
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2-fixation into cyclic and polymeric carbonates: principles and applications
Aeilke J.
Kamphuis
,
Francesco
Picchioni
and
Paolo P.
Pescarmona
 *
*
Chemical Engineering group, Engineering and Technology institute Groningen (ENTEG), Faculty of Science and Engineering, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.p.pescarmona@rug.nl
First published on 2nd January 2019
Abstract
The reaction between carbon dioxide and epoxides is an attractive pathway for CO2-utilisation as it can lead to the formation of two different, yet valuable, products: cyclic and polymeric carbonates. In this review, the advancements made within this field are critically discussed with special attention to the potential of these two classes of compounds as green chemical products. First, an overview is provided of the various types of homogeneous and heterogeneous catalytic systems developed for achieving the reaction of carbon dioxide with epoxides with high activity and selectivity towards either the cyclic or the polymeric carbonate products. Then, the chemical and physical properties of the cyclic and polycarbonate products are discussed, focussing on the correlation between such properties and the potential applications of each class of compounds. Finally, the most relevant applications of these materials, both potential and industrially implemented, are critically reviewed covering the fields of polymer products, energy storage devices, and biomedical and pharmaceutical applications.
 Aeilke J. Kamphuis | Aeilke J. (Arjen) Kamphuis was born in Marum, in the Netherlands, and studied Chemical Engineering at the University of Groningen where he obtained his BSc degree in 2012 and his MSc degree in 2014. In the same year he started his PhD research at the University of Groningen under the supervision of Prof. Dr. Paolo P. Pescarmona and Prof. Dr. Francesco Picchioni. His research interests lie in the development of novel and sustainable chemical products obtained from renewable resources. His PhD project focuses on the catalytic conversion of CO2 into cyclic and polymeric carbonates. |
 Francesco Picchioni | Francesco Picchioni was born in Terni, Italy. In 1996 he received his MSc degree in Chemistry from the University of Pisa, together with a diploma from the Scuola Normale Superiore. In 2000 he obtained his PhD in Polymer Chemistry from the same university. After 3 years as post-doc researcher at the Technical University of Eindhoven, he became Assistant Professor at the University of Groningen, where he was appointed as Full Professor in Chemical Product Technology in 2013. His research interests include thermally reversible networks, polymer processing in supercritical CO2, bio-based polymeric materials and water-soluble polymers for Enhanced Oil Recovery. |
 Paolo P. Pescarmona | Paolo P. Pescarmona was born in Torino, Italy. He received his MSc degree in Chemistry from the University of Torino in 1997 and his PhD from the Delft University of Technology in 2003. From 2005 to 2014 he worked at the University of Leuven, first as post-doc researcher and since 2008 as Assistant Professor. In 2014 he moved to the University of Groningen, where he is currently Associate Professor in Sustainable Chemical Products and Catalysis. His research interests are centred on the rational design and development of catalytic materials, with a special focus on applications in the field of green chemistry. Currently, he is the president of the Dutch Zeolite Association. |
1. Introduction
In current chemical product development, the focus of research lies increasingly on environmental friendliness and overall sustainability. Various ways exist to improve the green character of a chemical product, ranging from the substitution of hazardous components of an existing product with safer and more sustainable alternatives, to the design and development of completely novel green chemical products. Incentives for the development of these chemical products can stem from various origins, such as the ambition to design a safer manufacturing process, to decrease the dependence on depleting fossil resources or to reduce waste generation during the production and disposal stages. Criteria and guidelines for these ambitions are summarised by the ‘Twelve Principles of Green Chemistry’.1 The utilisation of carbon dioxide as a building block for chemical products has the potential to fulfil many of these principles and, accordingly, is receiving growing attention from the academic community, from industry and, more in general, from the whole society. CO2 is a renewable and abundant C1-feedstock and is inexpensive as it is being generated as a waste gas in enormous quantities globally2–4 (the total worldwide CO2 emissions were estimated to be 36.2 billion tonnes in 2015).5 Being the most oxidised form of carbon, it is extremely stable and hence non-toxic and non-flammable.3,4,6 Although chemically rather inert, CO2 can act as a greenhouse gas by absorbing a fraction of the radiation emitted by the Earth and then re-emitting it towards the planet.7 Therefore, the increasing emissions of carbon dioxide in the atmosphere have been identified as one of the main causes of global warming.8–10 Various strategies have been considered in order to counter the accumulation of anthropogenic CO2 in the atmosphere, including improving energy efficiency and minimising heat losses, increasing the usage of fuels with low or no carbon content (e.g. hydrogen) and of renewable energy sources, applying climate engineering approaches (e.g. reforestation and afforestation), carbon capture and storage/sequestration (CCS) and carbon capture and utilisation (CCU).10 The latter refers to processes aimed at capturing CO2 at the location where it is emitted and at reusing it, so that it will not enter the atmosphere. The capture is typically limited to localised industrial emissions with high CO2 concentration, whereas recovering carbon dioxide from, e.g., residential and transportation sources would be technologically, logistically and economically more demanding.10,11 The largest industrial source of CO2 emissions is represented by power plants based on fossil fuel combustion (Table 1). Other major sources for CCU include the cement industry (where CO2 is generated from the conversion of CaCO3 into CaO), the iron and steel industry, the oil and gas industry (where non-marketable gas is flared, and hence is converted into CO2) and ammonia production.5,8 Conceptually, CCU represents a very attractive option in the context of carbon neutrality and of a circular economy.12 However, a number of hurdles need to be overcome before this approach can be implemented. Although technologies are available to capture high amounts of CO2 with an efficiency of over 80% from large CO2 emitters such as power plants and cement factories, full-chain technologies including capture, separation, transport and utilisation are still quite costly.10 Additionally, the final utilisation step represents a relevant challenge because the conversion of CO2 is limited by its high thermodynamic and kinetic stability.13–15 The thermodynamic stability can be overcome by reacting CO2 with high-energy substrates such as hydrogen, amines or epoxides,3,14,16–19 or by providing an energy input in the form of heat,20 electric current21 or radiation;22 whereas the kinetic barrier can be overcome by employing a suitable catalyst for the chosen conversion reaction. The conversion of carbon dioxide into fuels is a very attractive option, in view of the large-scale applicability and because it would allow reconverting CO2 into the same kind of compounds from which it was generated, leading to a circular carbon economy and thus helping to prevent the accumulation of anthropogenic CO2 in the atmosphere. The possible products of CO2 reduction include, in increasing order of reduction degree, CO, formic acid, formaldehyde, methanol and higher alcohols, methane and higher hydrocarbons.23,24 All these products are industrially relevant, but only those with higher degree of reduction can be employed as fuels. The reduction of CO2 can be carried out either through direct reaction with H2 or through an electrochemical or photochemical route:• The reaction with H2 has been widely studied and suitable catalysts for this reaction have been developed.25–27 However, this route is not yet sustainable because the H2 needed for this reaction is currently produced from a fossil resource as CH4 through the steam reforming and water–gas shift reactions,28 whereas efficient and cost-competitive photochemical and/or electrochemical technologies for the production of H2 through H2O splitting are not yet mature.29–31
• The direct routes based on the electrochemical or photochemical reduction of CO2 with concomitant oxidation of H2O are very attractive from the point of view of sustainability as they can rely on a renewable form of energy (e.g. electric power from wind turbines or solar radiation), but they are still facing important challenges in terms of activity in the conversion of CO2 and product selectivity,32,33 particularly if the target product requires the exchange of several electrons (e.g. 6 e− for the reduction of CO2 to CH3OH; 8 e− for the reduction to CH4).
