||
After obtaining O-Ph cardanol, 24, 1-phenoxy-3-vinylbenzene, 25, can be synthesized in two steps (Step B and C in Scheme 8). Firstly, the O-Ph cardanol, 24, was metathesized to the corresponding 1-(non-8-en-1-yl)-3-phenoxybenzene, 26, using HG1 catalyst in high yield (97%) with 1-octene as the side product. 1-(Non-8-en-1-yl)-3-phenoxybenzene, 26, was then converted to 1-phenoxy-3-vinylbenzene, 25, by the isomerising metathesis reaction with palladium dimer and M1 catalysis, as discussed earlier for the synthesis of 3-vinylphenol. A one-pot isomerising metathesis of 1-(non-8-en-1-yl)-3-phenoxybenzene, 26, first afforded a mixture of 1-phenoxy-3-vinylbenzene, 25, and 1-phenoxy-3-(prop-1-en-1-yl)benzene, 27. The crude mixture was filtered through a plug of silica, and metathesized again with M1 catalyst. The desired 1-phenoxy-3-vinylbenzene, 25, was successfully obtained in 80% yield (Scheme 9).
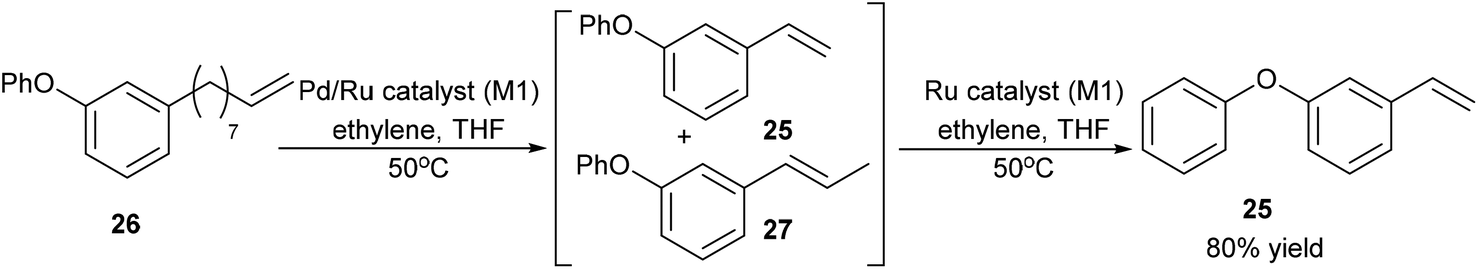 | ||
| Scheme 9 Synthesis of 1-phenoxy-3-vinylbenzene by isomerising metathesis. | ||
The next step is the synthesis of the branched ester 28 (Step D, Scheme 8). Branch selective methoxycarbonylation with Pd/DTBPMB system in the presence of racemic BINOL-phosphoric acid (rac-BNPA) afforded the desired product 28 in 82% yield together with 12% of the linear product, methyl 3-(3-phenoxyphenyl)propanoate, 29 (Scheme 10). Hydrolysis of 28 in 1,4-dioxane afforded fenoprofen, 5, in 91% yield. The hydrolysis of 28 can also be carried out in DMSO, which afford the desired fenoprofen, 5, in 92% yield. However, the hydrolysis reaction in 2-MeTHF did not give the desired product at all. With this method, biomass is successfully converted to fenoprofen drug in good yield (47.5% overall yield from cardanol, 2).
