
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-organocatalytic synthesis of acetals from aldehydes†
Nikolaos F.
Nikitas
,
Ierasia
Triandafillidi
and
Christoforos G.
Kokotos
 *
*
Laboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, Athens 15771, Greece. E-mail: ckokotos@chem.uoa.gr
First published on 16th January 2019
Abstract
A mild and green photo-organocatalytic protocol for the highly efficient acetalization of aldehydes has been developed. Utilizing thioxanthenone as the photocatalyst and inexpensive household lamps as the light source, a variety of aromatic and aliphatic aldehydes have been converted into acyclic and cyclic acetals in high yields. The reaction mechanism was extensively studied.
Introduction
The carbonyl moiety is one of the most popular and widely used functional groups in organic synthesis.1 In some cases, its high reactivity can pose a puzzling obstacle that has to be overpowered, in order to achieve the desired transformations. This is usually accomplished via the protection of carbonyl compounds as acetals. Except for this protecting character, acetals can be used as starting materials for new synthetic applications.2 Apart from their synthetic interest, acetals are also employed as flavouring additives and aroma enhancers in cosmetic and food products3 or as anti-freezing additives in biodiesel fuels.4 Due to their versatile use, a variety of methodologies have been reported for the synthesis of acetals. The most common approach includes the use of strong mineral acids,5 transition-metal Lewis acids or organic acids6–8 (Scheme 1A). The use of a stoichiometric amount of acids and/or the use of metals raise serious environmental concerns, clearly violating the principles of green chemistry.9 To address this problem, alternative methods for the acetalization of organic compounds have been developed. Acetals can be produced from carbonyl compounds utilizing basic10 or other non-acidic conditions employing iodine,11 trialkyl orthoformates12 or halohydrins13 (Scheme 1B). Very recently, a protocol for the synthesis of acetals, based on the principles of photochemistry,14 has been developed15 (Scheme 1C).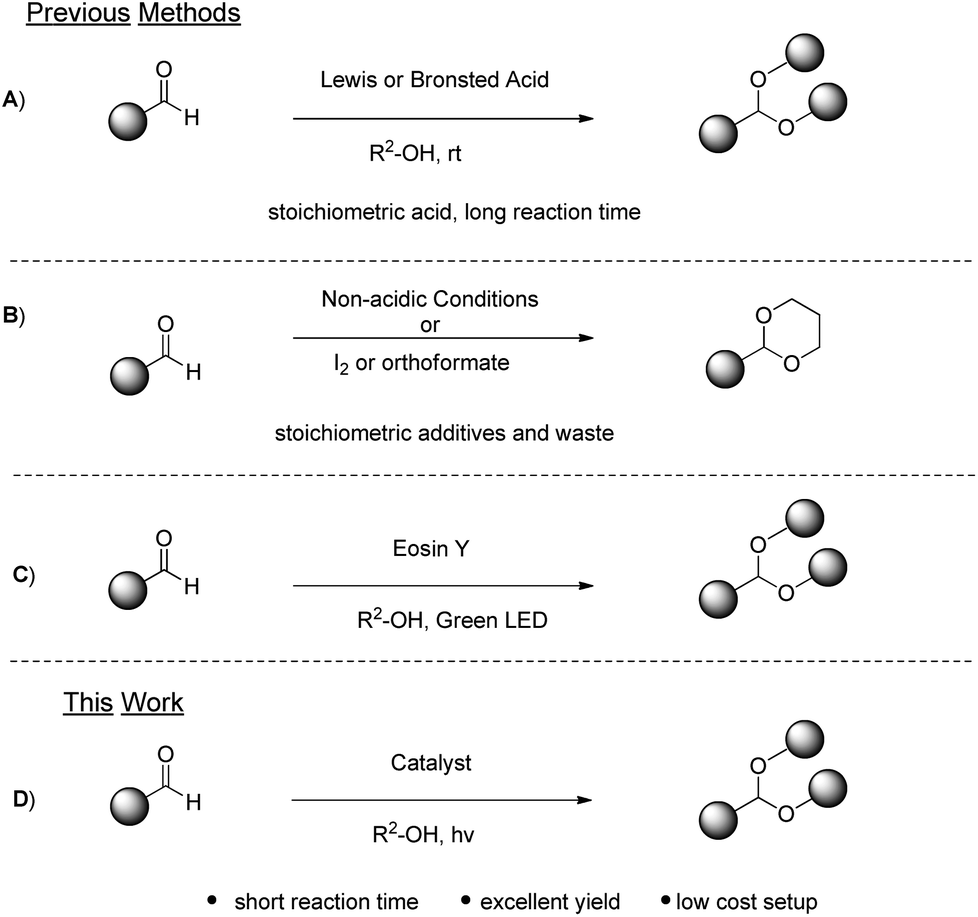 | ||
| Scheme 1 Common synthetic pathways for the acetalization of aldehydes. | ||
The authors proposed the use of an organic dye, Eosin Y, with the use of green LED irradiation for the promotion of the reaction. Eosin Y, an organic dye bearing acidic groups, was employed in that work and the crucial question, that was not addressed, was whether the acidic nature of the catalyst, in conjunction with heat deriving from the LED irradiation, could contribute or be the main reason for product formation, in addition to the photochemical pathway.
Our laboratory has a multiannual experience in photochemistry.16 Utilizing various photocatalysts, a variety of important and useful organic compounds were synthesized, studying exhaustively the mechanism of each photoreaction. Herein, we present a new, green, mild and fast photo-organocatalytic synthesis of acetals from aldehydes, utilizing thioxanthenone as the photo-organocatalyst and inexpensive and commercially-available household lamps as the irradiation source (Scheme 1D). Based on our mechanistic studies and understanding of the reaction mechanism, an inexpensive and organic catalyst is employed and not an organic dye, which requires additional cost for removing it from industrial scales. Also, inexpensive household lamps are employed as the irradiation source and no heating, special apparatus or an inert atmosphere are required. The reaction time is very short and the product can be obtained via solvent evaporation or distillation.
