
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical cross-coupling of biogenic di-acids for sustainable fuel production†
F. Joschka
Holzhäuser
a,
Guido
Creusen
b,
Gilles
Moos
ac,
Manuel
Dahmen
 d,
Andrea
König
e,
Jens
Artz
a,
Stefan
Palkovits
a and
Regina
Palkovits
*a
d,
Andrea
König
e,
Jens
Artz
a,
Stefan
Palkovits
a and
Regina
Palkovits
*a
aInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
bInstitut für Makromolekulare Chemie, Albert-Ludwigs-Universität Freiburg, Freiburg, Germany
cCurrently at: Max Planck Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany
dInstitut of Energy and Climate Research IEK-10: Energy Systems Engineering, Forschungszentrum Jülich, Jülich, Germany
eAachener Verfahrenstechnik - Process Systems Engineering, RWTH Aachen University, Aachen, Germany
First published on 2nd April 2019
Abstract
Direct electrocatalytic conversion of bio-derivable acids represents a promising technique for the production of value-added chemicals and tailor-made fuels from lignocellulosic biomass. In the present contribution, we report the electrochemical decarboxylation and cross-coupling of ethyl hydrogen succinate, methyl hydrogen methylsuccinate and methylhexanoic acid with isovaleric acid. The reactions were performed in aqueous solutions or methanol at ambient temperatures, following the principles of green chemistry. High conversions of the starting materials have been obtained with maximum yields between 42 and 61% towards the desired branched alkane products. Besides costly Pt electrodes also (RuxTi1−x)O2 on Ti electrodes exhibited a notable activity for cross-Kolbe electrolysis. As some of the products are insoluble in water, easy product isolation and reuse of the reaction solvent is enabled via phase separation. Several side products have been identified to evaluate the efficiency of the reaction and to elucidate the factors influencing the product selectivity. The yielded alkanes and esters were assessed with regard to their potential as fuels for internal combustion engines. While the longer alkanes constitute promising candidates for the compression–ignition engine, the smaller ester represents an interesting option for the spark-ignition engine.
Introduction
Today's world energy supply and basic organic chemical production are mainly based on fossil resources like coal, crude oil and natural gas. The depletion of these resources has fostered research on the use of renewable carbon sources such as lignocellulosic biomass, which displays a rich variety of chemical compounds.1–3 Besides furans, sugars and others, the US Department of Energy (DoE) highlighted organic acids as valuable bio-based platform chemicals.4 For instance, the DoE named levulinic, succinic and itaconic acid as three of twelve future major platform molecules derived from biomass feedstocks. Since these carboxylic acids are readily and abundantly available from biorefineries, they have received much attention in recent decades.5 Other acids such as methyl succinic acid are easily obtainable based on itaconic acid.6,7 In addition, aliphatic C2–C6 acids, e.g. acetic or isovaleric acid, become available from biomass via fermentation.8–10 In the last decades, research focussed on efficient processes for the production of alternative fuels and other value-added chemicals with the aforementioned acids as feedstocks.2,11,12 Since most studies dealt with thermo-catalytic biomass transformations, first examples of electricity driven reactions instead of heat have only recently been reported.13–18 These studies are also motivated by the fact that electrical energy production continues to shift from fossil to renewable resources such as wind and solar energies. This shift is inherently accompanied by an intermittent energy supply following temporal, seasonal and geographical fluctuations. This translates into a local and temporal deviation of electricity demand and supply. Accordingly, new technologies to transform surplus electricity into a storable form are required.19Besides water splitting and battery technologies, a new approach uses the electrical energy from renewable resources for electrochemical production of compounds with an enhanced calorific value and the potential to serve as fuel candidates.13–15,20–23 First examples of the opportunities offered by electrocatalysis were provided by the groups of Schröder and Li, who reported the electroreduction of levulinic acid in aqueous acidic solutions at room temperature. The selectivity towards valeric acid or γ-valerolactone was controlled by using Pb or glassy carbon electrodes, respectively.13,14,20,21 The obtained valeric acid could be converted to n-octane in a consecutive step via Kolbe electrolysis either in an aqueous or organic solvent.13,14
In 2017 Yin et al. reported the successful synthesis of three-dimensional membrane electrodes (Co3O4 nanowires on Ti) for the selective oxidation of alcohols.25Via stable nano-V2O5/Ti membrane electrodes cyclohexane could be efficiently oxidized into cyclohexanol and cyclohexanone only one year later.26 Another example for the successful electrochemical upgrading of bio-derived compounds into value-added products is the electroreduction of furfural and 5-(hydroxymethyl)furfural towards the respective furanes or even to 2,5-hexanedione presented by Choi and co-workers.15,27 The group of Schröder reported the production of olefin/ether mixtures and olefins with diesel-like properties via non-Kolbe electrolysis of fatty acids and triglycerides.16 A very recent study dealt with the synthesis of drop-in fuels or more specifically of alkanes and alcohols from biomass through fermenting corn beer into different carboxylic acids.22 The authors successfully demonstrated extraction of the respective acids using a pertraction system with mineral oil (3 wt% trioctylphosphine oxide) and an alkaline back solution and the coupling of the obtained acids via Kolbe electrolysis into alkanes.22
Noticeable, Kolbe electrolysis presents a key reaction to access alternative fuels from biomass-based carboxylic acids. The reaction itself is well understood nowadays. Its history goes back more than 180 years, when Faraday first reported the formation of hydrocarbons for the electrolysis of an aqueous acetate solution in 1834.28 Roughly 15 years later in 1849, Hermann Kolbe studied the electrochemical decarboxylation of organic acids and described the formation of an oily product (n-octane) at the anode upon electrolysis of a valeric acid solution.29 According to the reaction mechanism of Kolbe electrolysis proposed by Schäfer (Scheme 1),24 the deprotonated organic acid transfers an electron to the anode resulting in a carboxyl radical. A fast decomposition of this species into an alkyl radical and carbon dioxide is followed by a dimerization of two radicals, to yield the so-called Kolbe product. Besides this, the alkyl radical can also disproportionate into a saturated and unsaturated compound. Another electron abstraction from the alkyl radical leads to a carbocation, which reacts to the corresponding alcohol in the presence of water (Hofer–Moest reaction). A multitude of products is accessible through this reaction by substituting water by other nucleophiles. The formation of esters can be explained by the reaction of a carbocation with a deprotonated acid. Moreover, by eliminating a proton from the cationic species, an unsaturated compound is obtained (non-Kolbe electrolysis). Recently, we demonstrated the Kolbe and non-Kolbe electrolysis of succinic acid towards valuable monomers. Additionally, a cheap and facile synthesis of a Ru/TiO2-anode was established replacing the Pt-anode as the most common working electrode (WE) for Kolbe electrolysis.30 Despite several studies on Kolbe electrolysis of carboxylic acids31 and also other coupling techniques, such as C–H aryl-dimerization,32 strategies to access promising molecular structures with suitable fuel properties are not yet available.
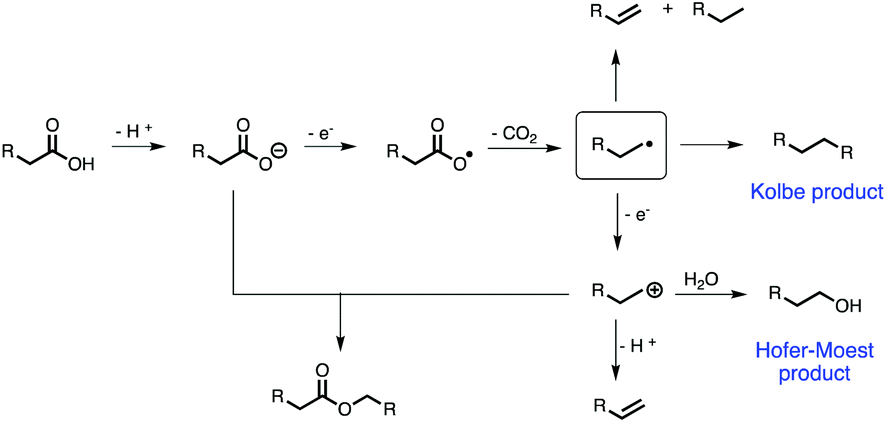 | ||
| Scheme 1 Reaction pathway of Kolbe electrolysis and main side reactions.24 | ||
The present study proposes a new methodology for the production of branched and long-chained alkanes as well as esters from bio-organic mono- and diacids. The concept is simple: Kolbe electrolysis is the required tool for the chosen biogenic acids in order to cross-connect them similar to bricks in a construction kit (Scheme 2). The most important benefit of this electrochemical approach lies in the facile methodology to synthesize fuels and fuel additives with unique and tailor made properties. The employed different bio-derived diacids and mono-acids depict model molecules to demonstrate the proposed methodology. Because of the bifunctional character of the diacids, significantly more combinations are possible than for carboxylic acids with only one acid-head function. In a first step, mono-ethyl succinic acid (HESA) or α-methyl methylsuccinic acid (MMSA) was cross-coupled with isovaleric acid (IVA) to yield a branched ester. In a second step, the resulting 5-methylhexanoic acid (MHO) was used as a model substrate for further Kolbe cross-coupling with IVA or itself. We studied the influence of various parameters on the transformations and confirmed the performance of the derived compounds and mixtures as fuel compounds.