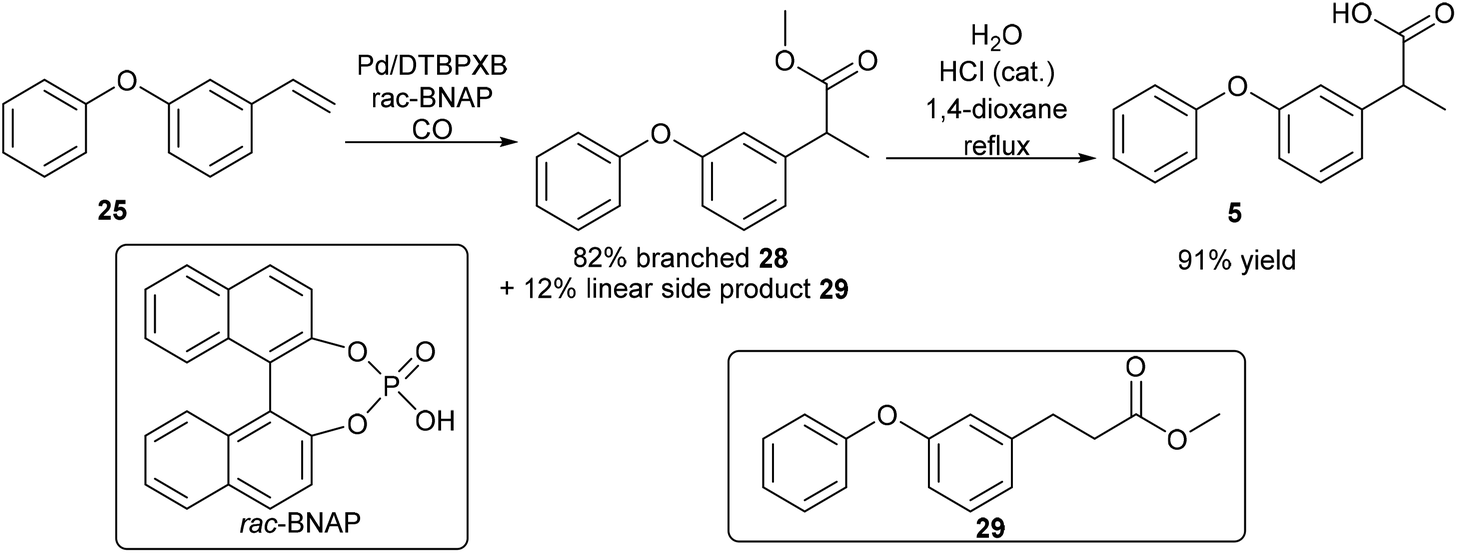 | ||
| Scheme 10 Synthesis of fenoprofen from 1-phenoxy-3-vinylbenzene. | ||
Enantioselective synthesis of the branched ester 28, from 1-phenoxy-3-vinylbenzene, 25, was also performed using a catalyst developed by Clarke,3030 (Scheme 11). After 17 h, 44% conversion was observed, with 69% branched to linear selectivity and 88% ee.
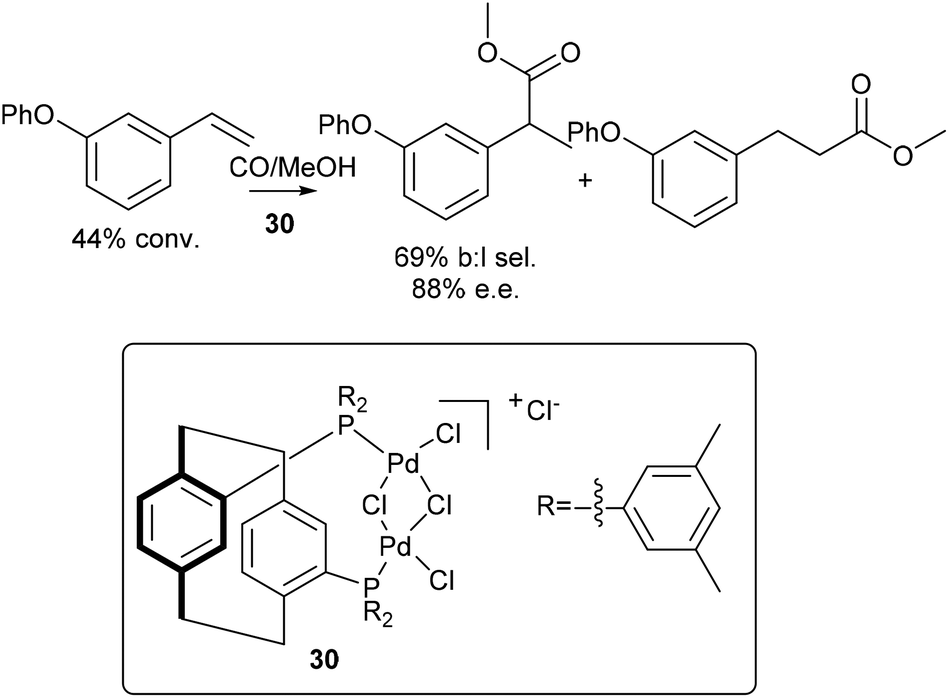 | ||
| Scheme 11 Enantioselective methoxycarbonylation of 1-phenoxy-3-vinylbenzene. Reagents: 1-Phenoxy-3-vinylbenzene (0.55 mmol), LiCl (2 mol%), para-toluenesulfonic acid monohydrate (2 equiv.), 30 (1 mol%), MeOH (0.75 mL), CO (30 bar), 40 °C, 17 h.30 | ||
Comparisons with literature processes
The main platform chemical for this study is 3-vinylphenyl, which we have synthesised from cardanol in 75% isolated yield after two steps. 3-Vinylphenol has been made in one step by pyrolysis of cardanol31,32 or from fossil fuel derived cyclohexanone.33 We note that the first step of our synthesis, ethenolysis of cardanol produces products as shown in Fig. 2.