Results and discussion
We began our investigations with the conversion of 3-phenyl propanal 1a (Table 1). A number of photo-organocatalysts were tested with thioxanthenone affording the highest yield (Table 1, entries 1–5). When phenylglyoxylic acid (among the best photo-organocatalysts that we had identified in other studies16) was employed as the photocatalyst, 2a was synthesized quantitatively; however, this was due to the acidic character of phenylglyoxylic acid and not due to the photochemical reaction (Table 1, entries 1 vs. 2). Thus, the acidic character of the photocatalyst must be taken into account, before concluding for its activity. Decreasing the catalyst loading to 10 mol% and the reaction time to 1.5 h, the desired acetal 2a was obtained in 95% yield (Table 1, entry 6).17 It must be noted that the product of the reaction is of enough purity and by just simple evaporation of the excess of methanol, no further purification is required. When the catalyst loading was decreased, the desired acetal 2a was formed in a lower yield (Table 1, entries 7 and 8). If the catalyst or visible light is omitted, no reaction took place (Table 1, entries 9 and 10 vs. 1).17 When the reaction was covered with foil, under the optimized reaction conditions, the desired acetal was formed in a very low yield, verifying that heating from the lamps does not contribute to the reaction outcome (Table 1, entry 11). When a mixture of methanol and acetonitrile (other solvents have been tested as well) was used as a solvent, the yield decreased (Table 1, entry 12).17|
|
||||
|---|---|---|---|---|
| Entry | Catalyst loading (mol%) | Catalyst | Time | Yieldb (%) |
a All reactions were carried out with 1a (0.50 mmol), catalyst (x mol%) and solvent (2 mL) under household bulb irradiation.
b Isolated yield.
c The reaction was performed in the dark.
d The reaction was performed under household bulb irradiation and the reaction tube was covered with foil.
e The reaction took place in a methanol–acetonitrile mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). :1).
|
||||
| 1 | 20 |
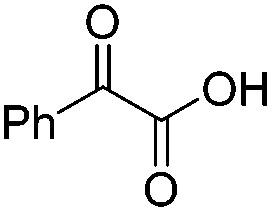
|
18 | 100 |
| 2c | 20 |
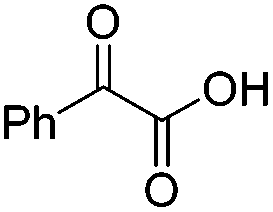
|
18 | 90 |
| 3 | 20 |
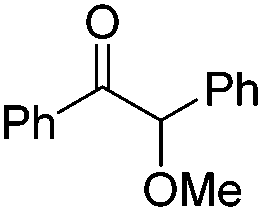
|
18 | 22 |
| 4 | 20 |
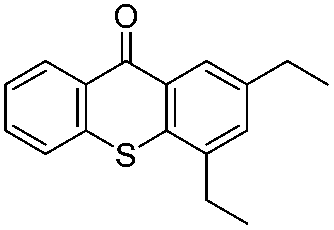
|
18 | 90 |
| 5 | 20 |
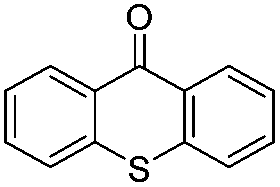
|
18 | 95 |
| 6 | 10 |
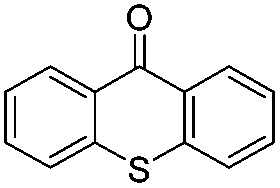
|
1.5 | 95 |
| 7 | 5 |
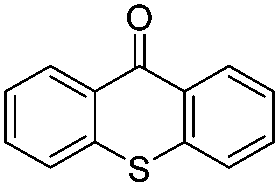
|
18 | 75 |
| 8 | 2 |

|
18 | 62 |
| 9 | — | — | 18 | 5 |
| 10c | 10 |
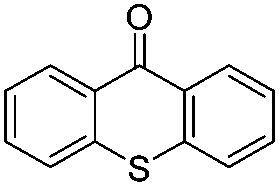
|
18 | 5 |
| 11d | 10 |
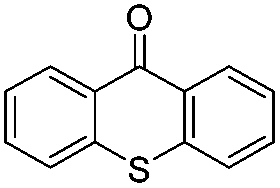
|
18 | 5 |
| 12e | 10 |
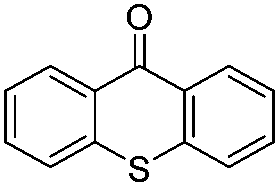
|
1.5 | 65 |
Having in hand the optimum reaction conditions, we turned our attention towards exploring the substrate scope (Schemes 2–5). Utilizing as the starting material 3-phenylpropanal (1a), a variety of alcohols were tested, providing the desired products in very good yields (Scheme 2). Primary alcohols, alcohols with functional groups, secondary alcohols or diols were applied successfully, affording acetals 2a–2l in high yields (Scheme 2). As expected, secondary alcohols provided the products (2c and 2g) in slightly lower yields, due to steric hindrance. Cyclic acetals, like 2h, can be produced by our methodology.