| Source of CO2 emission | Amount of emitted CO2 (Mtonnes per year) |
|---|---|
| a Such as commercial/institutional activities, agriculture/forestry, fishing and residential. b Data from 2010. | |
| Fuel combustion | 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 381 381 |
| Power and heat production |
13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 625 625
|
| Fuel used in the extraction and refining of fossil feedstock | 1683 |
| Manufacturing industries and construction | 6230 |
| Transport and other sourcesa |
10843
|
| Fugitive emissionsb | 483 |
| Industrial processesb | 2468 |
Alternatively to its conversion into fuels, CO2 can be used as feedstock for the production of polymers and fine chemicals.34 Examples include the reaction of CO2 with epoxides leading to either cyclic or polymeric carbonates;13,23,24,35 the formation of dimethyl carbonate via transesterification of CO2-derived ethylene carbonate or propylene carbonate with methanol, or via the direct reaction of carbon dioxide with methanol;23,35 the reaction of CO2 with glycerol to produce glycerol carbonate;23,35,36 carboxylation of ethylene, phenol salts and aryl and alkyl halides to produce, respectively, acrylic acid, salicylic acid and a variety of aryl and alkyl carboxylic acids;13,23,24,35 and the reaction of CO2 with ammonia to generate urea.23,35 The market for these CO2-based chemicals is considerable (see Table 2 for the estimated market of selected products). Therefore, the utilisation of carbon dioxide as substitute for conventional petroleum-based building blocks can result in a significant decrease in dependence on non-renewable fossil feedstock needed for product manufacturing. While this is a target of large societal and industrial relevance, it is important to realise that CO2 fixation into chemical products, such as those reported in Table 2, will most likely not have a major impact on mitigating the concentration of CO2 present in the atmosphere, as the amount of carbon dioxide that would be converted yearly is only a small fraction of the anthropogenic CO2 that is annually emitted into the atmosphere.2,23
| Chemical | Examples of applications | Estimated market size | |
|---|---|---|---|
| a Note that the synthesis method employing CO2 is not necessarily the (only) current route to produce these chemicals. | |||
| Cyclic carbonates |
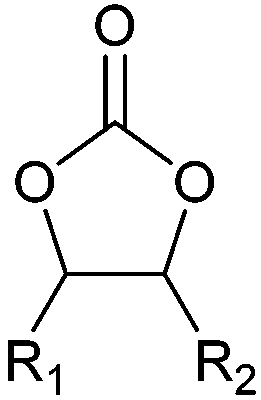
|
Battery electrolyte, reactive intermediate, solvent | 100 kt y−1 (in 2013) |
| Bisphenol-A-based polycarbonates |
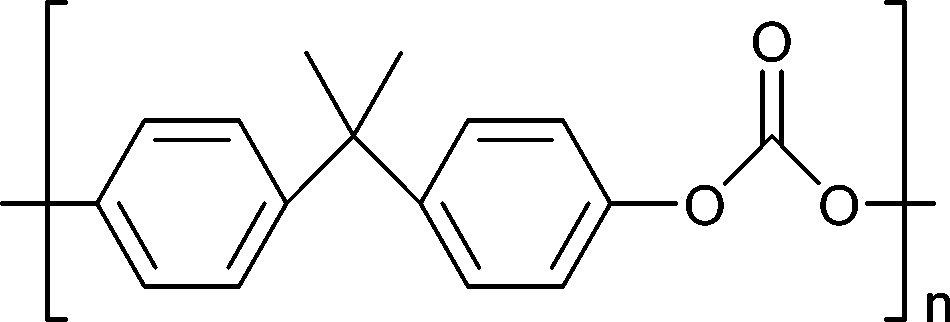
|
Car manufacturing, CDs and DVDs, optical lenses, construction | 5–6 Mt y−1 (in 2016) |
| Dimethyl carbonate |
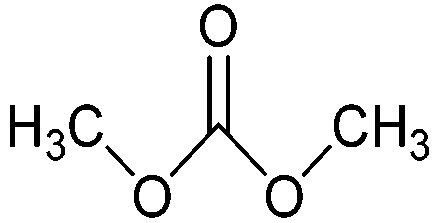
|
Reagent, solvent, battery electrolyte, gasoline additive | ≥200 kt y−1 (in 2013) |
| Acrylic acid |
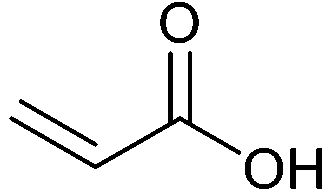
|
Monomer for polymers used as e.g. superabsorbents, detergents, adhesives and coatings | 2.5 Mt y−1 (in 2014) |
| Urea |
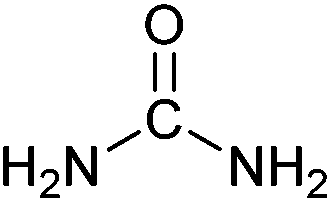
|
Liquid fertiliser | 240 Mt y−1 (in 2016) |
Among the above-mentioned routes for CO2 fixation into valuable chemical products, the 100% atom efficient reaction of CO2 with epoxides is an attractive CO2-valorisation pathway that has gained increasing attention during the past few decades.3 This review focuses on this route, by providing an overview of the progress made in the catalytic synthesis of cyclic and polymeric carbonates and in their applications. The major classes of catalysts that have been developed to enable the reaction of carbon dioxide with epoxides are reviewed in section 2. The physicochemical properties of the cyclic and polymeric carbonates directly derived from CO2 and their correlation with the potential applications of these compounds are discussed in the first part of section 3. The rest of this section provides a critical review of the existing and potential applications of these carbonate products, with special focus on polymeric products (e.g. coatings, adhesives and foams), electrolytes in energy storage devices and biomedical applications.
2. Catalysts for the fixation of CO2 into cyclic and polymeric carbonates
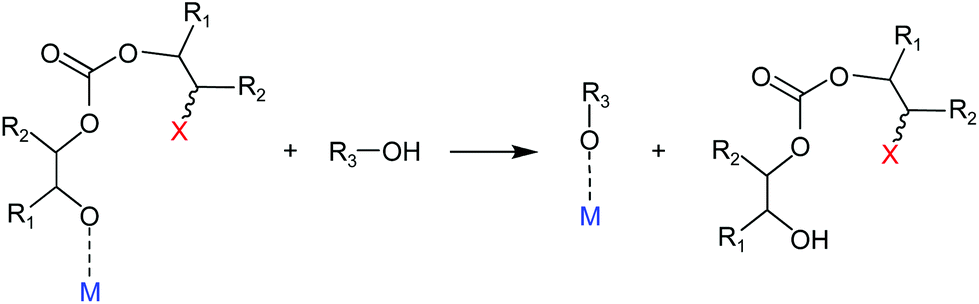