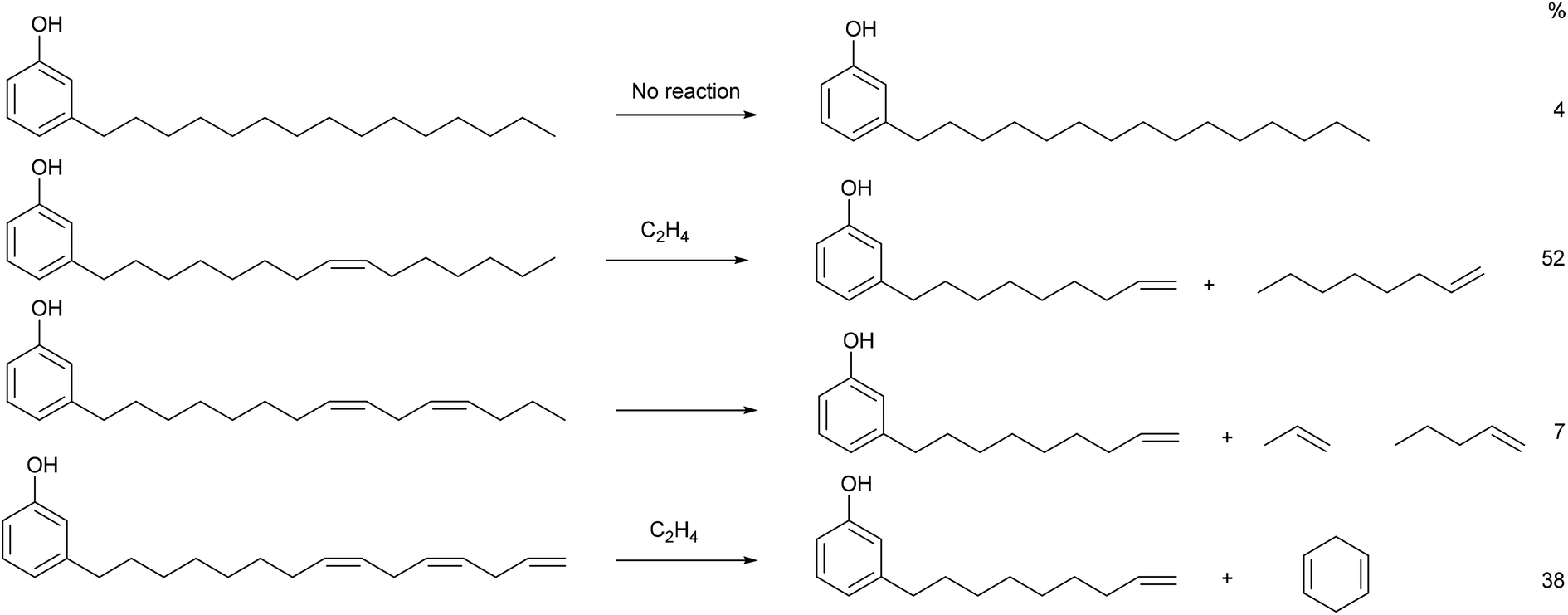 | ||
| Fig. 2 Products from the ethenolysis of each component of cardanol. The percentage figures on the right refer to the amount of each component in the batch used in this study. | ||
It is clear from analysis of Table 4 that our route is superior to pyrolysis because of the low temperatures used, the high yield and the side products. 1-Octene (from ethenolysis of cardanol containing 1 double bond is a valuable chemical used as a co-monomer for polyethylene and is required on 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonne per year in the US alone at a value of $1–2000 per tonne.34,35 1,4-Cyclohexadiene (self-metathesis of cardanol containing 3 double bonds; note that this does not require ethene) is a useful organic synthon and hydrogen donor or acceptor36 and we have shown that it can allow ethenolysis of a number of alkenes including methyl oleate to proceed cleanly using cheap first-generation ruthenium-based metathesis catalysts.16,37 Indeed, it is essential for the clean ethenoylsis of cardanol in the first step of the synthesis of 3-vinylphenol discussed here. Both 1-octene and cyclohexanone can be separated easily from one another and from 3-(non-8-enyl)phenol by distillation. The second step of the synthesis of 3-vinylphenol produces propene (>94%)15 as the by-product. This is also a valuable commodity chemical (∼100000 tonnes per year) used for making polymers and plasticisers and in the cumarin process for the manufacture of phenol.38 The price of propene39 is similar to that of ethene40 per mol, so making it as a side product in this way is an attractive proposition.