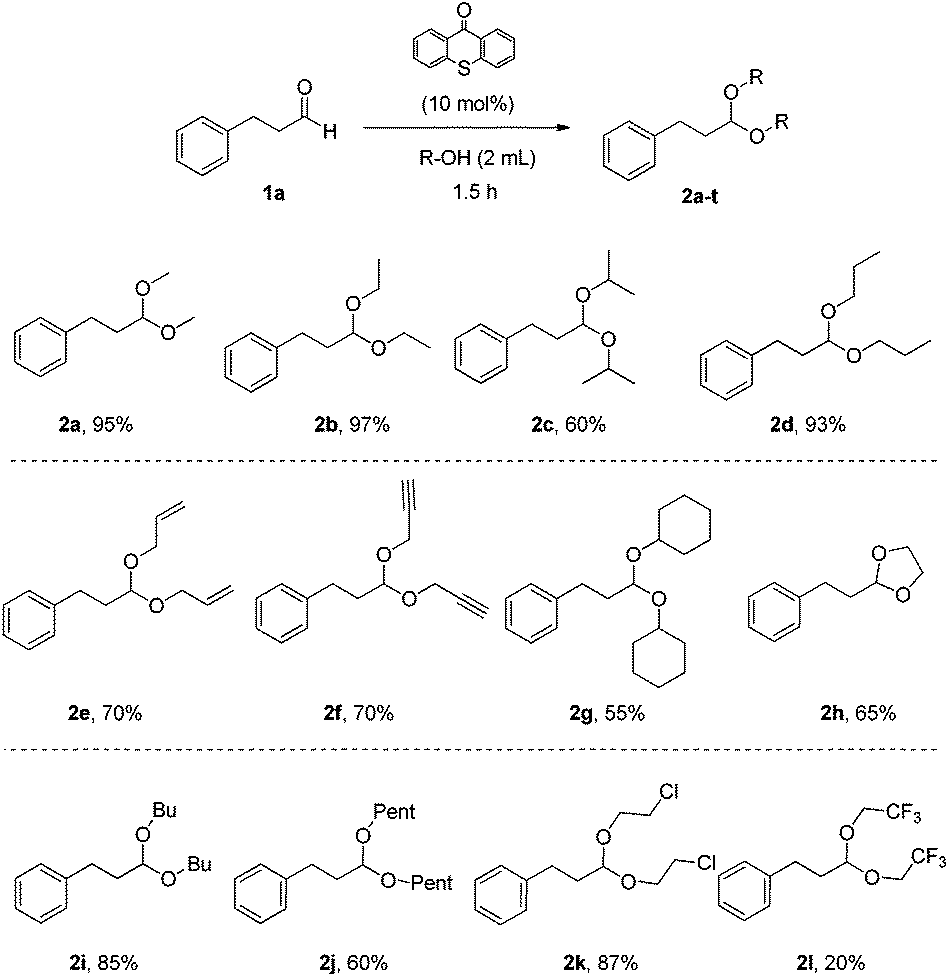 | ||
| Scheme 2 Substrate scope – alcohols. | ||
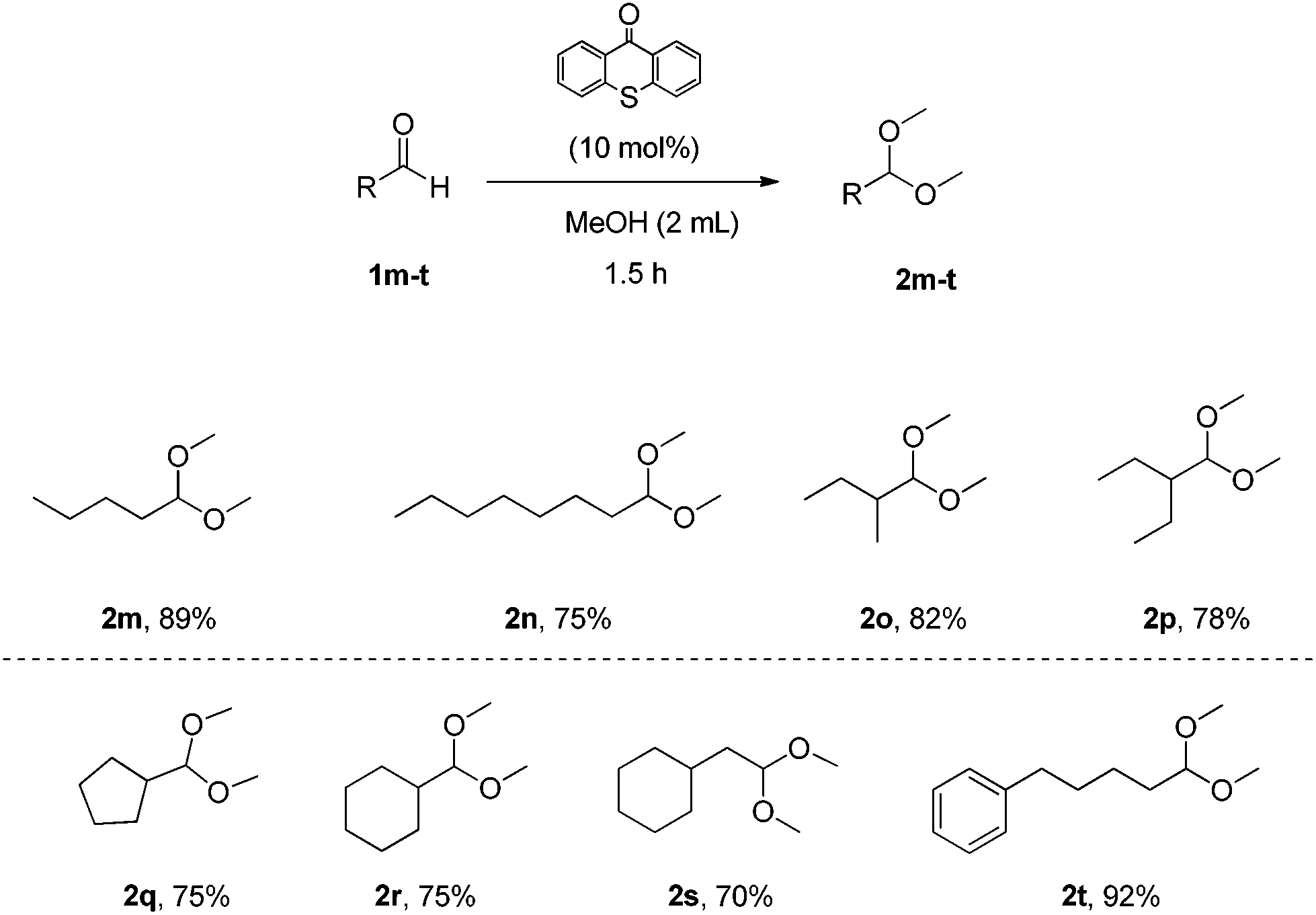 | ||
| Scheme 3 Substrate scope – aliphatic aldehydes. | ||
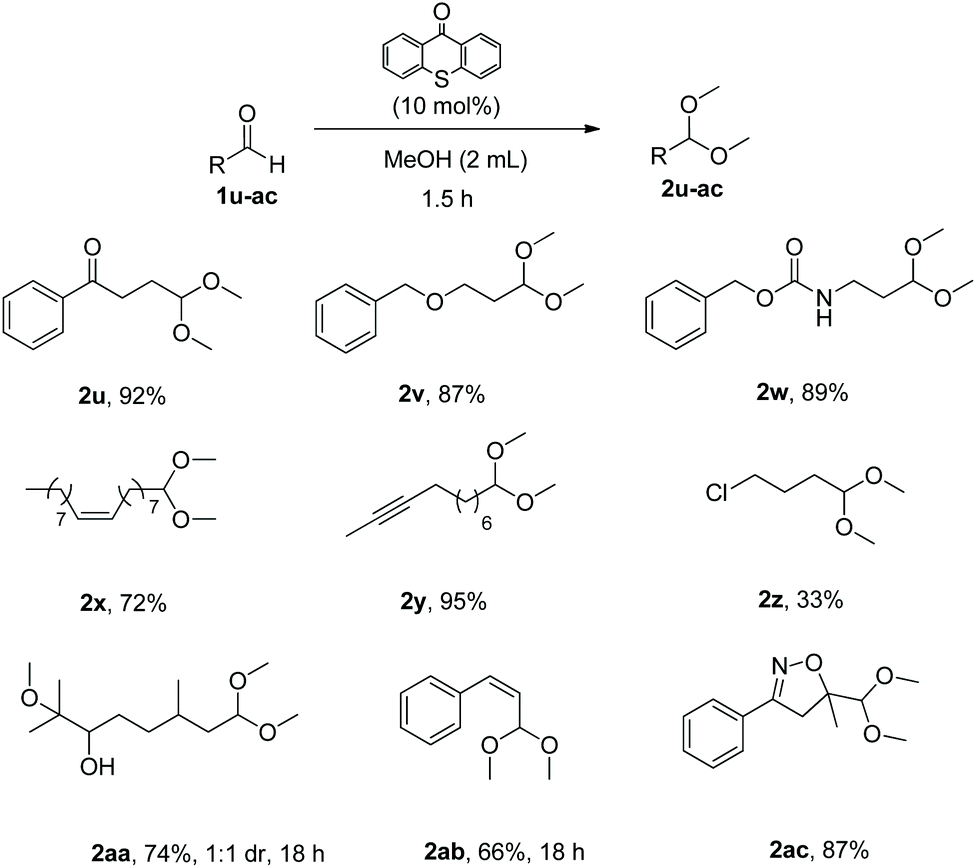 | ||
| Scheme 4 Substrate scope for the synthesis of acetals with functional groups. | ||
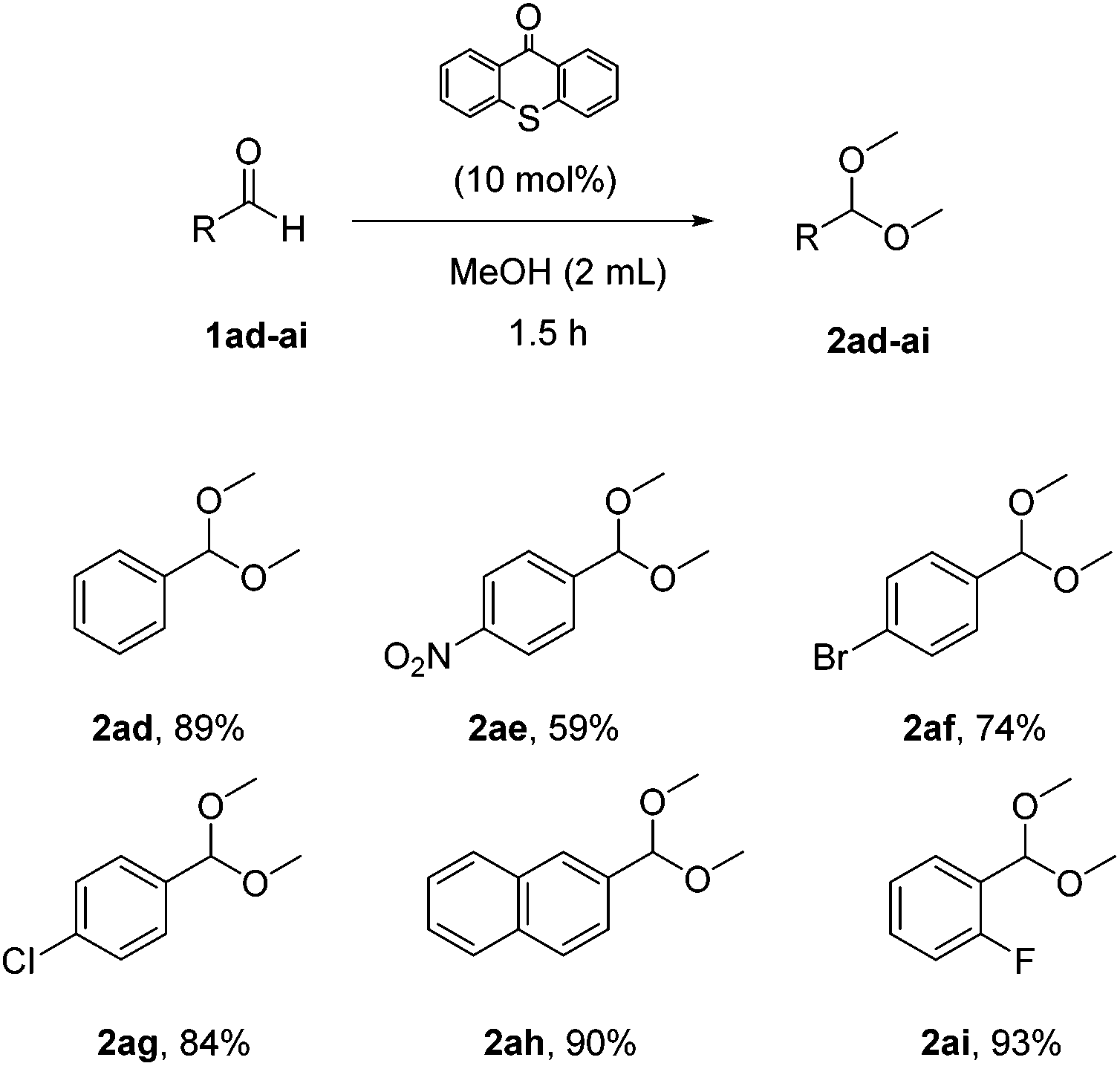 | ||
| Scheme 5 Substrate scope – aromatic aldehydes. | ||
Next, we explored the scope of the aliphatic aldehydes with methanol (Scheme 3). It proved to be also very broad. Linear aliphatic aldehydes or branched aliphatic aldehydes led to the desired acetals 2m–2p in high yields. Aldehydes that contain cyclic or aromatic moieties led to acetals 2q–2t in similarly high yields. In the case of aldehyde 1o, a larger scale experiment (10 mmol) was attempted and the desired product 2o was isolated by distillation (68% yield, bp 2o 132–134 °C, bp 1o 92–94 °C was not observed).
