
Thermosetting polyurethanes prepared with the aid of a fully bio-based emulsifier with high bio-content, high solid content, and superior mechanical properties†

Abstract
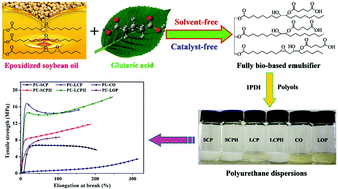
In this paper, a novel, fully bio-based emulsifier has been successfully prepared from epoxidized soybean oil and glutaric acid through a solvent-free and self-catalysis method. The effects of reaction time and carboxyl : epoxy molar ratios on the structures of the emulsifier were systematically investigated. This novel emulsifier exhibits properties similar to those of dimethylolpropionic acid (DMPA) and dimethylolbutanoic acid (DMBA), with hydroxyl groups serving as crosslinking agents and the carboxylic acid group acting as an ionic segment. In order to validate the robustness of this emulsifier, a series of anionic, waterborne polyurethane dispersions were prepared from typical polyols (vegetable oil- and petroleum-based). The structure of this emulsifier and its good compatibility with bio-based polyols confer the resulting dispersions excellent storage stability and high solid content (up to 45%). The prepared waterborne polyurethane films exhibited higher toughness and thermal stability than traditional solvent-based polyurethane films and waterborne polyurethane films prepared from DMPA and DMBA. Moreover, a high bio-based content of up to 74% was achieved for the prepared polyurethanes. This new environmentally friendly, liquid emulsifier, prepared using a solvent-free and self-catalysis synthetic route, can potentially replace typical petroleum-based emulsifiers for the production of waterborne polyurethanes.
 Please wait while we load your content...
Please wait while we load your content...