000 tonne per year in the US alone at a value of $1–2000 per tonne.34,35 1,4-Cyclohexadiene (self-metathesis of cardanol containing 3 double bonds; note that this does not require ethene) is a useful organic synthon and hydrogen donor or acceptor36 and we have shown that it can allow ethenolysis of a number of alkenes including methyl oleate to proceed cleanly using cheap first-generation ruthenium-based metathesis catalysts.16,37 Indeed, it is essential for the clean ethenoylsis of cardanol in the first step of the synthesis of 3-vinylphenol discussed here. Both 1-octene and cyclohexanone can be separated easily from one another and from 3-(non-8-enyl)phenol by distillation. The second step of the synthesis of 3-vinylphenol produces propene (>94%)15 as the by-product. This is also a valuable commodity chemical (∼100000 tonnes per year) used for making polymers and plasticisers and in the cumarin process for the manufacture of phenol.38 The price of propene39 is similar to that of ethene40 per mol, so making it as a side product in this way is an attractive proposition.
| Metathesis of cardanol | Pyrolysis of cardanol31,32 | From 3-vinylcyclohex-2-enone33 | ||
|---|---|---|---|---|
| Number of steps | 2 | 1 | 1 | |
| Step 1 | Step 2 | 6 from benzene | ||
| Other reagents | C2H4 | C2H4 | None | DMSO |
| Temperature /°C | 25 | 40–80 | 540–600 | 100 |
| Isolated yield/% | 96 | 70 | 12–15 | 45% (final step) |
| Side products | 1-Octene, 1,4-cyclohexadiene | Propene | 3-Ethylphenol, 3-methylphenol, phenol, alkenes and alkanes (C2–4 and higher), high boiling residue | Unknown |
| Small amount of volatile hydrocarbons from cardanol with 2 double bonds. | ||||
| Atom economy42,43 | 65 | 47 | ||
| Overall atom economy | 31 | 40 | 59.4 | |
| Atom economy (all usable products) | 92 | 100 | ||
| Over all atom economy (all usable products | 92 | 40 | 59.4 | |
| E Factor44,45/kg (waste) kg (products)−1 | 0.09 | 0.1 | 76 | |