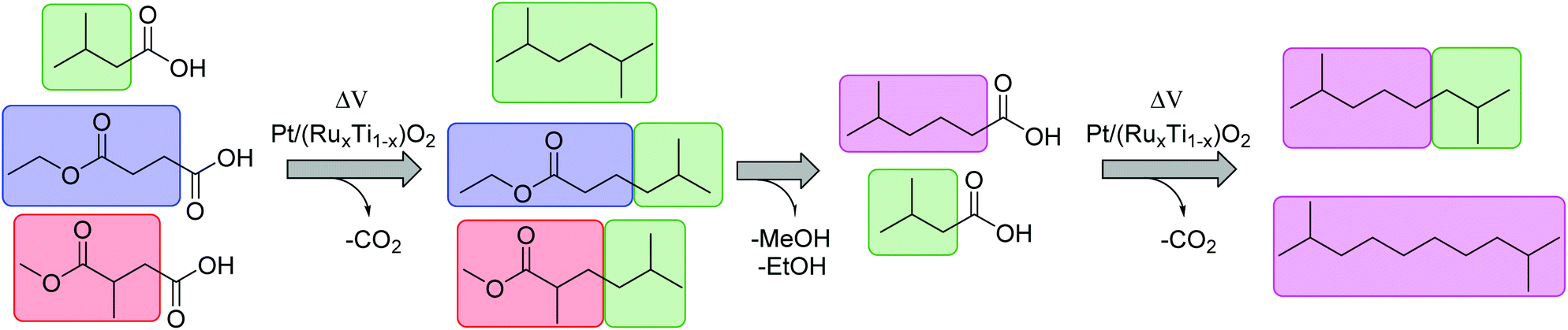 | ||
| Scheme 2 The cross-coupling of HESA, MMSA, MHO and IVA into potential fuel and diesel compounds. The carboxylic acids can be selected like small bricks (different colors) and connected to each other at the α-carbon atom. | ||
Results and discussion
The first step: HESA/MMSA coupling with IVA
The main products of both cross-coupling reactions investigated within this study are depicted in Scheme 3. Further potential side-products such as isobutyl isovalerate, propene, propane or iso-octane were either not observed or detected in trace amounts in the liquid phase. The compounds which can be potentially derived by cross-coupling of HESA or MMSA with IVA are of high industrial interest. The hydrolysed forms of diethyl adipate (DA)33 and dimethyl 2,5-dimethylhexanedioate (DDH)17 as well as the acrylates34,35 are known building blocks in polymer industry. Even the by-product tert-butanol is known as antiknock agent and can thus be used as fuel additive or as solvent.36 Moreover, ethyl propionate (EP) and methyl isobutyrate (MIB) may for instance serve as solvents for cellulose ethers and esters.37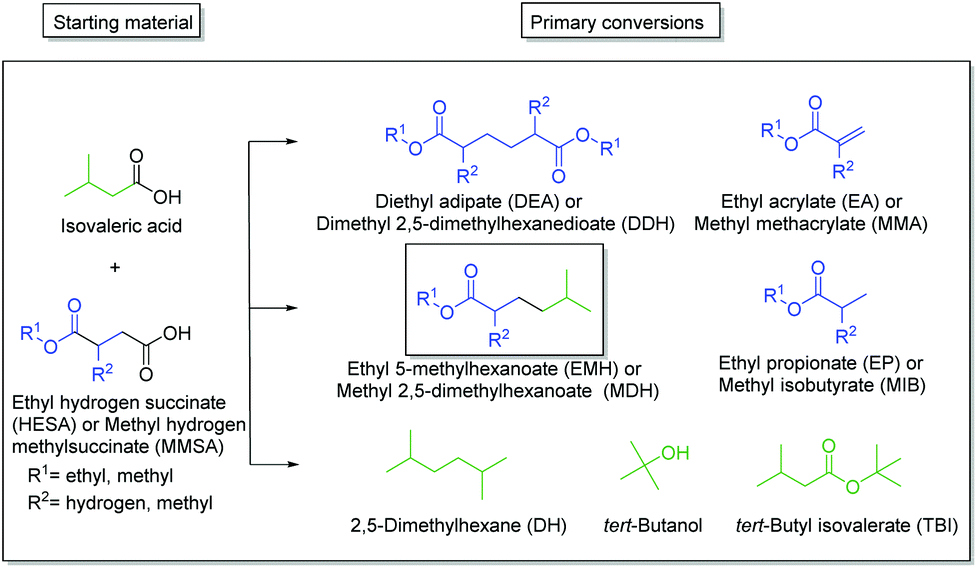 | ||
| Scheme 3 Schematic illustration of the different observed products after the electrochemical oxidation of HESA/MMSA and IVA. The colored molecule parts of the substrates are also depicted in the end-products. | ||
The only exception is tert-butyl isovalerate (TBI) for which there are no reported applications yet to the best of our knowledge. In order to optimize the electrochemical oxidative coupling of the monoester acids HESA and MMSA with the mono-acid IVA, various reaction parameters were separately investigated. All experiments were conducted with the consumption of one farad equivalent for all substrates exactly. One farad equivalent represents the charge amount which is required to electrochemically convert all substrates into the desired products (see ESI 1. Calculations†). The results for cross-coupling of MMSA with IVA follow a similar trend (see ESI Fig. 1a†). Therefore, the discussion herein focusses mainly on HESA as a substrate. At first, the optimal substrate ratio of HESA or MMSA to IVA was determined. Screening experiments were conducted within HESA–IVA ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 up to 1:16 (Fig. 1 and Fig. ESI 1a†). According to previous findings that higher current densities and low temperatures favour Kolbe dimerization, all experiments were conducted at 100 mA cm−2 and 0 °C.24 In addition, the use of an ice bath ensured that all reactions were carried out at constant temperatures. Preliminary tests showed good performance for a solvent mixture of 80% methanol and 20% water. An excessive amount of IVA was used to promote the formation of cross-coupled product. Though, it cannot completely suppress the dimerization of HESA or MMSA. Side products such as the acrylates (EA and MMA), tert-butanol, EP, TBI and MIB were in the same order of magnitude contributing between 1 and 5% in total for all parameter variations. The yield of the cross-coupled product EMH decreases with lower HESA–IVA ratios. While the yield of EMH reached 22% at a 1:1 ratio of both acids, a final yield of approximately 50% was achieved increasing the ratio to 1:8 or 1:16. For DH an analogue behaviour was observed and for lower HESA–IVA the yield increased to approximately 30%. As expected, the yield of DEA decreases with lower concentrations of HESA, as coupling with IVA becomes more likely. The results regarding MMSA and IVA showed a similar trend. The desired cross-coupling product yields of methyl 2,5-dimethylhexanoate (MDH) and DH decreased with increasing ratios of MMSA to IVA, whereas the yield of dimethyl 2,5-dimethylhexanoate (DDH) increased. Nevertheless, all product yields with MMSA were slightly lower compared to HESA as a substrate, yielding a maximum of 37% MDH and 21% DH, respectively. This behaviour was observed for all reactant ratios and most likely caused by a higher steric hindrance of MMSA in comparison to HESA.
:1 up to 1:16 (Fig. 1 and Fig. ESI 1a†). According to previous findings that higher current densities and low temperatures favour Kolbe dimerization, all experiments were conducted at 100 mA cm−2 and 0 °C.24 In addition, the use of an ice bath ensured that all reactions were carried out at constant temperatures. Preliminary tests showed good performance for a solvent mixture of 80% methanol and 20% water. An excessive amount of IVA was used to promote the formation of cross-coupled product. Though, it cannot completely suppress the dimerization of HESA or MMSA. Side products such as the acrylates (EA and MMA), tert-butanol, EP, TBI and MIB were in the same order of magnitude contributing between 1 and 5% in total for all parameter variations. The yield of the cross-coupled product EMH decreases with lower HESA–IVA ratios. While the yield of EMH reached 22% at a 1:1 ratio of both acids, a final yield of approximately 50% was achieved increasing the ratio to 1:8 or 1:16. For DH an analogue behaviour was observed and for lower HESA–IVA the yield increased to approximately 30%. As expected, the yield of DEA decreases with lower concentrations of HESA, as coupling with IVA becomes more likely. The results regarding MMSA and IVA showed a similar trend. The desired cross-coupling product yields of methyl 2,5-dimethylhexanoate (MDH) and DH decreased with increasing ratios of MMSA to IVA, whereas the yield of dimethyl 2,5-dimethylhexanoate (DDH) increased. Nevertheless, all product yields with MMSA were slightly lower compared to HESA as a substrate, yielding a maximum of 37% MDH and 21% DH, respectively. This behaviour was observed for all reactant ratios and most likely caused by a higher steric hindrance of MMSA in comparison to HESA.