The catalysed reaction of CO2 with an epoxide producing a polymer containing carbonate linkages was reported for the first time in 1969.37 Since then, growing research efforts have been dedicated to the development of catalytic systems for this reaction, as well as towards the employment of a variety of different epoxide substrates. These include petroleum-based epoxides such as ethylene oxide, propylene oxide and cyclohexene oxide as well as (potentially) bio-based epoxides such as limonene oxide and vinylcyclohexene oxide. The reaction of an epoxide with CO2 can in principle yield two possible products: a five-membered cyclic carbonate and/or a polymeric carbonate (Fig. 1). The catalytic systems that are used to promote the conversion of carbon dioxide and epoxides into these organic carbonates generally consist of a Lewis base acting as a nucleophile, often assisted by a Lewis acid species in the form of one or more metal centres. The most commonly proposed reaction mechanism is depicted in Fig. 2.14,17,19 The first step of the catalytic cycle is the activation of the epoxide ring by the Lewis acid (1 in Fig. 2), which promotes the nucleophilic attack by the Lewis base leading to the opening of the epoxide ring. The formed alkoxide intermediate (2) can in turn act as a nucleophile and attack CO2 to form a carbonate intermediate (3). The following step is either a ring closure (4a) to produce a cyclic carbonate (5a), or the alternating insertion of more epoxide and CO2 molecules (4b and 5b) to yield a polycarbonate (6b).14,17,19 The consecutive insertion of two epoxide molecules in the polymer chain may also occur, leading to the presence of ether linkages in the polycarbonate. Besides this, other reaction mechanisms have been proposed, in some cases specific to a type of catalyst (e.g. β-diiminate zinc complexes – see Fig. 3).39,40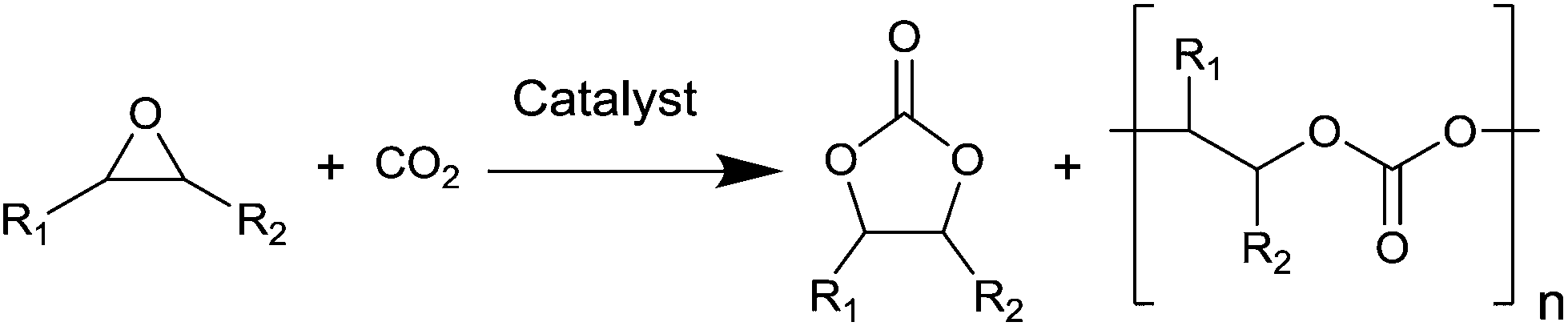 | ||
| Fig. 1 General scheme of the reaction of an epoxide with CO2. | ||
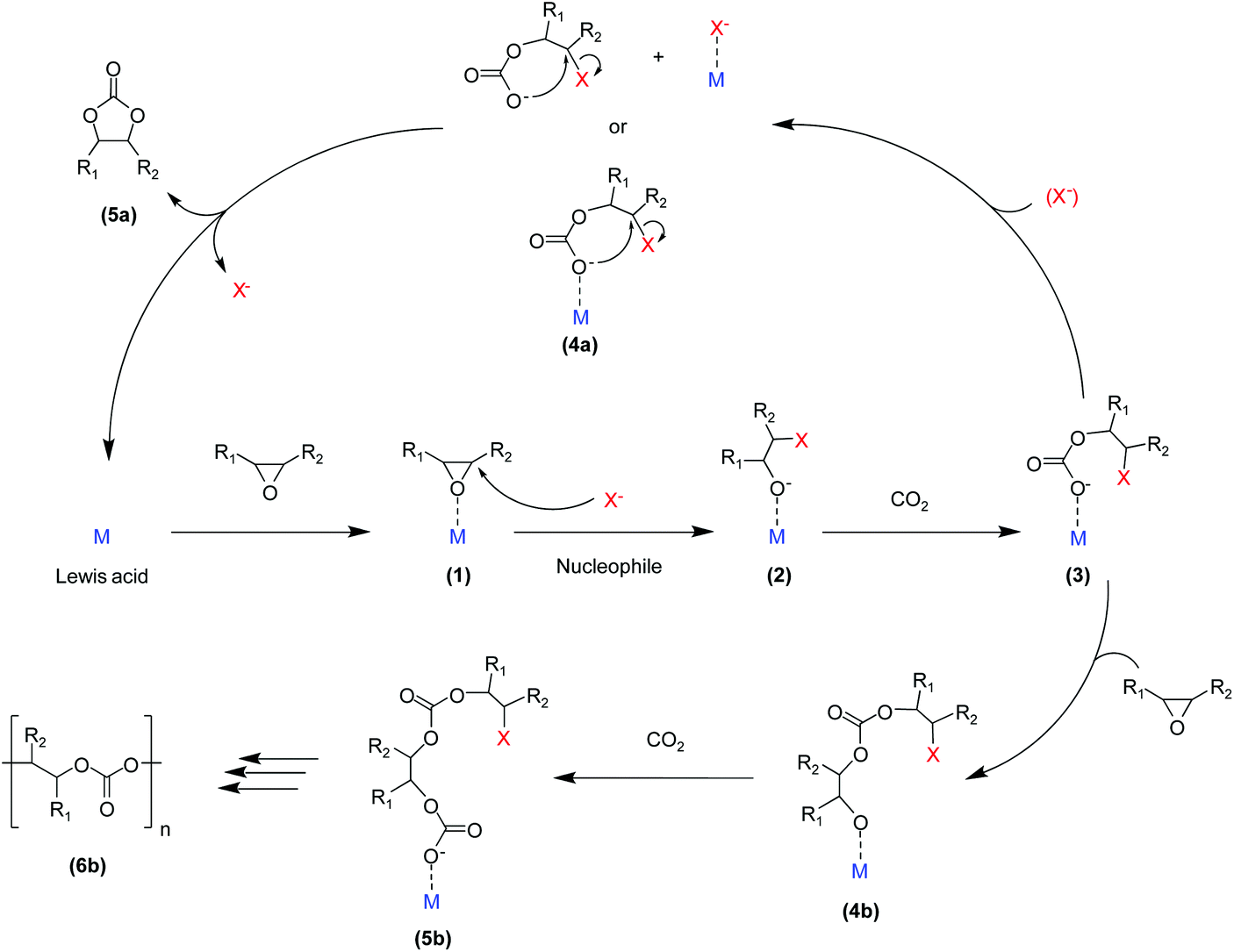 | ||
| Fig. 2 Proposed reaction mechanism for the reaction of CO2 with epoxides yielding either cyclic or polymeric carbonates, in the presence of a Lewis acid (typically a metal centre, M) that activates the epoxide, and a Lewis base (X−) acting as nucleophile. | ||
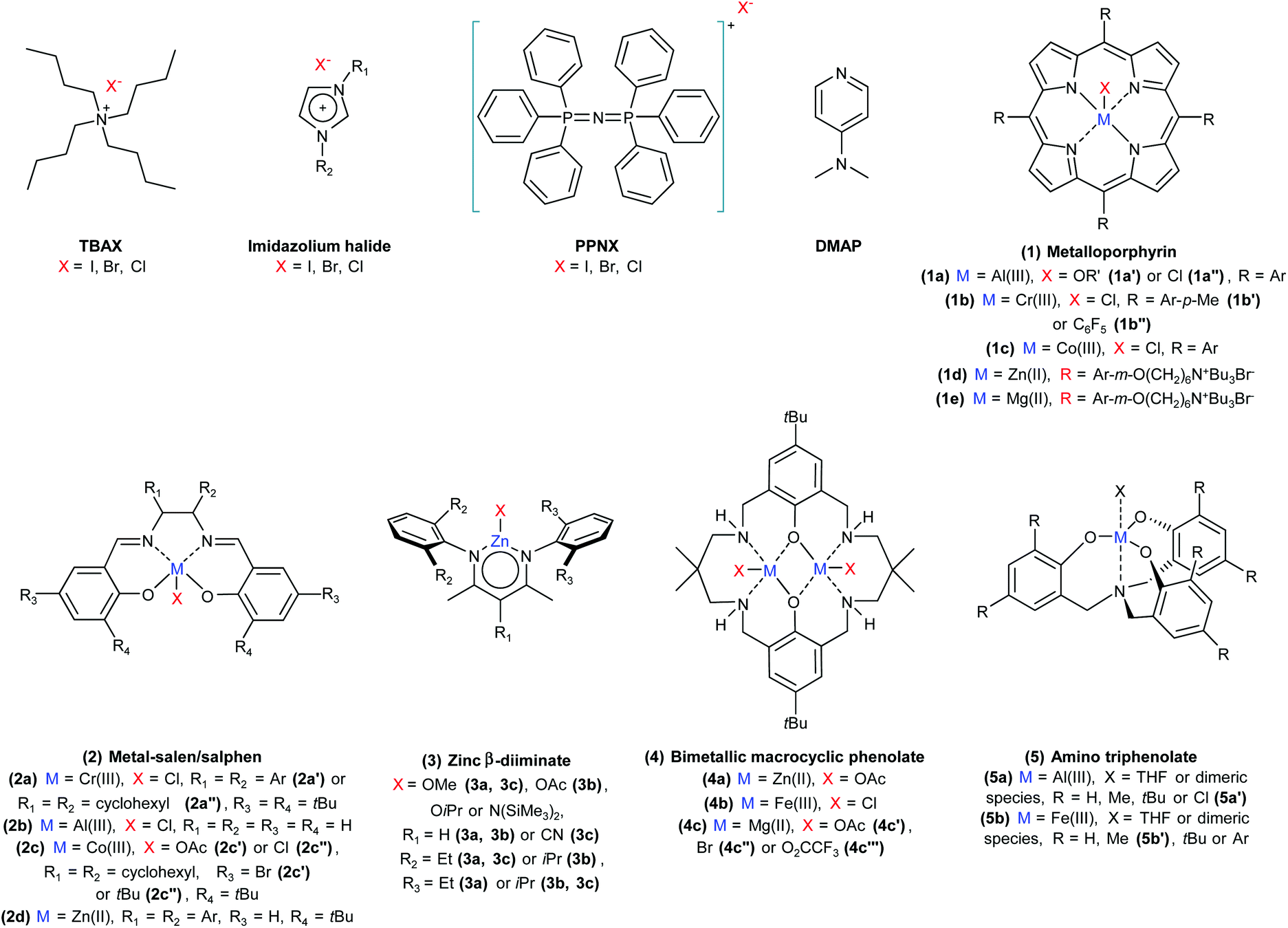 | ||
| Fig. 3 Selected homogeneous catalysts for the CO2/epoxide reaction. | ||
A wide variety of homogeneous and heterogeneous catalytic systems has been developed for the reaction of CO2 with epoxides, with the purpose of increasing the activity but also of maximising the selectivity towards either the cyclic carbonate or the polycarbonate product. Numerous excellent reviews exist on the catalysis of CO2/epoxide reactions,3,13,14,24,41,42 including few focussing only on the synthesis of cyclic carbonates16,43–50 or only on polycarbonates.2,4,6,15,17,51–55 The reader interested in a detailed overview of the numerous existing catalytic systems and/or in the various proposed reaction mechanisms is kindly referred to the above-mentioned references. Here, we provide a systematic and concise overview of the main classes of catalysts that have been studied for the CO2/epoxide reaction, by rationalising them on the basis of their nature and of the type of active sites. For this purpose, the catalysts were categorised into homogeneous and heterogeneous systems, which were further divided with respect to their catalytic species: systems containing Lewis base functionalities, or systems presenting a combination of Lewis acid and base species. For the latter group, a distinction is made between metal-based and metal-free catalysts. For each of these categories, we summarised the main assets and drawbacks (Table 3).
| Catalytic system | Code | Activity | Selectivity | Stability | Reusability | Cost | ||
|---|---|---|---|---|---|---|---|---|
| Homogeneous | Lewis base | Hom1 | ± | Cyclic | ± (issue: thermal stability under operating conditions) | − | ++ | |
| Lewis acid + Lewis base | Metal-based | Hom2 | ++ | Tuneable cyclic/polymeric | + (Al, Fe, Cr) | − | − | |
| − (Co porphyrin/salen, Zn β-diiminate) | ||||||||
| Metal-free | Hom3 | + | Cyclic | + | − | ++ | ||
| Heterogeneous | Lewis base | Het1 | − | Cyclic | ± (issue: thermal stability under operating conditions) | + | + | |
| Lewis acid + Lewis base | Metal-based | Het2 | + | Polymeric (Zn glutarate)/cyclic (others) | + (Zn glutarate) | ++± (MOFs) | + (Zn glutarate) | |
| ± (MOFs) | − (MOFs) | |||||||
| Metal-free | Het3 | + | Cyclic | + | ++ | + | ||
2.1. Homogeneous catalytic systems