As the demand for bio derived polymers rises, bio-1-octene and bio-propene will also be essential. The first step of our process produces 1-octene with 87.5% of the C derived from bioresources. If the ethenolysis were carried out using bio-ethene, from bioethanol, the 1-ocetene would be 100% bio-derived. Similarly, if bio-ethene were used for the isomerising metathesis, 100% bio-propene would be the by-product.
On the basis of atom economy, the synthesis of 3-vinylphenol from 3-vinylcyclohex-2-enone33 looks attractive. However, the oxidant is dimethylsulphoxide, which gives no useful side products and the yield is only 45%. For a direct comparison, we need to look at the synthesis from basic building blocks. 3-Vinylcyclohex-2-enone is derived from benzene, a non renewable resource in six steps via cumarin, cumarin peroxide, phenol, cyclohexan-2-one (Birch reduction) and finally 3-vinyl cyclohex-2-enone.41 Many of these steps are atom inefficient and or involve stoichiometric metal reagents.
Thus, on the basis of simplicity, mild conditions (important because 3-vinylphenol is unstable), yield, renewable resources that are of low value and not used in food and the value of the side products, the synthesis of 3-vinylphenol from cardanol seems to be very much more attractive than other available syntheses.
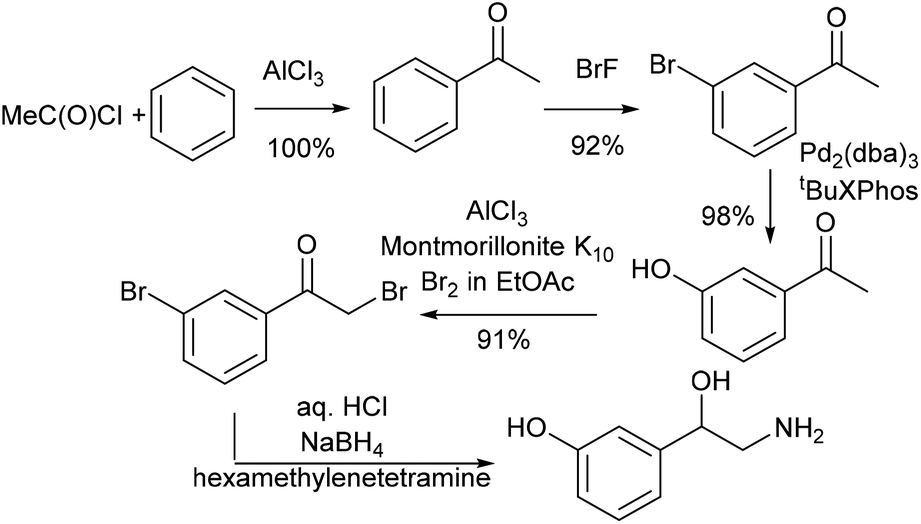 | ||
| Scheme 12 Synthesis of the norfenefrine in literature. | ||
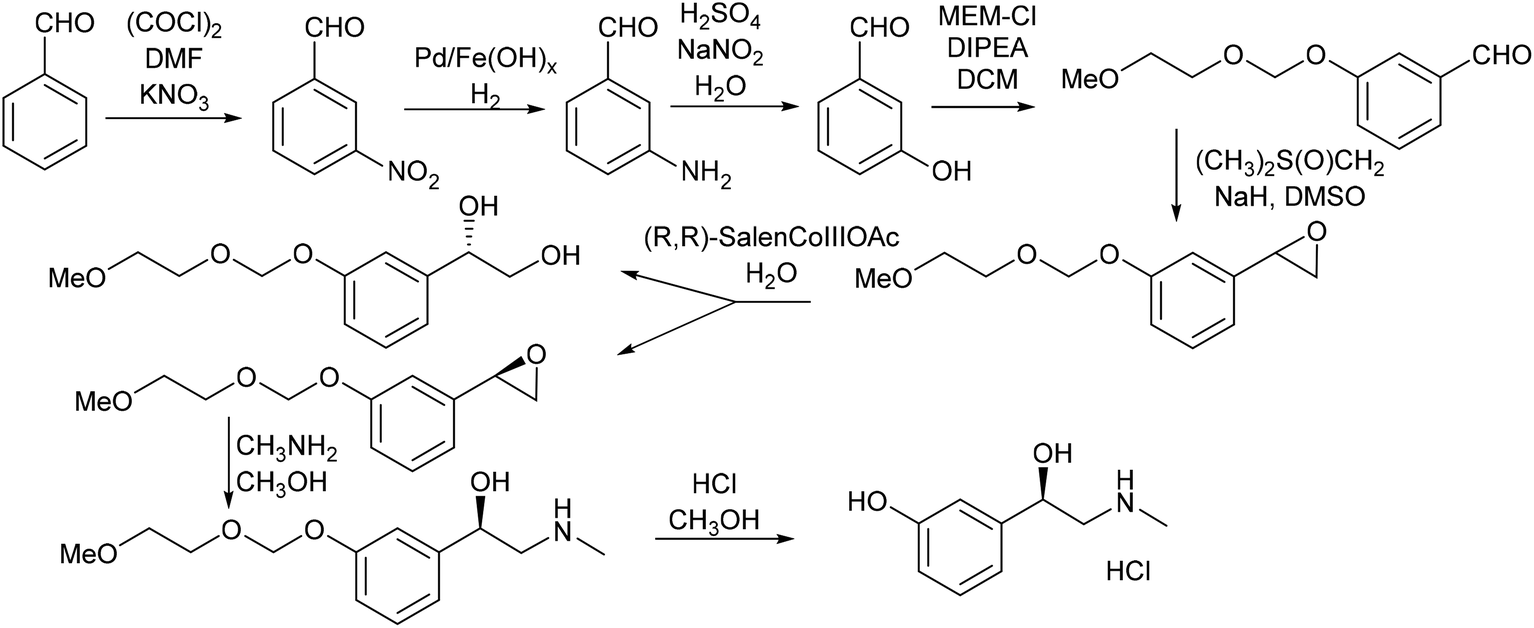 | ||
| Scheme 13 Original synthesis of phenylephrine. | ||
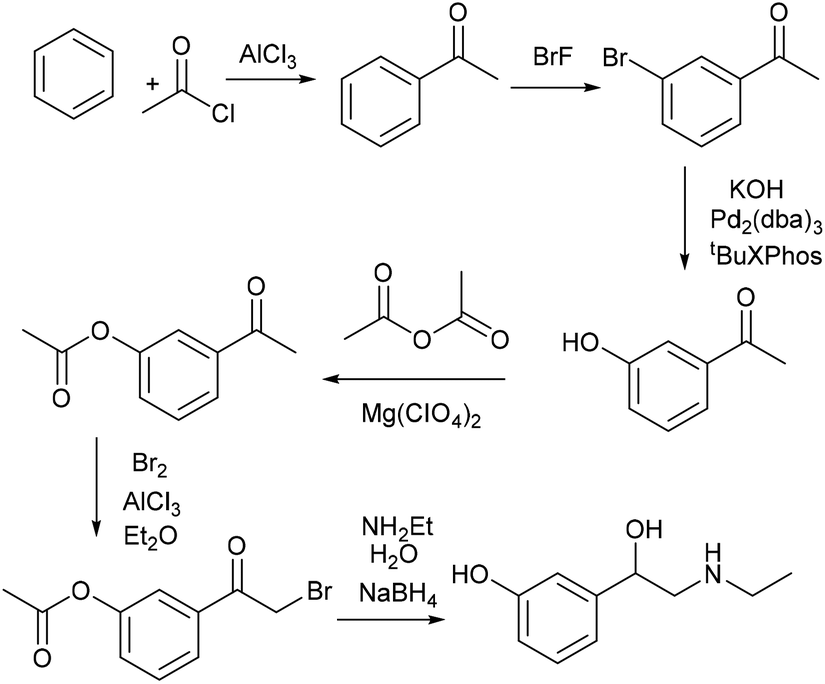 | ||
| Scheme 14 The original synthesis of etilefrine. | ||
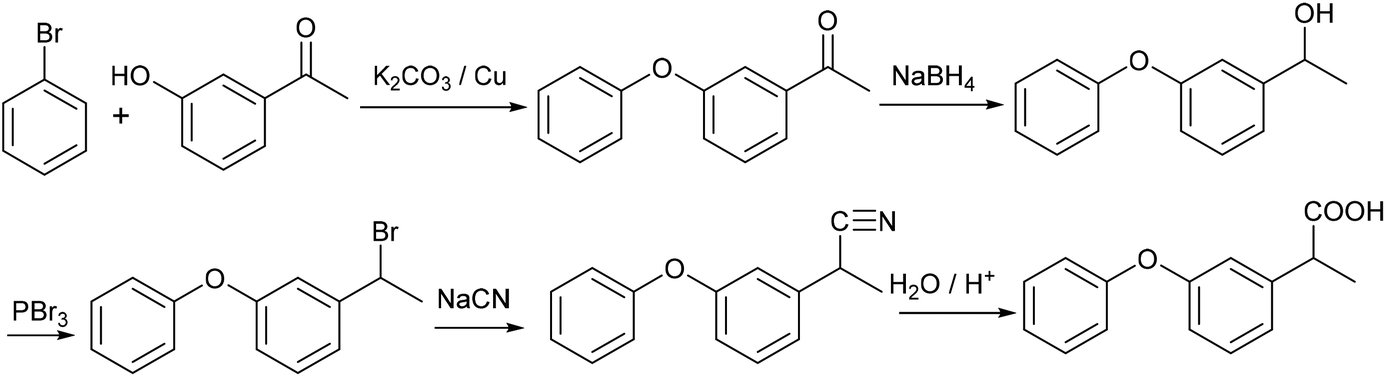 | ||
| Scheme 15 Original synthesis of fenoprofen. | ||
Although we have demonstrated significant advantages of the routes to certain pharmaceuticals described in this paper over conventional synthetic processes, we acknowledge that the synthesis of drugs and their precursors is tightly controlled by the United Nations Convention59 against illicit drugs and psychotropic substances, 1988 and EU60 legislation and that the high purity required of the final products is also enforced under the International Convention on Harmonisation (ICH) Guidelines.61 Further work on the process engineering by qualified API or drug manufacturers will be required before these routes from cardanol can be fully implemented.