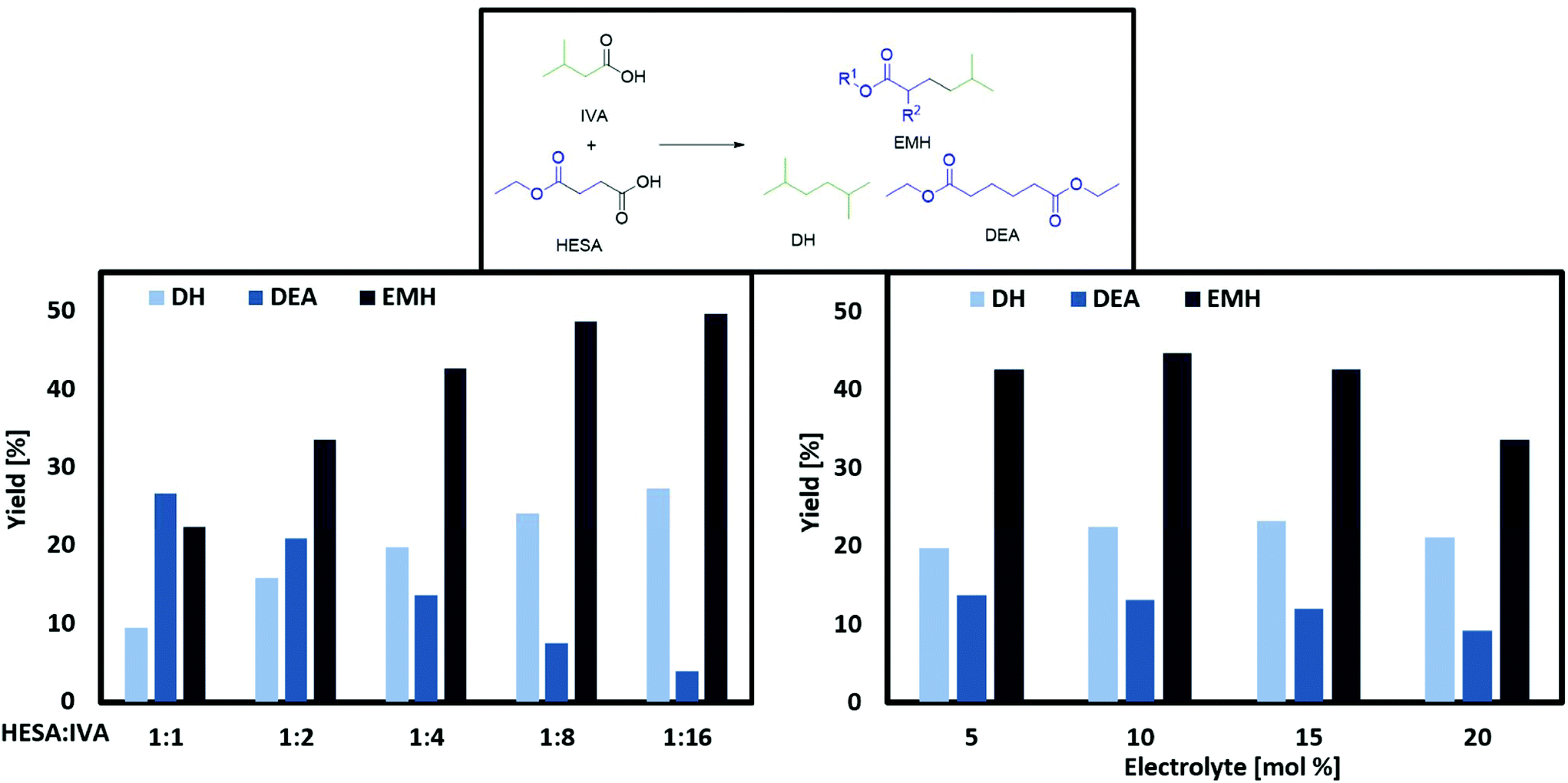 | ||
| Fig. 1 Variation of different ratios of HESA with IVA with 0.1 M NEt3 (left) and the electrolyte concentration with 0.33 M HESA and 1.3 M IVA (right). Conditions: 0 °C, MeOH:H2O 80:20, 1 farad equivalent, 100 mA cm−2, WE: Pt, CE: Ti. Yield of EMH/DEA related to HESA, DH related to IVA (total volume 5 mL). | ||
According to the state of the art, the carboxylates are most probably cleaved via an irreversible 1 e-transfer to the alkyl radical and CO2. However, for saturated alkyl radicals the dimerization takes place after desorption from the electrode. This is indicated by the statistical recombination of the alkyl radicals due to the radical mechanism of Kolbe electrolysis.24 Related to our study, there is a high probability that a radical species of the short-handed substrate couples with a radical of the substrate in excess. Furthermore, self-coupling is more likely for the radicals of the substrate in excess. For the following reactions, a ratio of 1:4 was chosen as technically feasible proportion. Furthermore, at this ratio similar yields for the cross-coupling product of HESA/MMSA with IVA were achieved as found for lower HESA/MMSA-IVA ratios. To assure good conductivity of the reaction solution, an electrolyte is generally added for Kolbe electrolysis. Although often KOH is used as electrolyte for Kolbe coupling reactions, in this report, NEt3 was chosen. NEt3 has the capability to deprotonate the starting material and simultaneously serve as an electrolyte. Contrary to KOH, it is not strong enough to hydrolyse ester functionalities under the applied reaction conditions, thus suppressing the formation of undesired side-products. From an economic and ecological point of view, the amount of added electrolyte should be as small as possible (Fig. 1 and ESI Fig. 1a + 2a†). The obtained results suggest that the cross-coupling of HESA/MMSA with IVA were almost independent of the electrolyte concentration. Surprisingly, large amounts of NEt3 reduce both the yields of EMH/MDH and DEA/DDH, whereas the yield of DH stays nearly constant. This might be due to the lower acidity of IVA in comparison to HESA and MMSA. A lower acidity would require a stronger deprotonating base in order to achieve a fast oxidation and according radical formation.24 These results coincide with the trends described by the group of Harnisch stating that an increase of the reactant concentrations facilitates Kolbe electrolysis rather than adding electrolytes.38 Beside the electrolyte concentration, the composition of the electrolysis solvent has a decisive impact on the success of Kolbe electrolysis. Often methanol is reported as one of the best solvents for Kolbe electrolysis, as high faradaic efficiencies can be achieved. In addition, it also shows notable fuel properties and might be used as a blend component.39 On the other hand, water possesses ecological and economic advantages, as it is non-toxic and cheap.24 In this regard, the cross-coupling reaction was studied in different water:methanol compositions (Fig. 2 and ESI Fig. 3a†). For both substrates (HESA and MMSA), the lowest conversion of IVA and product yields were observed in pure water as a solvent. In addition, due to the low solubility of the organic acids in water, the acid concentration had to be reduced to only one-third. Stang et al. raised the question whether the low solubility of organic acids and the insolubility of alkanes in water might be the key reason for decreased yields.38 In this regard, it cannot be completely excluded that the low yields in comparison with the experiments in solely methanol are caused by the lower concentration of the starting materials or/and by the insolubility of the yielded alkanes and esters. Accordingly, by increasing the volume percentage of methanol in the solvent composition, the yields of EMH, MDH and DH can be increased. For instance, 59% of EHM or 51% of MDH were obtained using pure methanol as solvent. This leads to the conclusion that approximately two to three times more product can be obtained by switching from water to methanol. Following this data, already small amounts of methanol (20 vol%) are sufficient to boost the reaction, as the yield of EMH or MDH was increased by 10 or 20%, respectively.
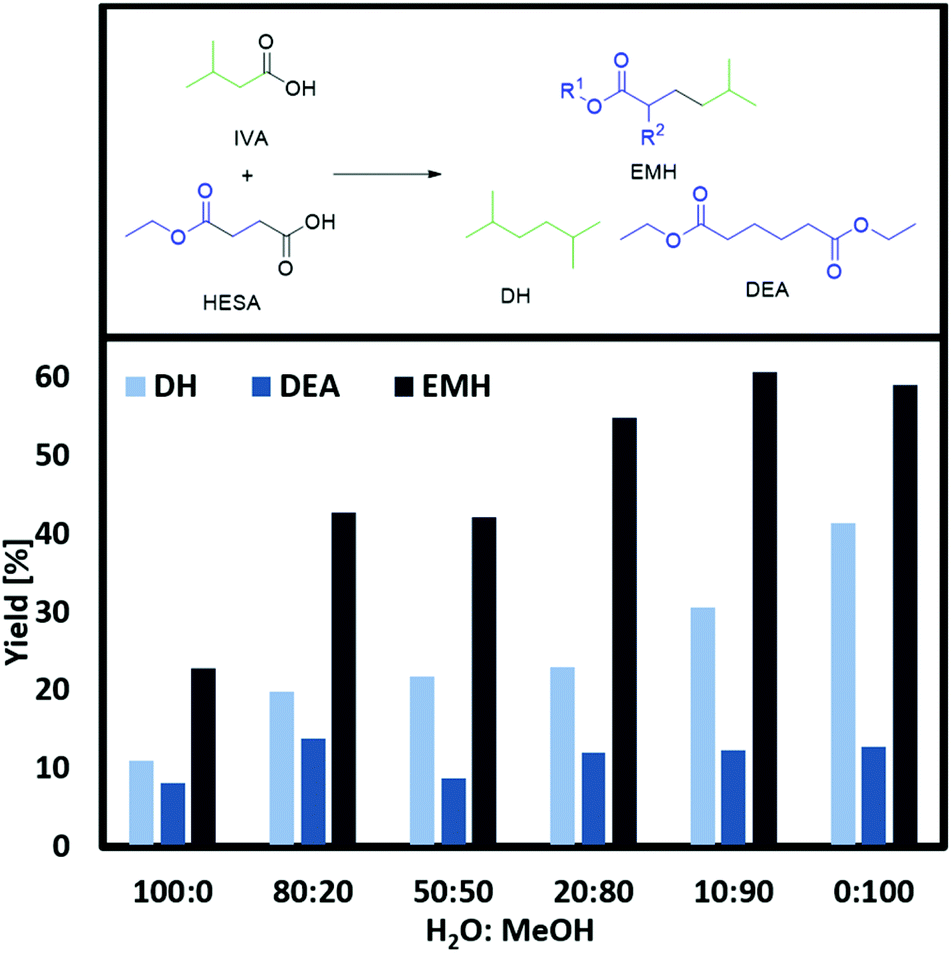 | ||
| Fig. 2 Variation of the solvent mixture in vol%. Conditions: 0 °C, 1 farad equivalent, 100 mA cm−2, WE: Pt, CE: Ti, 0.33 M HESA, 1.3 M IVA, 0.1 M NEt3 (for 100% water: 0.1 M HESA, 0.4 M IVA). Yield of EMH/DEA related to HESA, DH related to IVA (total volume 5 mL). | ||
Raising the percentage of alcohol to a 50:50 mixture did not improve the yield substantially. Even higher methanol ratios (80:20 or 90:10) further increased the yield of EMH or MDH and DH. Less than 10 vol% of water does not affect the formation of the desired product, while even small amounts of water seem to partly inhibit the formation of DH. Schäfer24 and Vijh40 reported that the adsorption of alkoxy or alkyl radicals on the electrode surface is a crucial factor for the suppression of water oxidation and the successful transformation of acids into Kolbe dimers. Thus, the addition of organic acids to a conductive aqueous solution shifts the onset potenial from the water oxidation potential to higher voltages, where the Kolbe decarboxylation takes place. Nevertheless, water oxidation poses a very challenging competitive side reaction and it already starts to lower the current efficiency and overall product yield for water amounts below 5 vol%.24
The solvent mixture does not affect the formation of tert-butanol, EA, EP, MIB and MMA as more or less constant values were found for those compounds. Despite the lower efficiency in aqueous solutions, using water or alcohol/water mixtures as a solvent system remains a promising approach: the coupling reaction of the acids leads to the formation of a water-insoluble organic phase mainly consisting of alkanes and esters. In this manner, addition of at least 50 vol% of water to the reaction solution enables facile isolation of the products by phase separation.