Lewis base homogeneous catalysts that only consist of an ionic nucleophile (Hom1 in Table 3) are typically organic halides such as quaternary ammonium salts, phosphonium salts and ionic liquids.16,42,44,46,56 Neutral Lewis bases such as strong organic bases are also used as homogeneous catalysts.42,44 A selection of commonly employed homogeneous Lewis base compounds is shown in Fig. 3: tetrabutylammonium halides (TBAX),57–60 imidazolium halides,46,61 bis(triphenylphosphine)iminium halides (PPNX)62 and 4-dimethylaminopyridine (DMAP).63,64 When these Lewis bases are employed as catalysts in the absence of a Lewis acid (that would coordinate the growing polycarbonate chain, see Fig. 2), the ring closure becomes the most favourable step after the formation of the carbonate intermediate, leading to the selective formation of the cyclic carbonate product. The major limitation in the use of Lewis base homogeneous catalysts is that the separation of the produced cyclic carbonates by distillation is quite energy-intensive, due to the fact that cyclic carbonates have high boiling points (see Table 7).63 Moreover, tetraalkylammonium halides tend to undergo thermal degradation trough a dequaternisation reaction, which would hamper their reuse if the removal of the carbonate is carried out by distillation at high temperature.65 Lewis base catalysts typically require relatively high reaction temperatures (≥80 °C) to achieve good carbonate yields.14,16 In order to increase the activity and reach high conversions also under milder conditions, the Lewis base catalysts are often combined with Lewis acids. The most widely employed type of Lewis acid sites is provided by metal cations complexed by suitable organic ligands. A wide range of metal complexes have been studied, with zinc, chromium, cobalt, aluminium and iron being the most common metal atoms.4,16,24,41,43,45,54,66 The metal-based Lewis acid complexes are usually combined with one of the aforementioned Lewis bases. Such homogeneous binary catalytic systems (Hom2 in Table 3) constitute the class of catalysts for the CO2/epoxide reaction that has been most actively studied so far. The best representatives of this type of catalytic systems are characterised by very high activity even under mild conditions (T ≤ 60 °C, p(CO2) ≤ 10 bar) and can display high selectivity towards either the cyclic or the polymeric carbonate. Often, the selectivity can be tuned by varying the type of nucleophilic species and the nucleophile-to-metal ratio. A nucleophile that is a better leaving group (as I−) will tend to favour the ring-closure step (4a in Fig. 2), thus leading to higher selectivity towards the cyclic carbonate product, whereas a worse leaving group (as Cl−) will tend to remain bound to the intermediate, thus promoting the growth of the polymer chain (4b and 5b).14,17 It was also observed that at a nucleophile-to-metal ratio >1, the nucleophile can tend to displace the carbonate intermediate from the metal centre, thus favouring the ring-closure step (Fig. 2, 4a).17,19 The selectivity between cyclic and polymeric carbonate can also be adjusted by changing the reaction temperature. Cyclic carbonates are the thermodynamically favoured products of the reaction, while the formation of the polycarbonates generally has lower activation energy. Therefore, higher reaction temperatures will promote the formation of the cyclic product, whereas lower reaction temperatures can lead to a kinetic control of the reaction, thus favouring the polymeric product.17 Additionally, higher CO2 pressure (up to carbon dioxide in the liquid or supercritical state) can increase the reaction rate by improving the contact between the reaction components, while also favouring the formation of the polycarbonate product by enhancing the rate of the CO2-insertion step (Fig. 2, 5b).17 The tuning of the above-mentioned parameters often allows to achieve the selective formation of either the cyclic or polymeric carbonate (Table 4, entries 1–7), and even to switch effectively the selectivity between the two products while using the same metal complex (Table 4, entries 23 and 24).19,67| # | Cat. | Cat. loadinga (mol %) | Epox. | Conv/yield (%) | SelPC:CCb (%) |
TONmetalc |
TONLBd |
Temp. (°C) | CO2 pressure (bar) | Time (h) | M n (g mol−1) | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Catalytic loading in molar percentage of epoxide substrate, based on moles of metal sites/based on moles of nucleophilic species added separately.