Addition of functional groups on the aldehyde did not affect the efficiency of the synthetic method and acetals 2u–2ac were isolated in up to 95% yield (Scheme 4). It is worth noting that this method is selective, as it converts only aldehydes and ketones are left untouched (2u). Substrates with functional groups, as ether, amide, double or triple bonds and halogen (low yield due to product volatility) gave the desired acetals in very good yields (2v–2z). It is important to mention that when substrate 1aa, bearing an epoxide group, was employed, the epoxide ring was opened under the reaction conditions, leading to acetal 2aa in 74% yield. When cinnamaldehyde was used as the substrate, the reaction conditions led to Z-acetal 2ab. However, upon heating or standing, a ratio of almost 1:1 of the E:Z product was observed. Utilizing as the starting material an isoxazoline skeleton (1ac), the desired acetal 2ac was isolated in 87% yield proving the general character of this method.
In an effort to expand the limits of the synthetic method a variety of aromatic aldehydes were also tested (Scheme 5). Para-, meta- and ortho-substituted aromatic aldehydes were utilized and the corresponding acetals were isolated in very good yields (2ad–2ai).
In order to study the reaction mechanism, a variety of tools were employed. Based on the literature,18 the quantum yield (Φ) of the photocatalytic reaction was calculated: [Φ = 96 (Φ > 1)], which hints that a radical propagation mechanism takes place.
Next, fluorescence quenching studies were performed (Fig. 1). Increasing the added amount of pentanal, a decrease in the fluorescence of thioxanthenone was observed (Fig. 1A), while increasing the added amount of methanol, no changes in the catalyst's fluorescence were seen (Fig. 1B). In an effort to further expand the applicability of this method, we employed benzyl mercaptan as a reagent and the corresponding thioacetal was obtained in 89% yield. Unfortunately, during the preparation of this manuscript, She, Wang and coworkers introduced a blue LED-mediated process.19 In that work, a single electron transfer to the thiol was proposed,19 while in the Eosin-catalyzed process, an energy transfer mechanism was proposed.15
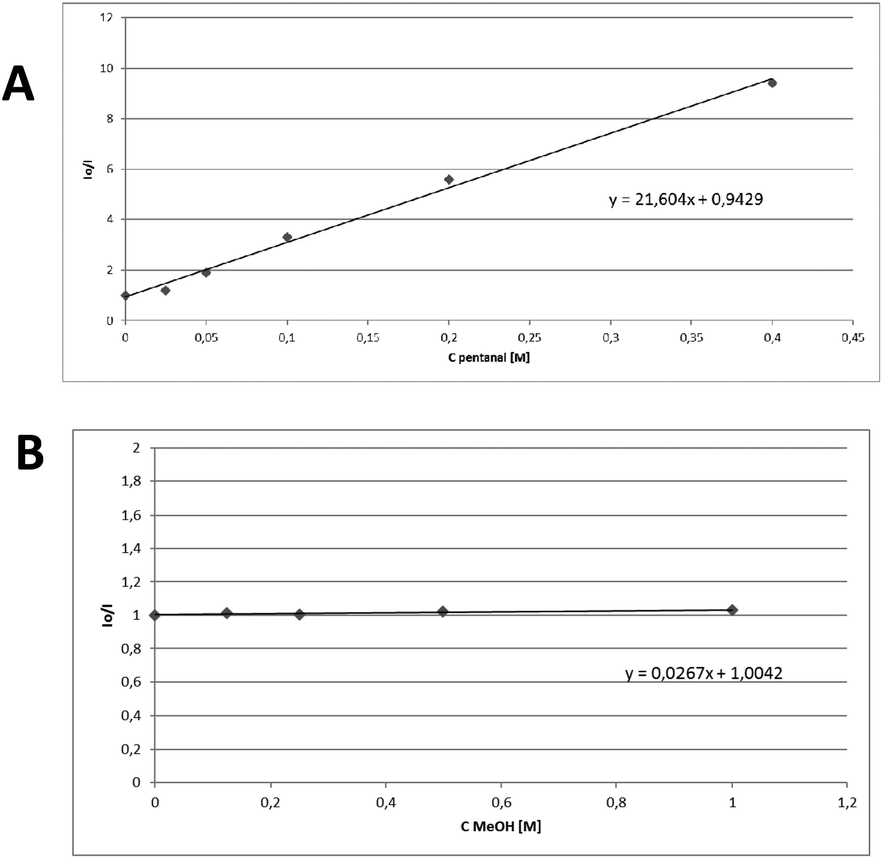 | ||
| Fig. 1 Stern–Volmer plots for the fluorescence quenching of thioxanthenone (1 mM in MeCN) with (A) pentanal and (B) methanol. | ||
Following the literature, a variety of solvents were then studied and the quenching of the catalyst presented similar behaviour, so an energy transfer pathway is hinted.20 This is in accordance with literature evidence that concurs that an electron transfer event is highly unlikely to happen [Eox,thiox = +1.69 V, Ered,thiox = −1.62 V, Eox,PhCHO = +2.71 V, Ered,PhCHO = −1.93 V].21