Generally, smooth Pt electrodes are used for the Kolbe electrolysis, as it is a highly active electrode material for the electrochemical oxidative coupling. One major drawback of Pt electrodes is their comparably high price, especially when it comes to larger setups, e.g., in industrial plants.
Recently, we have shown that using readily synthesized (RuxTi1−x)O2 on Ti plates as working electrodes for the self-coupling of HESA into DEA leads to comparable yields of the coupled HESA.30 As the best results were achieved for (Ru1Ti0)O2, (Ru0.75Ti0.25)O2 and (Ru0.5Ti0.5)O2, we tested these promising electrodes also for the cross-coupling of HESA or MMSA with IVA (Fig. 3 and ESI Fig. 4a†).
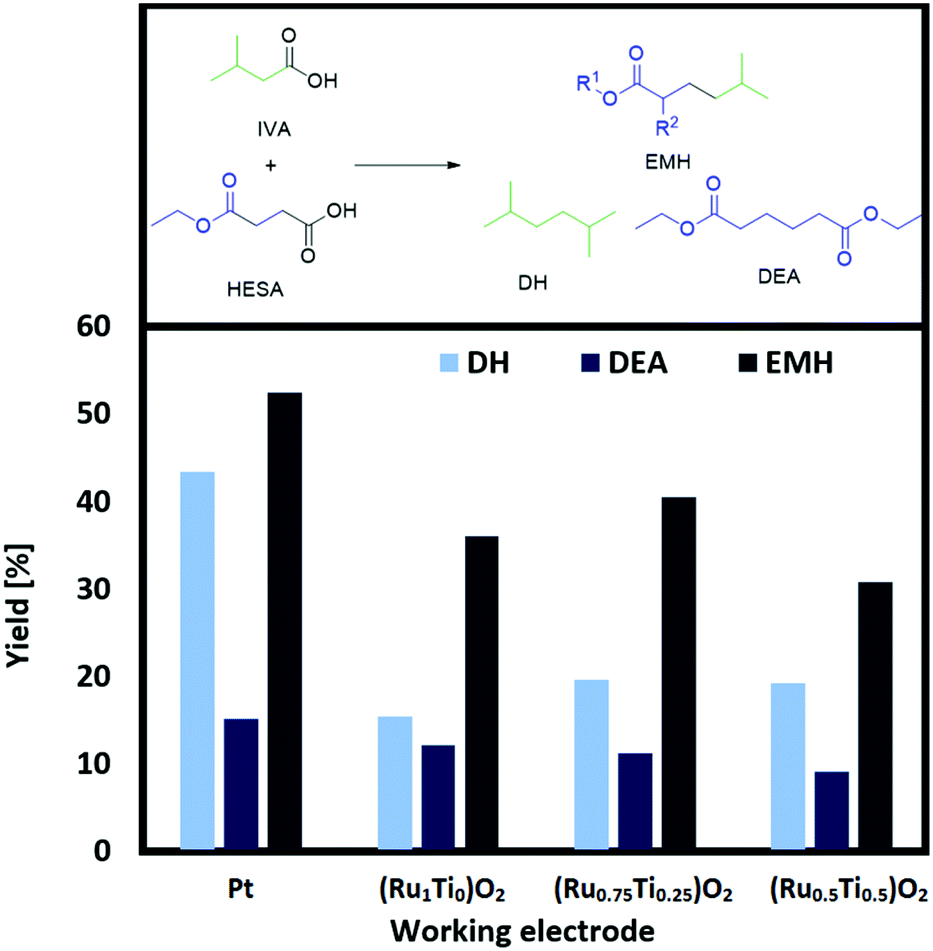 | ||
| Fig. 3 Screening of (RuxTi1−x)O2 on Ti plates. Conditions: 0 °C, 1 farad equivalent, 100 mA cm−2, MeOH as solvent, CE: Ti, 0.33 M HESA, 1.3 M IVA, 0.1 M NEt3. Yield of EMH/DEA related to HESA, DH related to IVA (total volume 2 mL). | ||
Full analytic descriptions of the freshly synthesized electrodes are given in our previous study.30 Although (RuxTi1−x)O2 electrodes of high Ru content also work in aqueous solutions, we chose methanol as a solvent to achieve high yields in the cross-coupling reactions. The obtained results show that ruthenium–titanium dioxide coatings as working electrodes are suitable alternatives to neat Pt. Although efficiency decreases slightly when lowering the Ru content, the results are in the same order of magnitude as Pt. For a full Pt plate a yield of 53% was achieved after 1 farad equivalent, whereas for (Ru0.75Ti0.25)O2 40% were achieved. The plain RuO2 showed lower efficiency compared to (Ru0.75Ti0.25)O2. These results were also observed when using MMSA as the substrate. The trend suggests that the activity of sole RuO2 and (RuxTi1−x)O2 in the rutile structure is not only influenced by RuO2 alone but also by the amount of TiO2 respectively. According to the raw material costs, (RuxTi1−x)O2 electrodes are clearly advantageous. Approximately 500 mg cm−2 Pt was used as bulk electrode while on the other hand only 0.8–1.6 mg cm−2 of Ru was coated on a Ti electrode. The new anode materials undercut the cost of solid Pt electrodes by several orders of magnitude. Additionally, the new electrodes have been used several times for the same reaction and thus can be considered as stable under the applied reaction conditions.
The conversion for HESA lies between 91 and 97%, for MMSA between 89 and 96%. Although there are several identified side products such as ethyl propionate and acrylate or methyl isobutyrate and methacrylate, respectively, the mass balances are not fully closed. In the worst cases, a mass loss of around 45% is found (e.g., for (Ru0.5Ti0.5)O2) whereas the results for Pt had a maximum mass loss of around 25%. The reason for the broad gap between yield and conversion might be due to the evaporation of volatile products (e.g., ethyl acrylate), the over-oxidation into CO2, several unidentified products in lower quantities and low extraction values from the aqueous phase to the organic n-heptane phase. A closed system and an optimized extraction procedure is believed to overcome these challenges.
Production of branched alkanes from MHO
In the second step, the hydrolysed form of ethyl-5-methylhexanoate (EMH) was used as starting material for the synthesis of the final products. One approach consisted of the coupling of IVA and MHO into 2,7-dimethyloctane while the second showed that MHO can also be coupled with itself to form 2,9-dimethyldecan (Scheme 4).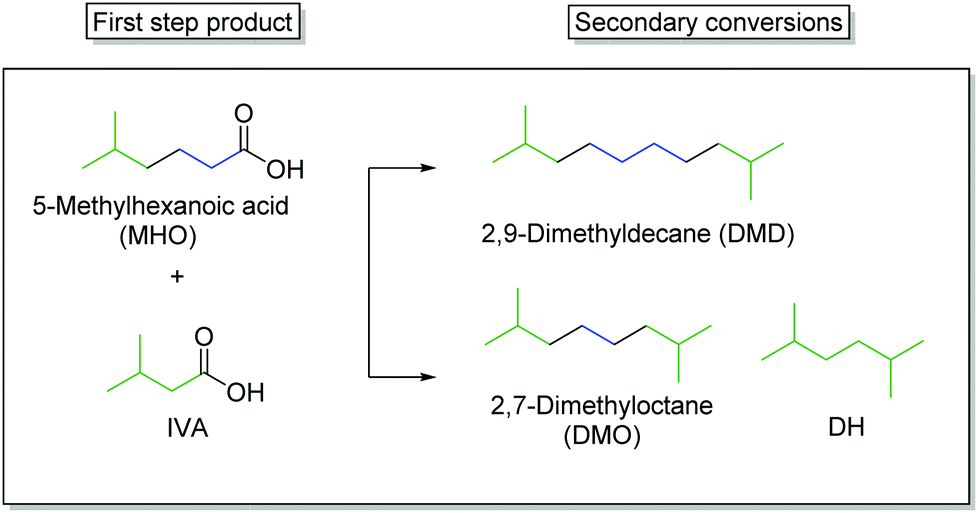 | ||
| Scheme 4 Illustration of the different observed products after the electrochemical oxidation of MHO alone and together with IVA. The coloured molecule parts of the substrates are also depicted in the end-products. | ||
Fig. 4 depicts the results of a systematic screening of (RuxTi1−x)O2 on Ti plates and Pt plates together with NEt3 or KOH as electrolyte.