b Selectivity towards polycarbonate:cyclic carbonate.
c Turnover number expressed as mol of converted epoxide (when the conversion was reported) or mol of produced carbonate (when the yield was reported) per mol of metal sites.
d Turnover number expressed as mol of converted epoxide or mol of produced carbonate per mol of nucleophilic species. n.a. = not available. CHO: cyclohexene oxide; EB: 1,2-epoxybutane; EH: 1,2-epoxyhexane; EO: ethylene oxide; PO: propylene oxide. PDI: polydispersity index, Mw/Mn.
|
|||||||||||||
| 1 | 1a′′ | 1/1 EtPH3PBr | CHO | 100 (C) | 100:0 |
100 | 50 | RT | 49 | 14 d | 6200 | 1.06 | 69 |
| 2 | 1b′ | 0.014/0.088 DMAP | EB | 100 (C) | 0:100 |
7073 | 984 | 50 | 54 | 100 | — | — | 70 |
| 3 | 1b′′ | 0.036/0.31 DMAP | CHO | 63 (Y) | 100:0 |
1743 | 291 | 110 | 228 | 8 | 3450 | 1.19 | 71 |
| 4 | 1c | 0.039/0.0772 DMAP | PO | 42 (Y) | 0:100 |
1102 | 367 | 120 | 17 | 1.33 | — | — | 72 |
| 5 | 1c | 0.2/0.15 DMAP | PO | 95 (Y) | 95:5 |
475 | 271 | 40 | 51 | 48 | 149000 |
1.18 | 73 |
| 6 | 1d | 0.005/0 | EH | 80 (Y) | 0:100 |
16000 |
4000 | 120 | 10 | 3 | — | — | 74 |
| 7 | 1e | 0.005/0 | EH | 88 (Y) | 0:100 |
17600 |
4400 | 120 | 10 | 3 | — | — | 74 |
| 8 | 2a′ | 0.075/0.075 DMAP | PO | 69 (Y) | n.a. | 916 | 458 | 100 | 6.9 | 1 | — | — | 76 |
| 9 | 2a′′ | 0.040/0 | CHO | 10 (C) | n.a. | 250 | 250 | 80 | 58.5 | 24 | 8900 | 1.2 | 77 |
| 10 | 2b | 0.02/0.02 TBABr | EO | 44 (Y) | n.a. | 2220 | 1110 | 110 | 150–160 | 1 | — | — | 78 |
| 11 | 2b | 0.1/0.1 TBABr | EO | 96 (C) | 0:100 |
960 | 480 | 30 | 25 | 72 | — | — | 78 |
| 12 | 2c′ | 0.2/0 | PO | 49 (Y) | >99:<1 |
243 | 243 | 25 | 55.2 | 3 | 15300 |
1.22 | 79 |
| 13 | 2c′′ | 0.067/0.133 DMAP | PO | 27 (Y) | n.a. | 400 | 133 | 100 | 20.7 | 0.33 | — | — | 80 |
| 14 | 2d | 2.5/2.5 TBAI | EH | 80 (C) | 0:100 |
32 | 32 | 45 | 10 | 18 | — | — | 81 |
| 15 | 3a | 0.1/0 | CHO | 45 (C) | 100:0 |
449 | — | 50 | 6.9 | 2 | 19100 |
1.07 | 83 |
| 16 | 3b | 0.1/0 | CHO | 48 (C) | 100:0 |
494 | — | 50 | 6.9 | 2 | 31000 |
1.11 | 83 |
| 17 | 3c | 0.1/0 | CHO | 38 (C) | 100:0 |
382 | — | 50 | 6.9 | 0.17 | 22900 |
1.09 | 85 |
| 18 | 4a | 0.2/0 | CHO | 53 (C) | 94:6 |
264 | 264 | 100 | 1 | 21 | 7360 | 1.21 | 88 |
| 19 | 4b | 0.02/0 | CHO | 25 (C) | 99:1 |
1285 | 643 | 80 | 10 | 24 | 17200 |
1.03 | 90 |
| 20 | 4b | 0.2/0.4 PPNCl | CHO | 41 (C) | 0:100 |
205 | 51 | 80 | 1 | 48 | — | — | 90 |
| 21 | 4c′ | 0.02/0 | CHO | 33 (C) | >99:<1 |
1650 | 1650 | 100 | 1 | 20 | 14600/3500 |
1.02/1.08 | 91 |
| 22 | 5a′ | 0.0005/0.05 TBAI | EH | 36 (C) | <1:>99 |
72000 |
720 | 90 | 10 | 2 | — | — | 92 |
| 23 | 5b′ | 0.5/5 PPNCl | CHO | 85 (C) | 0:96 |
170 | 17 | 85 | 80 | 3 | — | — | 19 |
| 24 | 5b′ | 0.5/0.5 PPNCl | CHO | 98 (C) | >99:<1 |
196 | 196 | 85 | 80 | 3 | n.a. | n.a. | 19 |
Among the classes of metal complexes employed as homogeneous catalysts for the synthesis of cyclic and polymeric carbonates from CO2 and epoxides, metal-porphyrins received considerable attention (1 in Fig. 3 and Table 4, entries 1–7). Their planar geometry makes them especially suitable for the coordination of terminal epoxides to the Lewis acidic metal centre.43 Though metal(III)-porphyrins can contain a nucleophilic species as axial ligand (1a, 1b and 1c in Fig. 3), the activity of these complexes is significantly enhanced by using them in combination with a Lewis base acting as nucleophile for the ring-opening of the epoxide.24 Used metal centres include aluminium,68,69 chromium70,71 and cobalt.72,73 More recently, a series of highly active bifunctional metalloporphyrin catalysts containing both Lewis acid sites (magnesium or zinc) and Lewis base functionalities has been reported (1d and 1e in Fig. 3).74,75 Another important class of homogeneous catalysts that has been investigated extensively for the synthesis of both cyclic carbonates as well as polycarbonates consists of metal complexes with salen/salphen-based ligands (Fig. 3, 2). Various metal centres have been examined, including chromium,76,77 aluminium78 and cobalt79,80 incorporated in salen complexes (Fig. 3, 2a–c and Table 4, entries 8–13) and zinc in salphen complexes81 (Fig. 3, 2d and Table 4, entry 14). An advantage of salen ligands is that their synthesis is easier and that they are less expensive than e.g. porphyrin ligands. Moreover, the synthesis method has the potential to be scaled-up, which is an important asset for a possible industrial use of these catalysts.43 Similarly to the case with porphyrins, salen/salphen-based catalysts with metal(III) centres contain an axial ligand that can act as a nucleophile, and can thus function as bifunctional catalysts in the CO2/epoxide reaction. However, they often require a Lewis base co-catalyst in order to reach sufficient product yields. In the case of zinc(II)-salphen, no axial ligand is present and including a separate Lewis base becomes strictly necessary to perform the opening of the epoxide ring. Although salen and salphen catalysts show high activity for the conversion of terminal epoxides, they are generally significantly less active towards internal epoxides.43 Furthermore, while Cr-based salen complexes were reported to be air-stable,77 many highly active Co-salen complexes are air and moisture sensitive.17 Moreover, Co-based salen and porphyrin catalysts were observed to deactivate during CO2/propylene oxide copolymerisation through reduction of the cobalt centre.2,82 Zinc β-diiminates represent another well-studied class of homogeneous catalysts that has found widespread application in polymerisations of CO2 with a broad range of epoxides (Fig. 3, 3).39,83,84 The catalytic behaviour of this type of complexes proved to be highly tuneable by modifying the substituents present on the N-aryl ring and the diamine backbone (Table 4, entries 15–17).14,85 The zinc β-diiminate complexes can exist both in dimeric (bimetallic) or monomeric (monometallic) form, with the former being most likely the main responsible for the catalytic activity. These metal-complexes do not need addition of a Lewis base to be able to catalyse effectively the reaction.39 Zinc β-diiminate catalysts are very selective towards the polycarbonate product and are very active with various epoxides, including challenging substrates as functionalised cyclohexene oxides86 and limonene oxide.87 A limitation of the zinc β-diiminates is their sensitivity to air and moisture, which imposes to prepare and use them under inert conditions.39,83,84 Another type of Zn-based catalyst is the bimetallic macrocyclic phenolate zinc complex 4a (Fig. 3), which