Conclusions
Relatively inexpensive cardanol, an inedible component cashew nut shell liquid has been successfully converted to important and high-value medicinal drugs, such as norfenefrine, phenylephrine, etilefrine and fenoprofen in good yields by mostly catalytic reactions. For fenoprofen, the key methoxycarbonylation step has been demonstrated with good enantioselectivity. The greener solvent, 2-methyl THF, gave very good results for the metathesis of cardanol. Our methods start from a readily available and relatively cheap bio-based resource, generally use catalytic reactions, have fewer steps and avoid unpleasant reagents. They have atom efficiencies better than or similar to those of literature syntheses. An added attraction of these synthetic procedures is that the valuable commodity chemicals, 1-octene (from monounsaturated cardanol) and 1,4-cyclohexadiene (from triunsaturated cardanol) are significant products of the first ethenolysis reaction and propene is the side product of the isomerising metathesis step. These valuable side products make the E factor for synthesis of 3-vinylphenol by our route very low compared with those of previously reported syntheses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank James Mgaya for the cashew nut shells and Dr José A. Fuentes for providing catalyst 30. We also thank the EPSRC for the critical mass grant ‘Clean Catalysis for Sustainable Development’ (EP/J018139/1), Sasol Technology, UK for a case studentship (Y. S.) and the EPSRC UK National Mass Spectrometry Facility at Swansea University for mass spectrometric analysis. We are grateful to the referees of an earlier version of this paper for suggesting the comparisons of our processes with standard literature methods for synthesis of the same compounds.Notes and references
- T. Rajeswari, B. Padmapriya, K. Teesha and P. K. Kumari, Int. J. Microbiol. Res., 2011, 2, 172 Search PubMed.
- D. Lomonaco, G. Mele and S. E. Mazzetto, in Cashew Nut Shell Liquid: A Goldfield for Functional Materials, ed. P. Anilkumar, Springer International Publishing, 1st edn, 2017, p. 19 Search PubMed.