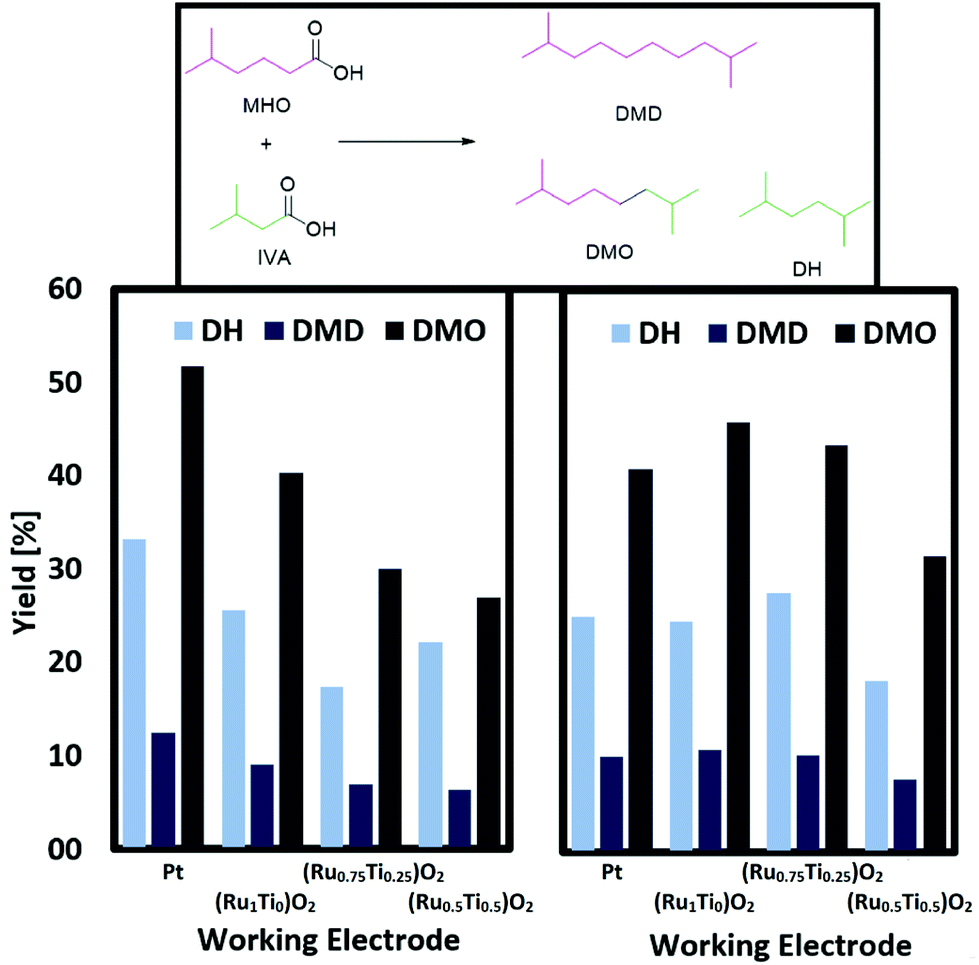 | ||
| Fig. 4 Screening of (RuxTi1−x)O2 on Ti and Pt plates with different electrolytes. Left: Using 0.1 M NEt3 as electrolyte and base. Right: Using 0.1 M KOH as electrolyte and base. General conditions: 0 °C, 1 farad equivalent, 100 mA cm−2, MeOH as solvent, CE: Ti, 0.33 M MHO, 1.3 M IVA. Yield of DMD/DMO related to MHO, DH related to IVA (total volume 2 mL). | ||
The conversion of MHO reaches between 79 and 90%. Since there were no challenges with ester groups, experiments with stronger bases as electrolytes are possible. Judging from the yield obtained when using NEt3 or KOH and a Pt plate as working electrode, the stronger base decreases the activity. The opposite case is observed when (RuxTi1−x)O2 on Ti electrodes are used. The stronger base KOH leads to a general increase in yield for the cross-coupling products DMO, DMD and DH. Reasons for this trend can be found in the low oxygen evolution activity of RuO2. The group of Hu reported that RuO2 coated Ti anodes show a much lower activity for oxygen formation in alkaline solutions while acidic solutions do not seem to suppress the formation of oxygen.41 In addition, it should not be neglected that the pH can have a large influence on adsorption and desorption processes. Hu reported a lower adsorption/desorption peak value in CV-measurements. Transferring these findings to the results reported in this work, undesired side reactions like the formation of over-oxidized products or the oxygen evolution reaction are suppressed while Kolbe electrolysis is favoured leading to higher yields of the cross-coupled products, when strong bases such as KOH are used. In case of Pt instead, the higher pH causes a shift towards non-Kolbe and over-oxidized products.
This assumption becomes more evident studying (RuxTi1−x)O2 on Ti electrodes for Kolbe electrolysis with MHO as single substrate. The yield of DMD can be increased by almost 10% when changing the electrolyte NEt3 (33%) to KOH (41%) and using RuO2 on Ti (Fig. 5). The group of Harnisch reported that they could not observe any effect due to a change in the pH while the group of Bandarenka stated a strong correlation between the pH and the oxidation activity of Pt for hydrogen and oxygen.42
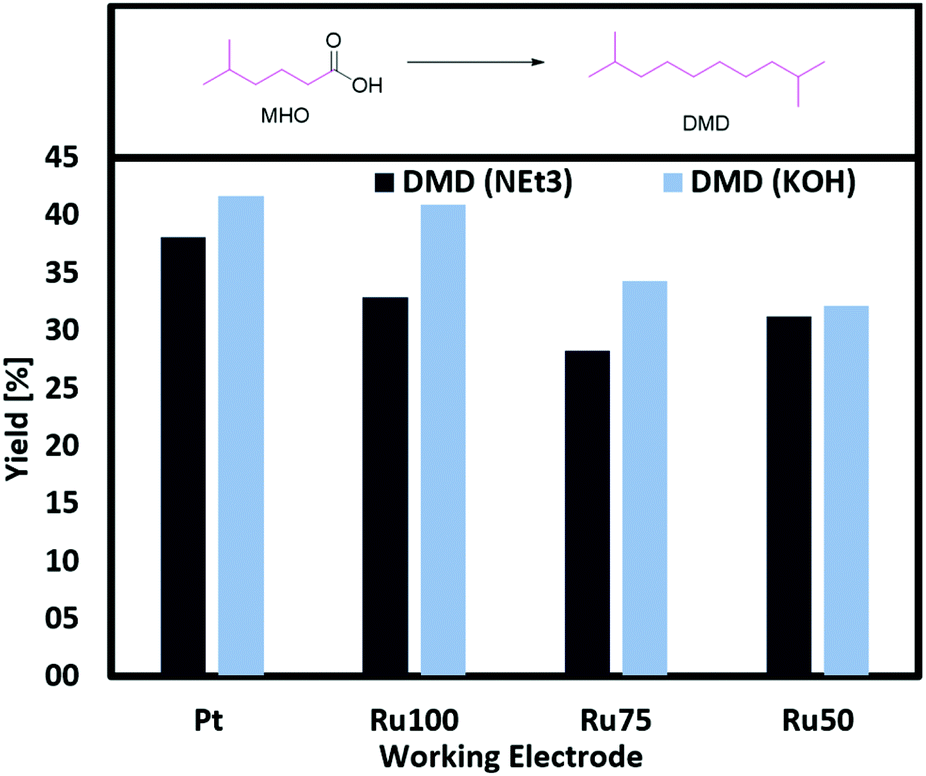 | ||
| Fig. 5 Screening of (RuxTi1−x)O2 on Ti and Pt plates with different electrolytes. General conditions: 0 °C, 1 farad equivalent, 100 mA cm−2, MeOH as solvent, CE: Ti, 1 M MHO (total volume 2 mL). | ||
In our study the obtained results do neither confirm the importance of a high pH for the oxidation activity nor the independence of the activity on the pH.