gained attention by showing high activity towards epoxide/CO2 polymerisation even at very low CO2 pressure (1 atm) (Table 4, entry 18).88,89 Moreover, this zinc complex was observed to be air-stable.88 In addition to zinc, other metals that have been investigated in this type of complex include iron90 and magnesium (Fig. 3, 4b and c).91 The iron bimetallic macrocyclic phenolate complex was able to catalyse the reaction of CO2 with cyclohexene oxide, propylene oxide and styrene oxide, although the addition of a Lewis base (PPNCl) was required with the latter two epoxides. Additionally, a shift in selectivity from complete selectivity towards poly(cyclohexene carbonate) formation to full selectivity towards cyclic cyclohexene carbonate formation was realised by increasing the catalyst loading (by a factor 10), lowering the CO2 pressure (from 1.0 to 0.1 MPa) and by addition of PPNCl (Table 4, entries 19 and 20).90 One of the most promising types of homogeneous catalysts for epoxide/CO2 conversion consists of an aluminium(III) centre combined with an amino triphenolate ligand (Fig. 3, 5a).18,92,93 The complex needs to be combined with a Lewis base, such as TBAX or PPNX: this binary catalytic system showed to be highly active for the reaction of CO2 with a wide range of epoxide substrates.92 Considerable variations in catalytic activity were observed between Al(III) amino triphenolate complexes with different substituents on position 2 and 4 of the phenolate rings.18 Recently, it was shown that the methyl-substituted aluminium amino triphenolate complex used in combination with PPNCl or PPNBr is able to efficiently catalyse the copolymerisation of CO2 with the highly challenging limonene oxide.94 In addition to aluminium-based complexes, iron-based amino triphenolate (Fig. 3, 5b; Table 4, entries 23 and 24) and amino bis(phenolate) complexes have also been used in CO2 conversion reactions, allowing to switch effectively the selectivity between cyclic and polymeric carbonates by tuning the type and relative amount of organic halide used as nucleophile source.19,67 When considering the potential for applicability of these catalysts, besides their activity, selectivity and stability, it is important to take into account the cost of the synthesis and the availability of the employed elements. In this context, only six metals are classified as abundant worldwide: aluminium, iron, calcium, sodium, potassium and titanium (in order of decreasing abundance).45 Of these metals, which have the additional advantage of displaying relatively low-toxicity,95 aluminium and iron were employed in highly active and selective catalysts for the synthesis of CO2-based carbonates and seem thus an optimum choice from a sustainability point of view.45
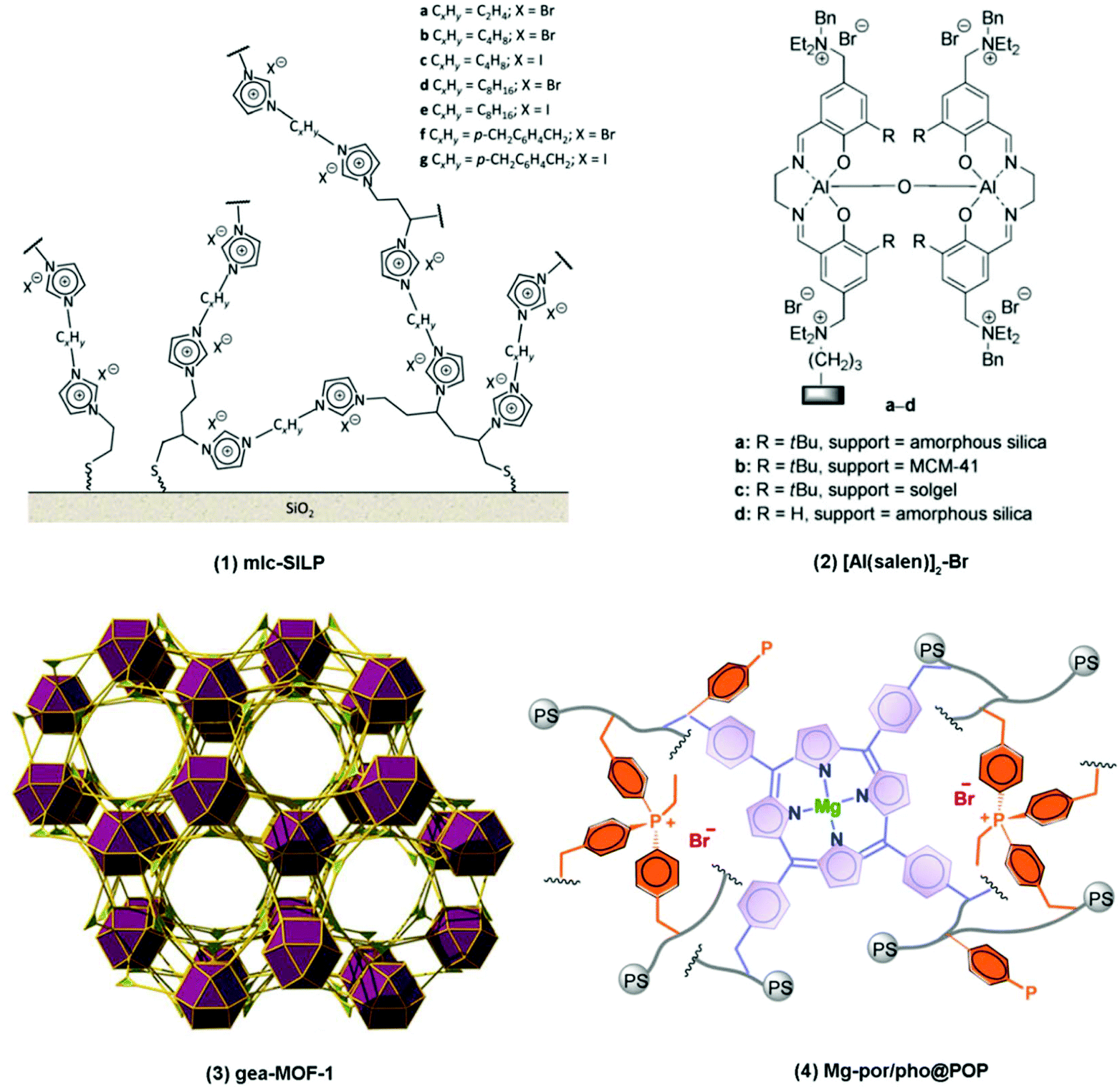 | ||
| Fig. 4 Selected heterogeneous catalysts for the CO2/epoxide reaction: (1) multilayered covalently supported ionic liquid phase (mlc-SILP); (2) immobilised metal complex; (3) metal–organic framework (MOF); (4) porphyrin-based metal-functionalised porous organic polymers (metal-POP). | ||
The metal-based complexes discussed above can reach excellent catalytic performance but their applicability can be hampered by their often costly and complex synthesis procedure. Against this backdrop, the development and investigation of metal-free catalysts for the CO2/epoxide reaction has gained increasing attention in recent years. Organocatalysts are considered as a non-toxic, low-cost and more sustainable alternative to metal-based catalysts.42,48,49 Additionally, organocatalysts generally possess good stability and inertness towards air and moisture and in some cases can be derived from renewable recources.42 Moreover, the use of organocatalysis instead of metal complexes has the potential to increase the safety of consumer products by minimising the amount of metal residues that may cause health hazards.42 On the other hand, the catalytic activity of organocatalysts is still significantly lower compared to the best metal-based homogeneous catalysts and cyclic carbonates are typically the only products, thus excluding polycarbonates from the scope of these catalysts.42 In addition to the previously discussed homogeneous catalysts that only possess a Lewis base functionality (e.g. TBAX, PPNX and DMAP in Fig. 3), the newest generation of metal-free homogeneous organocatalysts includes both Lewis acid and Lewis base functionalities (Hom3 in Table 3). These can be present in a bifunctional single-component catalyst or in a binary system consisting of two separate components. The first category typically consists of onium salts or ionic liquids containing hydroxyl, carboxylic acid or amino moieties, such as protic tetraalkylammonium halides (e.g.Table 5, entry 1),96–99 protic phosphonium halides (e.g.Table 5, entry 2),100,101 protic imidazolium compounds (e.g.Table 5, entry 3)102–104 and functionalised DMAP.105 In these systems, the protic functional groups can activate the epoxide substrate by interacting with its O atom through hydrogen bonding.42,48,65,106 In the second category, several protic compounds containing hydroxyl or carboxylic acid moieties have been employed as catalysts in combination with Lewis bases, including bio-based compounds such as pyrogallol (Table 5, entry 4),65,107 gallic acid,107 tannic acid (Table 5, entry 5)108 and water109 but also other phenolic compounds,110,111 silanediol complexes,112 fluorinated alcohols,107,113,114 boronic acids,115 carboxylic acid-containing amines,116 simple alcohols117 and cavitand-based polyphenols.118 The relative weakness of the hydrogen bonding between the protic groups of these catalysts and the epoxides compared to the typical strong coordination of an epoxide with a metal, explains why cyclic carbonates are the only products when these organocatalysts are employed. On the other hand, there exist a report of the synthesis of polycarbonates from CO2 and epoxides using metal-free catalysts (Table 5, entry 6).119 However, a strongly pyrophoric and water- and air-sensitive compound as triethylborane was employed as a Lewis acid, which limits the applicability of the catalytic system and diminishes the ‘green’ character typically associated with organocatalysts.
| # | Cat. | Cat. loadinga (mol %) | Epox. | Conv./yield (%) | SelPC:CCb (%) |
TONLAc |
TONLBd |
Temp. (°C) | CO2 pressure (bar) | Time (h) | M n (g mol−1) | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Catalytic loading in molar percentage of epoxide substrate, based on moles of Lewis acid sites/based on moles of nucleophilic species added separately.