We then turned our attention towards employing UV-Vis absorbance studies (Fig. 2).17 Based on the UV-Vis spectra, the absorption of thioxanthenone changes, after the addition of aldehyde (Fig. 2). Thus, an EDA-complex is formed. The mixing of two compounds can lead to the formation of an aggregate, called an electron donor–acceptor (EDA) complex. Though these complexes are known in the literature since the 1950s,22 it was not until recently, when Melchiorre and others have identified them as active photochemical species.23 An EDA complex can be recognized by an increase in the UV-Vis absorbance, upon mixing the two reaction components. The indication that an EDA-complex is formed is also supported by 19F-NMR.17 Also,13C-NMR, 1H-NMR and 19F-NMR studies were carried out.17
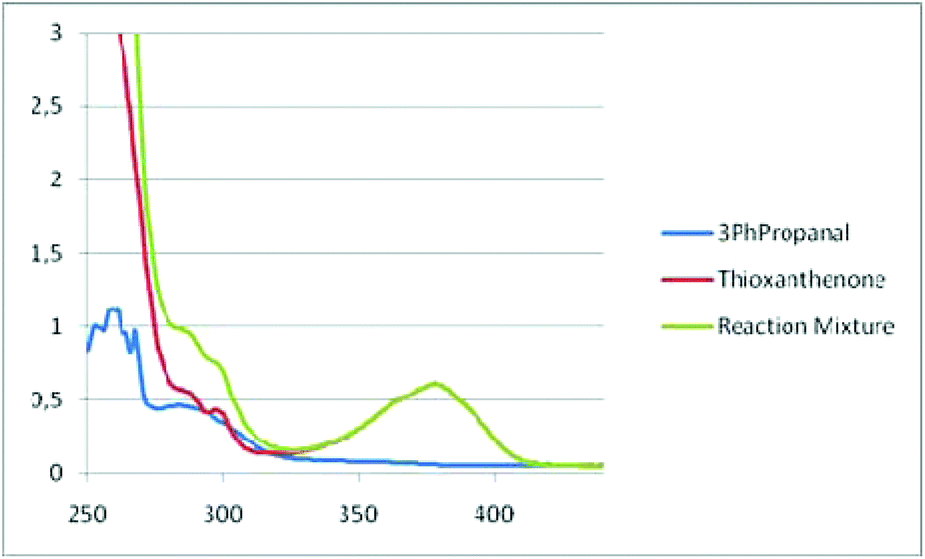 | ||
| Fig. 2 UV-Vis absorption spectra, recorded in a 1 cm path quartz cuvette, 3-phenylpropanal (1a) (3 × 10−4 M), thioxanthenone (2 × 10−5 M) and aldehyde with thioxanthenone in methanol. | ||
The crucial nature of the EDA-complex was highlighted, when a reaction with a cut-off filter (below 400 nm) was employed and the product was not formed.17 Furthermore, some additional control experiments were carried out. The addition of radical scavengers (Tempo, BHT) stopped the reaction, which means that besides the energy transfer, radicals are involved.17 The use of oxygen species quenchers proved that oxygen is involved in the process,17 only to accelerate the process, since the reaction under argon led to 44% yield of the desired acetal.17 Utilizing all these data, the following mechanism can be proposed (Scheme 6). The addition of the aldehyde to the photo-organocatalyst leads to the formation of an EDA-complex, which upon irradiation activates the aldehyde via energy transfer, similar to the activation that a Lewis acid performs, lowering the LUMO of the aldehyde and making it more prone to the upcoming nucleophilic attack of methanol, leading to the corresponding acetal. Then, the catalyst interacts with molecular oxygen and returns to its ground state, while the radical oxygen species, that are generated, lead to the propagation of radicals and ensure product formation.
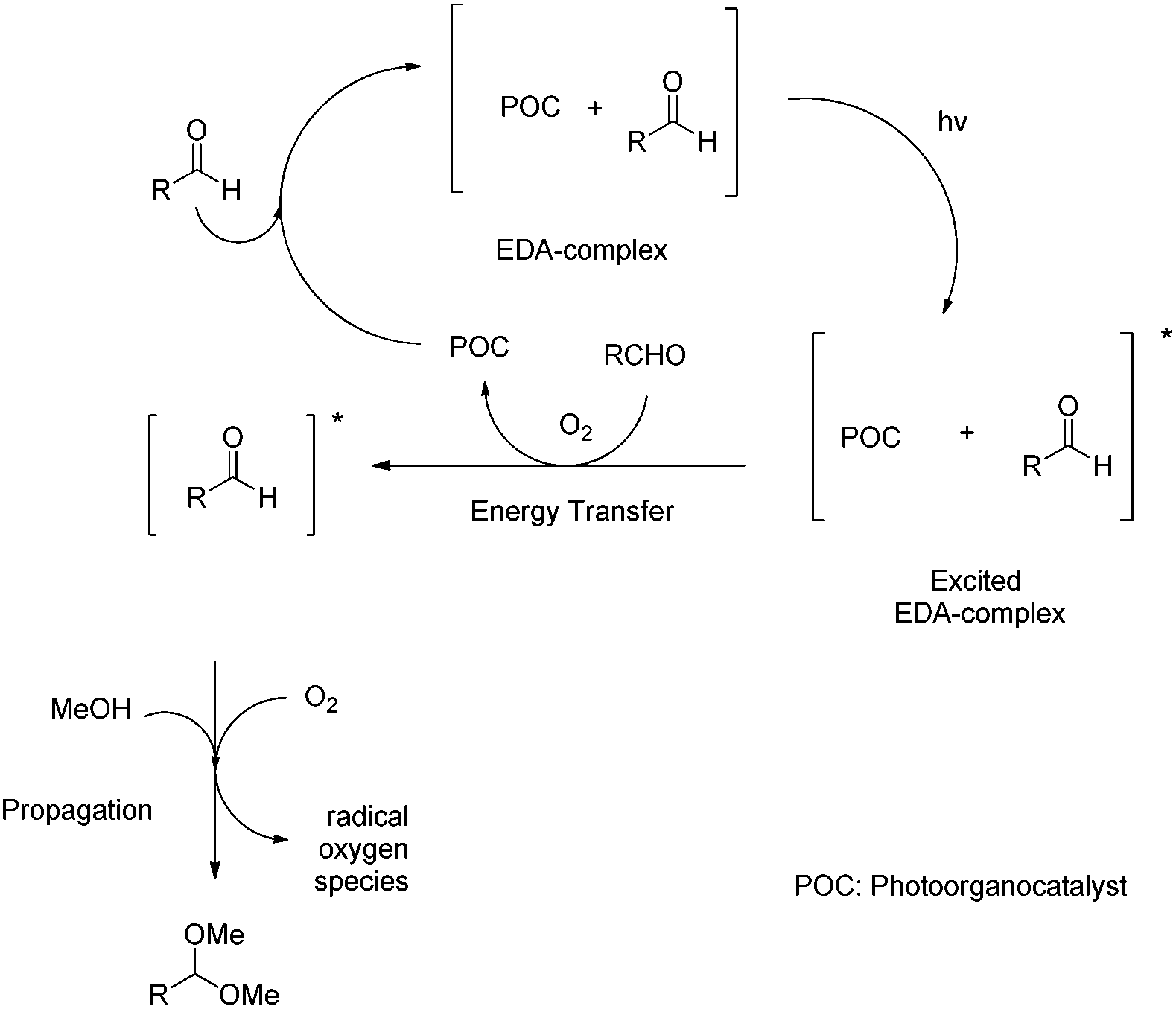 | ||
| Scheme 6 Proposed reaction mechanism. | ||
Conclusions
In conclusion, a simple, inexpensive, green and efficient photo-organocatalytic protocol was developed, activating aldehydes for their reaction with alcohols leading to acetals. Bypassing the need for transition metal-complexes and stoichiometric acids, this method relies on a small organic molecule and inexpensive household lamps activating aldehydes via EDA-complexes, which can react readily with alcohols leading to acetals in high yields, that can be isolated either by solvent evaporation or distillation. Based on extensive mechanistic studies, a proposed mechanism is presented.Experimental