Fuel assessment
Electrochemical conversion of MMSA and HESA can store electrical energy in a chemical form. From an energetic point of view, the branched esters and alkanes obtained in this study may thus be envisioned as renewable fuel components for use in internal combustion engines. To make a first assessment of their suitability as tailor-made fuels, we consider two properties of paramount importance to smooth engine operation: auto-ignition quality and volatility.43Table 1 compares the normal boiling points of the synthesized compounds to the boiling ranges of typical fossil-based gasoline and diesel fuels. In spark-ignition (SI) engines, low fuel volatility can lead to problems when starting the engine at cold conditions. Moreover, high boiling point components can cause engine oil dilution and excessive deposit formation.44,45 Generally, compression–ignition (CI) engines can burn less volatile fuel, but again, too heavy fuel components can result in cylinder wall impingement and hence oil dilution, particularly in case of low-temperature combustion strategies.46,47 By promoting the evaporation of injected liquid fuel, higher fuel volatility is thought to improve mixture homogeneity and thus to yield lower soot emissions.44 Fuel auto-ignition quality can be used to differentiate between fuels for SI and CI engines.43,48,49 Because in SI engines the ignition event is controlled by the spark plug, the fuel/air mixture has to withstand auto-ignition in the so-called end gas, i.e., those parts of the charge that have not yet been reached by the flame. The phenomenon of undesired auto-ignition in SI engines is referred to as engine knock and severely limits SI engine efficiency.50 On the contrary, the operating principle of the CI engine is based on fuel auto-ignition occurring briefly after injection of the fuel into compressed hot air.| Compound | DCN | RON | T Boil [°C] | Assessment |
|---|---|---|---|---|
| a Normal boiling point was predicted with Joback's method.58 | ||||
| 2,5-Dimethylhexane (DH) | 34.9 | 51.4 | 10851 | |
| tert-Butyl isovalerate (TBI) | 15.9 | 99.3 | 154–15652 | Potential blend component for SI engine fuels (octane improver) |
| Ethyl 5-methylhexanoate (EMH) | 26.7 | 71.2 | 181–18253 | |
| Methyl 2,5-dimethylhexanoate (MDH) | 20.5 | 87.7 | 190a | |
| 2,9-Dimethyldecane (DMD) | 59.6 | −5.0 | 200a | Potential blend components for CI engine fuels |
| 2,7-Dimethyloctane (DMO) | 47.8 | 21.9 | 16054 | |
| 2,5,6,9-Tetramethyldecane (TEMO) | 54.3 | 7.1 | 245a | |
| 2,4,7-Trimethyloctane (TMO) | 44.5 | 29.6 | 177a | |
| Petroleum diesel55,56 | 40–45 | — | 180–360 | |
| Petroleum gasoline55 | — | 91 | 25–210 | |
CI engines therefore require fuel that is prone to auto-ignition. The fuel's readiness to auto-ignite is often expressed as derived cetane number (DCN)59 or research octane number (RON).60 DCN and RON are inversely correlated, i.e., a higher value for DCN means higher auto-ignition tendency, whereas a higher value for RON means higher auto-ignition resistance.49,61 We employed the structural group contribution model proposed by Dahmen and Marquardt48 to predict DCN of the branched alkanes and esters obtained in this study. Furthermore, DCN was converted into RON by applying the correlation formula derived by Perez and Boehman.57 As can be seen from Table 1, TBI stands out with its high predicted RON. This suggests that TBI exhibits high knock-resistance. Due to its relatively high boiling point, we do not expect TBI to be a pure compound fuel, not even in a tailor-made SI combustion system.43,62 However, TBI could be attractive as an octane booster component for blending with fossil or renewable base fuels. Small esters, specifically methyl acetate and ethyl acetate, have been found to effectively increase the RON of gasoline.63 Furthermore, TBI/methanol blends might offer a promising fuel option both from a process and a product perspective. Methanol is already used as a solvent in the synthesis, thus, instead of removing it, it could be left as a blend component. This potentially leads to a more facile product separation and purification thereby improving an overall process. With respect to CI engines, the larger alkanes DMD, DMO, TEMO and TMO constitute promising candidates because they are associated with diesel fuel-like DCNs (>40)49 and volatilities. In fact, branched medium-sized alkanes like DMD, DMO, TEMO and TMO constitute a significant share of fossil-based diesel fuel.64 This structural similarity together with the favourable DCNs and boiling points suggest that DMD, DMO, TEMO and TMO can be blended with conventional fuels in substantial percentages without the need to change existing combustion systems or fuel distribution infrastructures. Finally, predicted DCNs and RONs of DH, EMH and MDH are poorly suited to the requirements of SI and CI engines. Adding these compounds to either gasoline or diesel fuel would deteriorate ignition quality. The rather high boiling points of EMH and MDH also hinder blending with gasoline. From a fuels perspective, the new synthetic approach thus is considered most promising with regard to its ability to yield either small, compact esters like TBI as octane improvers for SI engine fuels or medium-sized alkanes for blending with CI engine fuels. In future studies, an experimental assessment of a wider range of physico-chemical properties of different blends, followed by investigations in combustions engines are in the focus.
Future prospects
Besides the presented branched alkanes, the electrochemical system produces pure CO2 as by-product of Kolbe electrolysis and H2 on the cathode (Fig. 6). In this regard, the discussed Kolbe electrolysis could present an interesting alternative to oxygen evolution usually accompanying the electrochemical hydrogen generation. Also the released CO2 bears a high potential for further applications. For instance, it can be used in methanation processes65 or be recovered in an electrocatalytic reductive CO2 electrolysis.66–68 Particularly interesting is the idea of using H2 and CO2 as gaseous mixture in a molten carbonate fuel cell. Levy et al.69 reported in the report of the Department of Energy (DoE, USA) of 1980 that almost 50% of the necessary energy consumption of Kolbe electrolysis could be regained by applying this technique. In addition, two major advantages of molten fuel cell lie in the flexibility towards the used fuel (natural gas, hydrogen and CO) and that no Pt metal catalyst has to be employed. In this regard, a molten carbonate cell might be used to recover parts of the energy for the Kolbe electrolysis but in case of a lower demand of biofuels the cell can produce electrical energy with alternative fuels.69,70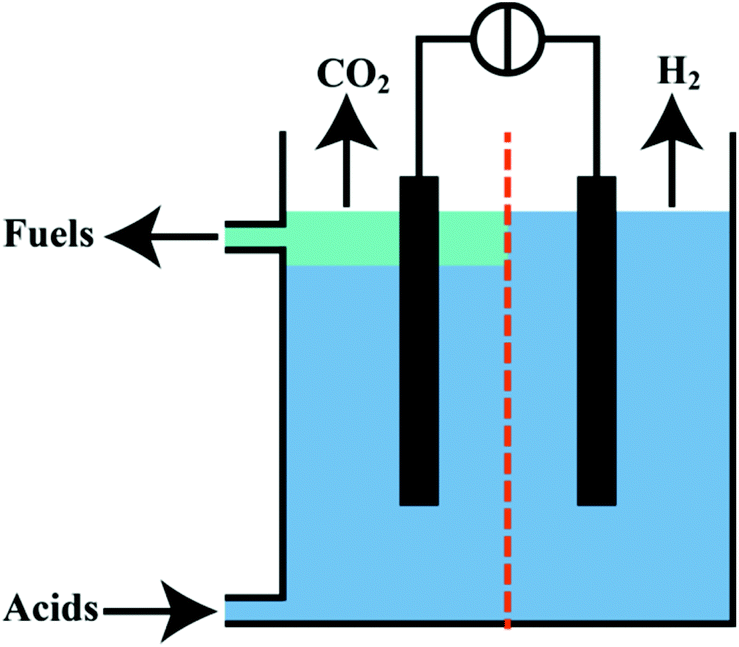 | ||
| Fig. 6 Concept of a continuous cell for production of fuels from organic acids and the separation of H2 and CO2. | ||
Conclusion
We could demonstrate a versatile electrochemical strategy to access tailored fuel compounds from biomass-based carboxylic acids. As case study, cross-coupling of ethyl hydrogen succinate, methyl hydrogen methylsuccinate and methyl hexanoic acid together with isovaleric acid by Kolbe electrolysis allows access to different esters and alkanes. The side products were identified and their potential applications discussed. Furthermore, a first assessment of the obtained products with regard to their use in internal combustion engines has been conducted. This yielded candidates for blending applications targeting both spark-ignition and compression–ignition engines.Notably, the proposed strategy follows the rules of green chemistry. Mild reaction conditions have been applied, only water and methanol were used as solvents and waste was minimized by using a heterogeneous electrode. The reactions conducted in aqueous solutions showed a phase separation of the main products and the reaction mixture. This might facilitate purification processes and enable the reuse of the reaction solvent. In addition, (RuxTi1−x)O2 on Ti electrodes are effective for the anodic decarboxylation and cross-coupling reaction with the promise to lower the content of precious metals from 107 mg cm−2 Pt (50 μm thick electrode) to up to 1.6 mg cm−2 Ru, while Ru in general is the less expensive metal to this date.
This proof of concept study confirmed cross-coupling via Kolbe electrolysis as a promising tool for the development of a completely new sets of tailor made chemicals and fuels. In the concept, we combine renewable carbon feedstocks with renewable energy providing a potential future technology for an electrified biorefinery.
Experimental
Electrochemical setup
In all electrolysis experiments, we used a potentiostat–galvanostat (Metrohm PGSTAT302N) as power supply. It was possible to choose a two electrode configuration in this work, as the focus lied on a constant current instead of an exact measurement of the potential. Throughout the electrolysis the potential was recorded (Metrohm Nova) and adapted in order to keep the current at a constant level. An undivided cell was used. Furthermore, smooth Pt or RuO2/TiO2 on Ti plates were used as working electrode (anode), whereas the counter electrode (CE) (cathode) was made from Ti material. The electrodes were gently washed with water and ethanol after every use. No decrease in activity could be observed after several runs with the same electrode. The electrolysis experiments were conducted in a 5 mL glass vial with an inhouse 3D-printed cover, holding the electrodes in place (see ESI†). All reported current densities are normalized to the geometric surface area of the working electrode (surface = 2 cm2).All chemicals and materials within this work if not stated otherwise were used as purchased.