b Selectivity towards polycarbonate:cyclic carbonate.
c Turnover number expressed as mol of converted epoxide (when the conversion was reported) or mol of produced carbonate (when the yield was reported) per mol of Lewis acid sites.
d Turnover number expressed as mol of converted epoxide or mol of produced carbonate per mol of nucleophilic species.
e Note that these compounds contain several potential Lewis acid sites, but that in this case the catalyst molar loading and TONLA are calculated based on the whole molecule. n.a. = not available. CHO: cyclohexene oxide; EB: 1,2-epoxybutane; EH: 1,2-epoxyhexane; PO: propylene oxide. PDI: polydispersity index, Mw/Mn.
|
|||||||||||||
| 1 | 2-Hydroxyethyl-TBAI | 0.1/0 | EB | 39 (Y) | n.a. | 392 | 392 | 150 | 20 | 1 | — | — | 96 |
| 2 | 2-Hydroxyethyl-TBPI | 2/0 | EB | 92 (Y) | n.a. | 46 | 46 | 90 | 10 | 2 | — | — | 100 |
| 3 | HEMIMBr | 1.6/0 | PO | 99.2 (C) | 0:>99 |
62 | 62 | 125 | 20 | 1 | — | — | 102 |
| 4 | Pyrogallol/TBABr | 3e/3 | PO | 79 (Y) | n.a. | 26e | 26 | 60 | 20 | 1.67 | — | — | 107 |
| 5 | Tannic acid/TBAI | 0.05e/2.0 | EH | 24 (Y) | <1:>99 |
472e | 12 | 80 | 10 | 2 | — | — | 108 |
| 6 | Et3B/PPNCl | 0.050/0.025 | CHO | 90 (Y) | 95:5 |
1800 | 3600 | 80 | 10 | 6 | 28300 |
1.9 | 119 |
2.2. Heterogeneous catalytic systems
The most extensively studied type of catalysts for the fixation of CO2 through the reaction of CO2 with epoxides are homogeneous systems, but the importance of an efficient and straightforward separation and reutilisation of the catalyst stimulated the investigation of several heterogeneous systems. Various Lewis base heterogeneous catalysts have been developed (Het1 in Table 3, for a selected example see catalyst 1 in Fig. 4),120 with the active sites being provided by immobilised ionic liquids,120–128 onium salts129–131 and non-ionic organic bases.132–134 Polymers121,128,129,131 and silica-based materials,120,122–126,130,132–134 including ordered mesoporous silica with high specific surface area such as MCM-41126,134 and SBA-15,122,125 have been studied as supports to immobilise these actives species. The immobilisation is realised through robust, covalent bonding to prevent the leaching of active species into the reaction mixture. These heterogeneous catalysts solve the separation problems associated with homogeneous catalytic systems, but they typically need high temperatures (≥100 °C) in order to reach high conversion rates of the epoxides.135,136 The performance of a selection of heterogeneous Lewis base catalysts for the CO2/epoxide reaction is displayed in Table 6, entries 1–3. Similarly to the case of homogeneous catalysts, the presence of a Lewis acid that activates the epoxide towards the nucleophilic attack by the Lewis base enhances the activity of the heterogeneous systems (Het2 in Table 3, for a selected example see catalyst 2 in Fig. 4).137 Examples of these systems include supported metal catalysts functionalised with Lewis base moieties, such as Zn/SBA-15 functionalised with ammonium groups,138 and the immobilisation of metal complexes that had previously proven to be promising homogeneous catalysts, such as bifunctional Al–salen complexes supported on polystyrene139 or silica (Table 6, entry 4).137 A different class of materials that can present Lewis acid sites and that has been increasingly studied as heterogeneous catalysts for the CO2/epoxide reaction are metal–organic frameworks (MOFs).140 These highly porous crystalline materials consist of metal nodes interconnected through organic linkers so that a crystalline structure is generated. MOFs can be prepared with different (micro)pore sizes and display very high surface area (typically SBET > 1000 m2 g−1). The metal nodes can contain coordinatively unsaturated sites displaying Lewis acid behaviour.141 Many MOF structures show high affinity towards CO2 adsorption. This feature and the possible presence of Lewis acid sites render them potentially suitable candidates as heterogeneous catalysts for CO2 fixation.49,141 This type of MOF (e.g. MOF-5,135 MOF-505,136 Cr-MIL-100,142 Cr-MIL-101,142,143 UMCM-1-NH2,144 In2(OH)(btc)(Hbtc)0.4(L)0.6·3H2O,145 USTC-253-TFA146 and gea-MOF-1147 (Table 6, entry 5, and catalyst 3 in Fig. 4)) can act as heterogeneous catalysts for the CO2/epoxide reaction in combination with an organic halide. In this case the catalytic system is partially homogeneous, which is a drawback in terms of catalyst separation and reutilisation. An alternative approach to introduce metallic Lewis acid sites into MOFs is based on the use of organic linkers that, besides connecting the metal nodes, are able to form complexes with single metal sites. This strategy has been employed to develop zirconium-based porphyrin MOFs148 (Table 6, entry 6) and copper-based MOFs incorporating azamacrocyclic ligands.136 In addition to MOFs solely containing Lewis acid sites, the organic linkers of specific MOFs possess Lewis basic moieties (e.g. ZIF-8,149 ZIF-67,150 ZIF-68,151 ZIF-90152 (Table 6, entry 7) and Co-MOF-74153) or can be functionalised with a nucleophile source (e.g. amine moieties,149 pyridinium iodide,154 quaternised hydrazine155 (Table 6, entry 8) and quaternised ammonium156,157 and phosphonium groups157) so that both Lewis acid and Lewis base active sites are present in the solid catalyst. Functionalisation of the organic linkers with bulky nucleophilic species can however cause a decrease in pore volume and surface area, with possible limitations for the diffusion of reactants and products.154–158 Although promising results have been achieved with MOFs as catalysts for the CO2/epoxide reaction, many of these materials still suffer from low stability under the employed reaction conditions, which leads to deactivation of the catalyst, thereby limiting reusability.142 Further limitations that can be encountered with MOF catalysts reported in the literature include the need to use very high loading of homogeneous organic halides,136,159,160 high reaction temperature,147,150–152 and diffusion limitations of reactants, products and organic halides.142| # | Cat. | Cat. loadinga (mol %) | Epox. | Conv/yield (%) | SelPC:CCb (%) |
TONLAc |
TONLBd |
Temp (°C) | CO2 pressure (bar) | Time (h) | M n (g mol−1) | PDI | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Catalytic loading in molar percentage of epoxide substrate, based on moles of Lewis acid sites/based on moles of nucleophilic species.