General procedure for the synthesis of acetals
In a glass vial with a screw cap containing thioxanthene-9-one (10.6 mg, 0.05 mmol) in alcohol (2 mL), aldehyde (0.50 mmol) was added. The vial was sealed with a screw cap and left stirring under household bulb irradiation (2 × 80 W household lamps) for 1.5 h. The desired product was isolated either by solvent evaporation, distillation or after purification by column chromatography.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support through the Latsis Foundation programme “EPISTHMONIKES MELETES 2015” (PhotoOrganocatalysis: Development of new environmentally-friendly methods for the synthesis of compounds for the pharmaceutical and chemical industries). The authors would also like to thank Prof. V. Constantinou from the Agricultural University of Athens for access to the fluorescence spectrometer and Dr Maroula Kokotou for her assistance in acquiring HRMS data. I. T. would like to thank the Hellenic Foundation for Research and Innovation (EΛIΔEK) and N. F. N. would like to thank the State Scholarships Foundation (IKY) for financial support. Also, COST Action C–H Activation in Organic Synthesis (CHAOS) CA15106 is acknowledged for helpful discussions.Notes and references
- (a) P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2004, 126, 7450–7451 CrossRef CAS PubMed; (b) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065–1068 CrossRef CAS PubMed; (c) T. Sandmeier, S. Krautwald, H. F. Zipfel and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 14363–14367 CrossRef CAS PubMed; (d) L. R. Malins, J. N. de Gruyter, K. J. Robbins, P. M. Scola, M. D. Eastgate, M. R. Ghadiri and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 5233–5241 CrossRef CAS PubMed.
- For selected examples, see: (a) P. A. Bartlett, W. S. Johnson and J. D. Elliott, J. Am. Chem. Soc., 1983, 105, 2088–2089 CrossRef CAS; (b) A. Mori and H. Yamamoto, J. Org. Chem., 1985, 50, 5444–5446 CrossRef CAS; (c) T. Xu, Z. Yu and L. Wang, Org. Lett., 2009, 11, 2113–2116 CrossRef CAS PubMed; (d) Z.-B. Zhu, Y. Wei and M. Shi, Chem. – Eur. J., 2009, 15, 7543–7548 CrossRef CAS PubMed; (e) I. Suzuki, M. Yasuda and A. Baba, Chem. Commun., 2013, 49, 11620–11622 RSC; (f) Q. Yang, T. Xu and Z. Yu, Org. Lett., 2014, 16, 6310–6313 CrossRef CAS PubMed.
- H. Maarse, Volatile Compounds I Foods and Beverages, Marcel Dekker, Inc., New York, 1991 Search PubMed.
- P. H. R. Silva, V. L. C. Gonçalves and C. J. A. Mota, Bioresour. Technol., 2010, 101, 6225–6229 CrossRef CAS PubMed.
- (a) T. H. Fife and L. K. Jao, J. Org. Chem., 1965, 30, 1492–1495 CrossRef CAS; (b) S. Sugai, T. Kodama, S. Akaboshi and S. Ikegami, Chem. Pharm. Bull., 1984, 32, 99–105 CrossRef CAS; (c) M. Kaufhold and E.-C. Moustafa, Patent, DE 4404515 A4404511 19950817, 1995 Search PubMed; (d) R. A. Ugarte and T. W. Hudnall, Green Chem., 2017, 19, 1990–1998 RSC.