General reaction procedure
The substrates and the electrolyte (triethylamine (NEt3 or KOH) were dissolved in methanol and/or water. The amount of solvent was adapted to obtain a total volume of 2 or 5 mL. In order to maintain a constant low temperature the electrochemical cell was placed in an ice-bath. A current of 100 mA cm−2 was applied, and a total charge of 1 farad equivalent was passed (calculations see ESI†). After electrolysis, n-heptane (2 mL) was added and the two-phasic system was intensely agitated. After phase separation, the n-heptane phase was analyzed by GC using ethyl heptanoate as internal standard.Metal oxide coated titanium electrodes
Mixed metal oxide on Ti electrodes are prepared following a modified literature procedure.71 The grade 2 Ti sheets (0.5 mm) are cut, sanded with 400 grit paper and rinsed with acetone and water prior to coating. (RuxTi1−x)O2 coatings are prepared for x = 1.00, 0.75, 0.50. Stock solutions of 1.0 M TiCl3 and 1.0 M RuCl3 both in 10% aqueous HCl are prepared, mixed in the appropriate ratio and diluted with isopropanol 2:1 by volume. Precursor solutions are applied to the surface and dried in an oven at 80 °C for 10 minutes prior to calcination at 470 °C for 10 min. This procedure is repeated four times, and the last coating was calcined at the same temperature for 4 h.
Synthesis of MMSA
The MMSA was synthesized following a method of Parker and coworkers with small modifications.72 20 g of itaconic acid dimethyl ester (127 mmol) were dissolved in a mixture of 72 mL formic acid, 8 mL water and 10 mL methanesulfonic acid. The reaction mixture was heated to 100 °C for 10 min and then poured into 200 mL of ice-water, followed by an extraction with ca. 400 mL dichloromethane. The organic phase was dried over MgSO4 and the solvent removed under reduced pressure. The obtained itaconic acid mono-ethyl ester was dissolved in 14 mL methanol and reduced under 60 bar H2 at 60 °C for 90 min in a steel autoclave, using 300 mg of Pd on carbon (5% Pd). After the reduction, the catalyst was removed by filtration and the solvent was evaporated. The crude MMSA was dissolved in water, the pH adjusted to 8 by addition of NaOH and then washed with Et2O. The pH-value of the aqueous phase was then adjusted to 2.5 and the product was extracted with Et2O. The solution was dried over MgSO4 and the solvent removed under reduced pressure to yield in a colorless liquid.1H NMR (400 MHz, (CDCl3)): δ = 3.69 (d, J = 1.7 Hz, 3H), 2.96–2.85 (m, 1H), 2.83–2.41 (m, 2H), 1.23 (dd, J = 7.2, 1.9 Hz, 3H).
Synthesis of HESA
30 g mono-ethyl sodium succinate (178 mmol) were dissolved in 50 mL water and the pH was adjusted to 2 with an aqueous hydrogen chloride solution. The solution was extracted with diethylether and the organic phase was dried over MgSO4, before the solvent was removed under reduced pressure. The product is a clear colorless oil.1H NMR (400 MHz, (CDCl3)): δ = 4.09 (q, J = 7.2 Hz, 2H), 2.62 (m, 2H), 2.55 (m, 2H), 1.20 (t, J = 7.2, 3H).
Synthesis of tert-butyl isovalerate
A solution of IVA (91 mmol, 9.3 g, 1 eq.), tert-butanol (182 mmol, 13.5 g, 2 eq.) and 98% fuming sulfuric acid (1.2 g, 5 wt% of solution) were combined in a microwave reaction vial. The reaction mixture was stirred under microwave irradiation (100 W) at 120 °C for 10 min in a microwave synthesis oven (CEM Discover SP). After the reaction was finished, a phase separation and color change of clear to orange-brown was observable. The upper phase was washed with an aqueous Na2CO3 solution before the pH was adjusted to 8 by adding further Na2CO3 solution. The organic phase was dried over MgSO4 and remaining traces of tert-butanol were removed under reduced pressure to yield in an orange-brown oil. Purity = crude product, determined by NMR.1H NMR (400 MHz, (CD3OD)): δ = 2.10 (d, J = 6.4 Hz, 2H), 2.04 (m, 1H), 1.47 (s, 9H), 0.96 (d, J = 6.5 Hz, 6H). MS (70 eV); m/z (%) = 129 (1) [M+ − CH3, –CH3], 103 (20) [M+ − C2H5], 85 (95), 60 (22), 57 (100), 43 (17), 41 (45).
Isolation of ethyl 5-methylhexanoate (EMH)
1.2 g of HESA (8.3 mmol, 1 eq.) were combined with 3.4 g IVA (33.4 mmol, 4 eq.), 170 mg NEt3 (4% of total acid amount), 3.8 mL methanol and 16.7 mL water. The reaction solution was portioned into five 5 mL electrochemical cells, and the reaction conditions were the following: 0 °C, 200 mA, 2 cm2, 1.0 farad equivalent. After electrolysis, the (upper) product phase was separated from the water methanol solution and submitted to liquid chromatography. As stationary phase silica was used, while the eluent mixture was n-hexane and ethylacetate (9:1). After solvent evaporation a colorless liquid was obtained.
R
f (TLC, n-hexane/ethylacetate 9:1) = 0.5
1H NMR (400 MHz, (CD3OD)): δ = 3.66 (s, 6H), 2.47–2.35 (m, 2H), 1.71–1.57 (m, 2H), 1.48–1.32 (m, 2H), 1.14 (d, J = 7.0 Hz, 6H). MS (70 eV); m/z (%) = 101 (15), 87 (15), 74 (100), 69 (25), 59 (42), 55 (22), 43 (60), 41 (47), 39 (25).
Isolation of dimethyl 2,5-dimethylhexanedioate (DDH)
2.37 g of MMSA (16.2 mmol) were combined with 100 mg NEt3 (6% of amount MMSA), 1.5 mL methanol and 6 mL water. The reaction solution was portioned into two 5 mL electrochemical cells, and the reaction conditions were the following: 0 °C, 200 mA, 2 cm2, 1.5 farad equivalent. After electrolysis, the upper product phase was separated from the water–methanol solution and submitted to liquid chromatography. As stationary phase silica was used, and the eluent mixture was n-hexane and ethylacetate (4:1). After solvent evaporation a colorless liquid was obtained.
R
f (TLC, n-hexane/ethylacetate 2:1) = 0.5
1H NMR (400 MHz, (CD3OD)): δ = 3.66 (s, 6H), 2.47–2.35 (m, 2H), 1.71–1.57 (m, 2H), 1.48–1.32 (m, 2H), 1.14 (d, J = 7.0 Hz, 6H). MS (70 eV); m/z (%) = 171 (5) [M+ − OCH3], 143 (20) [M+ − CHO].
Isolation of methyl 2,5-dimethylhexaneoate (MDH)
0.73 g of MMSA (5 mmol, 1 eq.) were combined with 1.63 g IVA (16 mmol, 4 eq.), 80 mg NEt3 (10% of amount MMSA) and 7.2 mL methanol. The reaction solution was portioned into four 2 mL electrochemical cells, and the reaction conditions were the following: 0 °C, 100 mA, 1 cm2, 1.0 farad equivalent. After electrolysis the mixture was concentrated and dried under high vacuum. The material was purified by distillation (25–150 °C) and the distillate was clear but still contaminated oil.1H NMR (400 MHz, (CD3OD)): δ = 3.66 (s, 3H), 2.42 (m, 1H), 1.63 (m, 1H), 1.40 (m, 2H), 1.14 (d, J = 7.0 Hz, 3H). MS (70 eV); m/z (%) = 143 (1) [M+ − CH3], 127 (3) [M+ − OCH3], 115 (10) [M+ − OCH3, –CH3], 101 (24), 88 (100).
Isolation of 2,9-dimethyldecane (DMD)
1.04 g of MHO (8 mmol, 1 eq.) were combined with 50 mg KOH (10% of amount MHO) and 8 mL methanol. The reaction solution was portioned into two 4 mL electrochemical cells, and the reaction conditions were the following: 0 °C, 200 mA, 2 cm2, 1.0 farad equivalent. After electrolysis, the mixture was concentrated and the organic phase was separated. The alkane was yielded as clear oil.1H NMR (400 MHz, (CD3OD)): δ = 1.58 (non, J = 5.2 Hz, 2H), 1.33 (m, 8H), 1.24 (m, 4H), 0.93 (d, 12H). MS (70 eV); m/z (%) = 127 (3) [M+ − C3H7], 113 (3) [M+ − CH2], 99 (5) [M+ − CH2], 85 (20) [M+ − CH2], 71 (25), 57 (50), 43 (100).
Analytics
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support by the Cluster of Excellence “Tailor-Made Fuels from Biomass” (TMFB; EXC 236) funded by the Excellence Initiative of the German federal and state governments to promote science and research at German universities and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center”. The completion of this project strongly benefited from the discussions and support of Joel Mensah und Stefanie Kerner.Notes and references
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, in Top Value Added Chemicals from Biomass, 2004, p. 76 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- F. J. Holzhauser, J. Artz, S. Palkovits, D. Kreyenschulte, J. Buchs and R. Palkovits, Green Chem., 2017, 19, 2390–2397 RSC.
- R. Luque and J. H. Clark, Catal. Commun., 2010, 11, 928–931 CrossRef CAS.
- M. Sauer, D. Porro, D. Mattanovich and P. Branduardi, Trends Biotechnol., 2008, 26, 100–108 CrossRef CAS PubMed.
- P. Schönicke, R. Shahab, R. Hamann and B. Kamm, in Microorganisms in Biorefineries, ed. B. Kamm, Springer, Berlin, Heidelberg, 2015, pp. 21–49 Search PubMed.