- R. N. Singh, U. Jena, J. B. Patel and A. M. Sharma, Renewable Energy, 2006, 31, 481 CrossRef CAS.
- R. Peter, V. R. Vijay, S. Ramakrishnan, R. Sukumar and A. R. R. Menon, Appl. Clay Sci., 2015, 105–106, 186 CrossRef CAS.
- S. K. Sanjeeva, M. P. Pinto, M. M. Narayanan, G. M. Kini, C. B. Nair, P. V. SubbaRao, P. K. Pullela, S. Ramamoorthy and C. J. Barrow, Renewable Energy, 2014, 71, 81 CrossRef CAS.
- L. F. B. Moreira, E. F. Lucas and G. Gonzalez, J. Appl. Polym. Sci., 1998, 73, 29 CrossRef.
- K. C. Dohhen, K. K. Swami, P. K. Mondal, S. Parkash, R. Sarin, D. K. Tuli and A. K. Bhatnagar, United States PatUS6339052B1, 2002 Search PubMed.
- A. Velmurugan, M. Loganathan and E. J. Gunasekaran, Fuel, 2014, 132, 236 CrossRef CAS.
- M. A. de Sousa Rios and S. E. Mazzetto, 13th Int. Electron. Conf. Synth. Org. Chem., 2009, vol. 13, p. c021.
- P. Peungjitton, P. Sangvanich, S. Pornpakakul, A. Petsom and S. Roengsumran, J. Surfactants Deterg., 2009, 12, 85 CrossRef CAS.
- S. Wahyuningsih, A. H. Ramelan, P. Rahmawati, B. P. N. Tamtama, P. P. Sari, P. L. Sari, S. Ichsan, Y. R. Kristiawan and F. N. Aini, IOP Conference Series: Materials Science and Engineering, International Conference on Advanced Materials for Better Future 2016, 2017, vol. 176, UNSP 012051.
- Y. C. Guo, G. Mele, F. Martina, E. Margapoti, G. Vasapollo and W. J. Xiao, J. Organomet. Chem., 2006, 691, 5383 CrossRef CAS.
- T. Shinde, V. Varga, M. Polášek, M. Horáček, N. Žilková and H. Balcar, Appl. Catal., A, 2014, 478, 138 CrossRef CAS.
- G. Vasapollo, G. Mele and R. Del Sole, Molecules, 2011, 16, 6871 CrossRef CAS PubMed.
- S. Baader, P. E. Podsiadly, D. J. Cole-Hamilton and L. J. Goossen, Green Chem., 2014, 16, 4885 RSC.
- J. Julis, S. a. Bartlett, S. Baader, N. Beresford, E. J. Routledge, C. S. J. Cazin and D. J. Cole-Hamilton, Green Chem., 2014, 16, 2846 RSC.
- J. A. Mmongoyo, Q. A. Mgani, S. J. M. Mdachi, P. J. Pogorzelec and D. J. Cole-Hamilton, Eur. J. Lipid Sci. Technol., 2012, 114, 1183 CrossRef CAS.
- J. E. Mgaya, S. A. Bartlett, E. B. Mubofu, Q. A. Mgani, A. M. Z. Slawin, P. J. Pogorzelec and D. J. Cole-Hamilton, ChemCatChem, 2016, 8, 751 CrossRef CAS.
- J. E. Mgaya, E. B. Mubofu, Q. A. Mgani, D. B. Cordes, A. M. Slawin and D. J. Cole-Hamilton, Eur. J. Lipid Sci. Technol., 2015, 117, 190 CrossRef CAS.
- J. Julis, S. Bartlett and D. J. Cole-Hamilton, WO2015114323 A1, 2015 Search PubMed.
- J. Pollini, V. Bragoni and L. J. Gooßen, Beilstein J. Org. Chem., 2018, 14, 2737 CrossRef CAS PubMed.
- M. R. L. Furst, R. Le Goff, D. Quinzler, S. Mecking, C. H. Botting and D. J. Cole-Hamilton, Green Chem., 2012, 14, 472 RSC.
- S. Pandey, D. R. Shinde and S. H. Chikkali, ChemCatChem, 2017, 9, 3997 CrossRef CAS.
- L. Legnani and B. Morandi, Angew. Chem., Int. Ed., 2016, 55, 2248 CrossRef CAS PubMed.
- C. R. Ganellin and D. J. Triggle, Dictionary of Pharmacological Agents, CRC Press, illustrate., 1996 Search PubMed.
- V. R. Kee, Crit. Care Nurse., 2003, 23, 79 Search PubMed.
- L. W. Retief, J. A. Roelofse and B. H. Meyer, S. Afr. Med. J., 1984, 66, 526 CAS.
