
Selective formylation or methylation of amines using carbon dioxide catalysed by a rhodium perimidine-based NHC complex†

Abstract
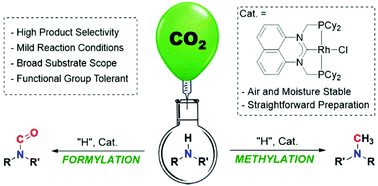
Carbon dioxide can play a vital role as a sustainable feedstock for chemical synthesis. To be viable, the employed protocol should be as mild as possible. Herein we report a methodology to incorporate CO2 into primary, secondary, aromatic or alkyl amines catalysed by a Rh(I) complex bearing a perimidine-based NHC/phosphine pincer ligand. The periminide-based ligand belongs to a class of 6-membered NHC ligand accessed through chelate-assisted double C–H activation. N-Formylation and -methylation of amines were performed using a balloon of CO2, and phenylsilane as the reducing agent. Product selectivity between formylated and methylated products was tuned by changing the solvent, reaction temperature and the quantity of phenylsilane used. Medium to excellent conversions, as well as tolerance to a range of functional groups, were achieved. Stoichiometric reactions with reactants employed in catalysis and time course studies suggested that formylation and methylation reactions of interest begin with hydrosilylation of CO2 followed by reaction with amine substrates.
- This article is part of the themed collection: 2018 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