- (a) J.-Y. Qi, J.-X. Ji, C.-H. Yueng, H.-L. Kwong and A. S. C. Chan, Tetrahedron Lett., 2004, 45, 7719–7721 CrossRef CAS; (b) R. Kumar and A. K. Chakraborti, Tetrahedron Lett., 2005, 46, 8319–8323 CrossRef CAS; (c) H. Mansilla and M. M. Afonso, Synth. Commun., 2008, 38, 2607–2618 CrossRef CAS; (d) A. Dutta Chowdhury and G. Kumar Lahiri, Chem. Commun., 2012, 48, 3448–3450 RSC.
- A. Thurkauf, A. E. Jacobson and K. C. Riee, Synthesis, 1988, 233–234 CrossRef CAS.
- B. Procuranti and S. J. Connon, Org. Lett., 2008, 10, 4935–4938 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and practice, University Press, Oxford, New York, 1998 Search PubMed.
- J. Grabowski, J. M. Granda and J. Jurczak, Org. Biomol. Chem., 2018, 16, 3114–3120 RSC.
- (a) M. K. Basu, S. Samajdar, F. F. Becker and B. K. Banik, Synlett, 2002, 319–321 CrossRef CAS; (b) B. Karimi and B. Golshani, Synthesis, 2002, 784–788 CrossRef CAS.
- K. Kumamoto, Y. Ichikawa and H. Kotsuki, Synlett, 2005, 2254–2256 CAS.
- M. Barbasiewicz and M. Mąkosza, Org. Lett., 2006, 8, 3745–3748 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS PubMed; (c) M. D. Kärkäs, J. A. Porco Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (d) K. L. Scubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef PubMed; (e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (f) D. Cambie, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noel, Chem. Rev., 2016, 116, 10276–10341 CrossRef CAS PubMed; (g) J. Schwarz and B. Konig, Green Chem., 2018, 20, 323–361 RSC; (h) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC.
- H. Yi, L. Niu, S. Wang, T. Liu, A. K. Singh and A. Lei, Org. Lett., 2017, 19, 122–125 CrossRef CAS PubMed.
- (a) G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Chem. – Eur. J., 2014, 20, 13811–13814 CrossRef CAS PubMed; (b) G. N. Papadopoulos and C. G. Kokotos, Chem. – Eur. J., 2016, 22, 6964–6967 CrossRef CAS PubMed; (c) G. N. Papadopoulos and C. G. Kokotos, J. Org. Chem., 2016, 81, 7023–7038 CrossRef CAS PubMed; (d) N. Kaplaneris, A. Bisticha, G. N. Papadopoulos, D. Limnios and C. G. Kokotos, Green Chem., 2017, 19, 4451–4456 RSC; (e) D. Limnios and C. G. Kokotos, Adv. Synth. Catal., 2017, 359, 323–328 CrossRef CAS; (f) G. S. Koutoulogenis, M. G. Kokotou, E. Voutyritsa, D. Limnios and C. G. Kokotos, Org. Lett., 2017, 19, 1760–1763 CrossRef CAS PubMed; (g) I. Triandafillidi, M. G. Kokotou and C. G. Kokotos, Org. Lett., 2018, 20, 36–39 CrossRef CAS PubMed; (h) G. N. Papadopoulos, E. Voutyritsa, N. Kaplaneris and C. G. Kokotos, Chem. – Eur. J., 2018, 24, 1726–1731 CrossRef CAS PubMed; (i) E. Voutyritsa, N. F. Nikitas, M. K. Apostolopoulou, A. D. D. Gerogiannopoulou and C. G. Kokotos, Synthesis, 2018, 50, 3395–3401 CrossRef CAS; (j) E. Voutyritsa, I. Triandafillidi and C. G. Kokotos, ChemCatChem, 2018, 10, 2466–2470 CrossRef CAS; (k) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Synlett, 2018, 29, 1324–1328 CrossRef CAS.
- For more information on optimisation studies and mechanistic investigations, see the ESI.†.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- Z. Xing, M. Yang, H. Sun, Z. Wang, P. Chen, L. Liu, X. Wang, X. Xie and X. She, Green Chem., 2018, 20, 5117–5122 RSC.
- (a) F. S. Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (b) M. Teders, C. Henkel, L. Anhauser, F. S. Kalthoff, A. Gomez-Suarez, R. Kleinmans, A. Kahnt, A. Rentmeister, D. Guldi and F. Glorius, Nat. Chem., 2018, 10, 981–988 CrossRef CAS PubMed.
- (a) H. J. Timpe and K. P. Kronfeld, J. Photochem. Photobiol., A, 1989, 46, 253–267 CrossRef CAS; (b) http: //www.cosmologic.de/files/downloads/white-papers/WP_redox_summary.pdf .
- (a) R. S. Mulliken, J. Phys. Chem., 1952, 801–822 CrossRef CAS; (b) I. R. Gould and S. Farid, Acc. Chem. Res., 1996, 522–528 CrossRef CAS; (c) S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young and D. J. Shukla, J. Am. Chem. Soc., 2011, 133, 4791–4801 CrossRef CAS PubMed; (d) S. Farid, J. P. Dinnocenzo, P. B. Merkel, R. H. Young, D. Shukla and G. Guirado, J. Am. Chem. Soc., 2011, 133, 11580–11587 CrossRef CAS PubMed; (e) M. Koch, G. Licari and E. Vaunthey, J. Phys. Chem. B, 2015, 119, 11846–11857 CrossRef CAS PubMed.
- (a) E. Arceo, I. D. Junberg, A. Alvarez and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (b) L. Wozniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678–5681 CrossRef CAS PubMed; (c) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassini and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed; (d) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed; (e) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed; (f) M. L. Spell, K. Devenaux, C. G. Bresnahan, B. L. Bernard, W. Sheffield, R. Kumar and J. R. Ragains, Angew. Chem., Int. Ed., 2016, 55, 6515–6519 CrossRef CAS PubMed; (g) For a review, see: C. G. S. Lima, T. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixao, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, 1H, 13C and 19F NMR data, UV-Vis, fluorescence quenching and mechanistic studies. See DOI: 10.1039/c8gc03605e |
| This journal is © The Royal Society of Chemistry 2019 |