- https://www.drugs.com/price-guide/fenoprofen . Date accessed: 11/04/2018.
- S. Kumar, A. Kumar, B. S. Bhakuni, C. D. Prasad and S. Kumar, Tetrahedron, 2013, 69, 5383 CrossRef.
- T. M. Konrad, J. T. Durrani, C. J. Cobley and M. L. Clarke, Chem. Commun., 2013, 49, 3306 RSC.
- E. E. Michael and W. J. E. Seager, US2698868A, 1955 Search PubMed.
- J. Bendig, M. Nickoleit and U. Schedler, DE19645287A1, 1998 Search PubMed.
- Y. F. Liang, S. Song, L. Ai, X. Li and N. Jiao, Green Chem., 2016, 18, 6462 RSC.
- https://www.alibaba.com/showroom/1–octene.html . Date accessed: 26 Nov.
- https://view.joomag.com/north-america-1-octene-market-is-expected-to-reach-497-thousand-mt-in-north-america-1-octene-market-is-expected-to-reach-497-thousand-mt-in/0935565001420237383 . Date accessed 26 Nov 2018.
- J. C. Walton and F. Portela-Cubillo, in ‘1,4-Cyclohexadiene’ Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, 2007, DOI:10.1002/047084289X.rn00806.
- J. Julis, D. J. Cole-Hamilton and C. Cazin, WO2014041344, 2014 Search PubMed.
- http://www.essentialchemicalindustry.org/chemicals/propene.html . Date accessed: 26 Nov 2018.
- https://www.platts.com.es/newsfeature/2014/petrochemicals/pgpi/propylene . Date accessed: 26 Nov 2018.
- https://www.platts.com.es/newsfeature/2014/petrochemicals/pgpi/ethylene Date accessed: 26 Nov 2018.
- J. L. Luche, C. Pétrier, A. L. Gemal and N. Zikra, J. Org. Chem., 1982, 47, 3806 CrossRef.
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- R. A. Sheldon, C. R. Acad. Sci., Ser. IIc: Chim., 2000, 3, 541 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- S. R. D. George, T. D. H. Frith, D. S. Thomas and J. B. Harper, Org. Biomol. Chem., 2015, 13, 9035 RSC.
- S. Rozen, M. Brand and R. Lidor, J. Org. Chem., 1988, 53, 5545 CrossRef CAS.
- K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694 CrossRef CAS PubMed.
- V. R. Uchil and V. Joshi, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2003, 42, 408 Search PubMed.
- A. Chatterjee, S. K. Srimany and B. Chaudhury, J. Chem. Soc., 1961, 4576 RSC.
- M. S. Kumar, K. R. Reddy, K. C. Rajanna, P. Venkanna and G. Krishnaiah, Synth. React. Inorg., Met. – Org., Nano-Met. Chem., 2013, 43, 977 CrossRef CAS.
- C. Zhang, X. Cui, Y. Deng and F. Shi, Tetrahedron, 2014, 70, 6050 CrossRef CAS.
- R. N. Icke, C. E. Redemannn, B. B. Wisegarver and G. A. Alles, Org. Synth., 1949, 29, 72 CrossRef CAS.
- M. K. Gurjar, L. M. Krishna, B. V. N. B. S. Sarma and M. S. Chorghade, Org. Process Res. Dev., 1998, 2, 422 CrossRef CAS.
- G. Bartoli, M. Bosco, R. Dalpozzo, E. Marcantoni, M. Massaccesi, S. Rinaldi and L. Sambri, Synlett, 2003, 39 CAS.
- Y.-J. Zhang, L.-L. Shen, H.-G. Cheon, Y.-N. Xu and J.-H. Jeong, Arch. Pharmacal Res., 2014, 37, 588 CrossRef CAS PubMed.
- T. Goto, Yakugaku Zasshi, 1954, 74, 318 CrossRef CAS.
- R. Vardanyan and V. Hruby, Synthesis of Essential Drugs, Elsevier, Amsterdam, 1st edn, 2006 Search PubMed.
- http://www.unodc.org/pdf/convention_1988_en.pdf . Date accessed: 05/12/2018.
- https://ec.europa.eu/taxation_customs/business/customs-controls/drug-precursors-control/legal-base-drug-precursors_en . Date accessed: 05/12/2018.
- https://www.ich.org/products/guidelines/quality/article/quality-guidelines.html . Date accessed: 05/12/2018.
Footnotes |
| † Dedicated to Ernesto Carmona, and outstanding chemist and great friend, on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc03823f |
| § Current address: Bioinspired Homo- & Heterogeneous catalysis, Leibniz Institute for Catalysis, Albert-Einstein-Straβe 29 a, 18059 Rostock, Germany. |
| This journal is © The Royal Society of Chemistry 2019 |