- A. Thierry, M.-B. Maillard and M. Yvon, Appl. Environ. Microbiol., 2002, 68, 608–615 CrossRef CAS PubMed.
- W. de Jong and J. R. van Ommen, Biomass as a Sustainable Energy Source for the Future: Fundamentals of Conversion Processes, Wiley, 2014 Search PubMed.
- D. L. Klass, Biomass for Renewable Energy, Fuels, and Chemicals, Elsevier Science, 1998 Search PubMed.
- T. R. dos Santos, P. Nilges, W. Sauter, F. Harnisch and U. Schröder, RSC Adv., 2015, 5, 26634–26643 RSC.
- P. Nilges, T. R. dos Santos, F. Harnisch and U. Schröder, Energy Environ. Sci., 2012, 5, 5231–5235 RSC.
- P. Nilges and U. Schröder, Energy Environ. Sci., 2013, 6, 2925–2931 RSC.
- T. R. dos Santos, F. Harnisch, P. Nilges and U. Schröder, ChemSusChem, 2015, 8, 886–893 CrossRef CAS PubMed.
- L. Wu, M. Mascal, T. J. Farmer, S. P. Arnaud and M.-A. Wong Chang, ChemSusChem, 2017, 10, 166–170 CrossRef CAS PubMed.
- Y. Zhang, G. Liu and J. Wu, J. Electroanal. Chem., 2018, 822, 73–80 CrossRef CAS.
- H. Hahn, K. Hartmann, L. Bühle and M. Wachendorf, Bioresour. Technol., 2015, 179, 348–358 CrossRef CAS PubMed.
- Y. Qiu, L. Xin, D. J. Chadderdon, J. Qi, C. Liang and W. Li, Green Chem., 2014, 16, 1305–1315 RSC.
- L. Xin, Z. Zhang, J. Qi, D. J. Chadderdon, Y. Qiu, K. M. Warsko and W. Li, ChemSusChem, 2013, 6, 674–686 CrossRef CAS PubMed.
- C. Urban, J. Xu, H. Strauber, T. R. dos Santos Dantas, J. Muhlenberg, C. Hartig, L. T. Angenent and F. Harnisch, Energy Environ. Sci., 2017, 10, 2231–2244 RSC.
- V. N. Andreev, V. A. Grinberg, A. G. Dedov, A. S. Loktev, I. I. Moiseev and A. Y. Tsivadze, Prot. Met. Phys. Chem. Surf., 2013, 49, 32–39 CrossRef CAS.
- H.-J. Schäfer, in Electrochemistry IV, ed. E. Steckhan, Springer Berlin Heidelberg, Berlin, Heidelberg, 1990, pp. 91–151 Search PubMed.
- Z. Yin, Y. Zheng, H. Wang, J. Li, Q. Zhu, Y. Wang, N. Ma, G. Hu, B. He, A. Knop-Gericke, R. Schlögl and D. Ma, ACS Nano, 2017, 11, 12365–12377 CrossRef CAS PubMed.
- Y. Zhang, Y. Qi, Z. Yin, H. Wang, B. He, X. Liang, J. Li and Z. Li, Green Chem., 2018, 20, 3944–3953 RSC.
- J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 2956–2960 RSC.
- M. Faraday, Philos. Trans. R. Soc. London, 1834, 124, 55–76 CrossRef.
- H. Kolbe, Liebigs Ann. Chem., 1849, 69, 257–294 CrossRef.
- G. Creusen, F. J. Holzhäuser, J. Artz, S. Palkovits and R. Palkovits, ACS Sustainable Chem. Eng., 2018, 6, 17108–17113 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CAS.
- Y. Gnanou and M. Fontanille, in Organic and Physical Chemistry of Polymers, John Wiley & Sons, Inc., 2007, pp. 19–48 Search PubMed.
- Y. Gnanou and M. Fontanille, in Organic and Physical Chemistry of Polymers, John Wiley & Sons, Inc., 2007, pp. 49–88 Search PubMed.
- Y. Gnanou and M. Fontanille, in Organic and Physical Chemistry of Polymers, John Wiley & Sons, Inc., 2007, pp. 213–248 Search PubMed.
- K. Frech and J. Tazuma, US Pat, US3822119A, 1974 Search PubMed.
- D. Stoye, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- C. Stang and F. Harnisch, ChemSusChem, 2016, 9, 126–126 CrossRef.
- S. Liu, E. R. Cuty Clemente, T. Hu and Y. Wei, Appl. Therm. Eng., 2007, 27, 1904–1910 CrossRef CAS.
- A. K. Vijh and B. E. Conway, Chem. Rev., 1967, 67, 623–664 CrossRef CAS.
- T. C. Wen and C. C. Hu, J. Electrochem. Soc., 1992, 139, 2158–2163 CrossRef CAS.
- V. Colic, M. D. Pohl, D. Scieszka and A. S. Bandarenka, Catal. Today, 2016, 262, 24–35 CrossRef CAS.
- M. Dahmen and W. Marquardt, Energy Fuels, 2016, 30, 1109–1134 CAS.
- W. De Ojeda, T. Bulicz, X. Han, M. Zheng and F. Cornforth, SAE Int., 2011, 4, 188–201 Search PubMed.
- M. Thewes, M. Muether, A. Brassat, S. Pischinger and A. Sehr, SAE Int. J. Fuels Lubr., 2012, 5, 274–288 CrossRef CAS.
- L. M. Pickett, S. Kook and T. C. Williams, SAE Int., 2009, 2, 785–804 Search PubMed.
- B. T. Fisher, G. Knothe and C. J. Mueller, Energy Fuels, 2010, 24, 5163–5180 CrossRef CAS.
- M. Dahmen and W. Marquardt, Energy Fuels, 2015, 29, 5781–5801 CrossRef CAS.
- G. T. Kalghatgi, Fuel/engine interactions, SAE International, Warrendale, PA, 2014 Search PubMed.
- G. T. Kalghatgi, Int. J. Engine Res., 2014, 15, 383–398 CrossRef CAS.
- D. Ciubotariu, M. Medeleanu, V. Vlaia, T. Olariu, C. Ciubotariu, D. Dragos and S. Corina, Molecules, 2004, 9, 1053 CrossRef CAS PubMed.
- J. B. Cloke and R. M. Wolff, J. Am. Chem. Soc., 1943, 65, 986–987 CrossRef CAS.
- J. J. Ritter and T. J. Kaniecki, J. Org. Chem., 1962, 27, 622–623 CrossRef CAS.
- S. Kyoichi and W. Shoji, Bull. Chem. Soc. Jpn., 1967, 40, 1257–1259 CrossRef.
- H. Bauer, K.-H. Dietsche and T. Jäger, Kraftfahrtechnisches Taschenbuch, 2003 Search PubMed.
- S. K. Hoekman, A. Broch, C. Robbins, E. Ceniceros and M. Natarajan, Renewable Sustainable Energy Rev., 2012, 16, 143–169 CrossRef CAS.
- P. L. Perez and A. L. Boehman, Energy Fuels, 2012, 26, 6106–6117 CrossRef CAS.
- K. G. Joback and R. C. Reid, Chem. Eng. Commun., 1987, 57, 233–243 CrossRef CAS.
- ASTM D 6890 , American Society for Testing and Materials.
- ASTM D 2699 , American Society for Testing and Materials.
- G. T. Kalghatgi, SAE Int., 2005 DOI:10.4271/2005-01-0239.
- F. Hoppe, B. Heuser, M. Thewes, F. Kremer, S. Pischinger, M. Dahmen, M. Hechinger and W. Marquardt, Int. J. Engine Res., 2015, 17, 16–27 CrossRef.
- H. A. Dabbagh, F. Ghobadi, M. R. Ehsani and M. Moradmand, Fuel, 2013, 104, 216–223 CrossRef CAS.
- C. J. Mueller, W. J. Cannella, T. J. Bruno, B. Bunting, H. D. Dettman, J. A. Franz, M. L. Huber, M. Natarajan, W. J. Pitz, M. A. Ratcliff and K. Wright, Energy Fuels, 2012, 26, 3284–3303 CrossRef CAS.
- M. Götz, J. Lefebvre, F. Mörs, A. McDaniel Koch, F. Graf, S. Bajohr, R. Reimert and T. Kolb, Renewable Energy, 2016, 85, 1371–1390 CrossRef.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- Y. Wang, J. Liu, Y. Wang, A. M. Al-Enizi and G. Zheng, Small, 2017, 13, 1–15 CAS.
- S. Hernandez, M. Amin Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326–2346 RSC.
- P. F. Levy, J. E. Sanderson, E. Ashare, D. L. Wise and M. S. Molyneaux, Liquid fuels production from biomass. Final report, Dynatech R/D Co., Cambridge, MA (USA), 1980.
- D. Pletcher, A First Course in Electrode Processes, The Royal Society of Chemistry, 2009 Search PubMed.
- L.-Å. Näslund, C. M. Sánchez-Sánchez, Á. S. Ingason, J. Bäckström, E. Herrero, J. Rosen and S. Holmin, J. Phys. Chem. C, 2013, 117, 6126–6135 CrossRef.
- K. Achiwa, P. A. Chaloner and D. Parker, J. Organomet. Chem., 1981, 218, 249–260 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the products, additional experimental results, calculations, photography's. See DOI: 10.1039/c8gc03745k |
| This journal is © The Royal Society of Chemistry 2019 |