b Selectivity towards polycarbonate:cyclic carbonate.
c Turnover number expressed as mol of converted epoxide (when the conversion was reported) or mol of produced carbonate (when the yield was reported) per mol of Lewis acid sites (note: in the case of metal-containing catalysts, the TON is expressed as mol of converted epoxide per mol of metal sites, though this might lead to an underestimation of the actual TON values because not all metal sites are necessarily acting as Lewis acid active sites).142
d Turnover number expressed as mol of converted epoxide or mol of produced carbonate per mol of nucleophilic species.
e In grams of polymer per moles of zinc. n.a. = not available. AGE: allyl glycidyl ether; CHO: cyclohexene oxide; EB: 1,2-epoxybutane; PO: propylene oxide; SO: styrene oxide. PDI: polydispersity index, Mw/Mn.
|
|||||||||||||
| 1 | SiO2-p-xylene-I (mlc-SILP) | 0/0.42 | SO | 99 (C) | <1:>99 |
— | 237 | 150 | 80 | 3 | — | — | 120 |
| 2 | n-Bu4NBr/SiO2 | 0/1 | PO | 97 (Y) | n.a.:98 |
— | 97 | 150 | 80 | 10 | — | — | 130 |
| 3 | TBD@silica | 0/0.27 | PO | 100 (C) | 0:100 |
— | 367 | 150 | 50 | 24 | — | — | 132 |
| 4 | [Al(salen)]2-Br/amorphous silica | 5/10 | SO | 78 (C) | n.a. | 16 | 8 | RT | 1 | 20 | — | — | 137 |
| 5 | gea-MOF-1/TBABr | 0.15/0.15 | EB | 94 (Y) | n.a. | 627 | 627 | 120 | 20 | 6 | — | — | 147 |
| 6 | PCN-224(Co)/TBACl | 0.090/0.20 | PO | 42 (C) | n.a. | 461 | 194 | 100 | 20 | 4 | — | — | 148 |
| 7 | ZIF-90 | 0.59/n.a. | PO | 88 (C) | n.a.:92 |
151 | n.a. | 120 | 12 | 8 | — | — | 152 |
| 8 | F-ZIF-90 | 0.088/n.a. | AGE | 75 (Y) | 0:100 |
855 | n.a. | 120 | 11.7 | 6 | — | — | 155 |
| 9 | Mg-por/pho@POP | 0.005/n.a. | PO | 78 (Y) | n.a.:>98 |
15600 |
n.a. | 140 | 30 | 1 | — | — | 161 |
| 10 | FDU-HEIMBr | n.a./0.5 | PO | 99 (Y) | <1:>99 |
n.a. | 198 | 110 | 10 | 3 | — | — | 177 |
| 11 | mQC-1.I | 0.8/0.4 | PO | 98 (C) | <1:>99 |
121 | 242 | 120 | 12 | 3 | — | — | 185 |
| 12 | Zn glutarate | 0.33/0 | PO | n.a. | 96:n.a. |
7146e | n.a. | 80 | 40 | 20 | 103000 |
2.8 | 190 |
| 13 | Zn3[Co(CN)6]2/tBuOH | 0.028/0 | CHO | 95 (C) | 43.5:0 |
3340 (Zn) | n.a. | 100 | 38 | 2 | 47250 |
3.4 | 191 |
Another class of materials containing metal centres in a porous structure that has gained increasing attention in recent years as heterogeneous catalysts for the CO2/epoxide reaction is represented by metal-functionalised porous organic polymers (metal-POPs).161–166 One of the key features of these porous organic polymers is that they can be synthesised in numerous types of amorphous porous structures incorporating various organic functional groups,161,163 thereby introducing a large degree of freedom in tuning properties such as type and amount of active sites, surface area, pore volume and pore size.162,164 The ability to tune the pore volume and pore size of POP materials could prevent diffusion limitations of reactants and products,164 which on the other hand is a common drawback with functionalised MOFs. The Lewis acid metal centres can be incorporated in the material via different strategies, e.g. crosslinking of pre-synthesised metal–organic complexes by organic linkers,161,162,164,166–168 or synthesis of a microporous organic network, in which metal ions are introduced afterwards.163,165,168 Examples of metal-POP catalytic systems that have been evaluated for the CO2/epoxide reaction include binary systems where the metal-POP was combined with an organic halide165–168 as well as fully heterogeneous bifunctional systems where the metal-POP contained both Lewis acid and Lewis base sites.161–164 A bifunctional porphyrin-based metal-POP system containing magnesium metal sites and quaternary phosphonium bromide species displayed one the highest turnover numbers for heterogeneous catalysts for the conversion of CO2 and epoxides into cyclic carbonates (Table 6, entry 9, Fig. 4).161
The activation of the epoxide can also be achieved without metal sites if protic groups are present at the surface of the heterogeneous catalyst (Het3 in Table 3). One of the first examples of this type of catalysts are the silica-supported phosphonium salts, for which it was proposed that the surface silanols can activate the epoxide and thus enhance the activity of the immobilised salt in their proximity.169 Typically, metal-free heterogeneous catalysts with Lewis acid and Lewis base functionalities are prepared by tailored synthesis of the immobilised active groups as in polymer-supported ionic liquids with hydroxyl or carboxylic acid moieties,170–174 hydroxyl- or carboxylic-acid-containing ionic liquids grafted on silica,125,175,176 hydroxyl- and carboxylic-acid-functionalised ionic liquids immobilised on a mesoporous polymeric substrate177 (Table 6, entry 10) and hydroxyl-functionalised quaternary ammonium moieties grafted on polystyrene or silica.178–180 Metal-free covalent organic frameworks (COFs) have also been reported as heterogeneous catalysts for the reaction of CO2 with epoxides.181,182 Being constructed from molecular organic building blocks, the type of functional groups (e.g. Lewis bases182 or Lewis acids181) as well as the size and shape of the pores of these catalyst can be tailored. Other examples of materials that possess metal-free Lewis acid moieties include bio-based compounds such as cellulose and chitosan. Cellulose has been investigated as a bio-based heterogeneous catalyst for the CO2/epoxide reaction both in the presence of a homogeneous Lewis base183,184 and as bifunctional single-component system with immobilised quaternary ammonium moieties (Table 6, entry 11).185 Chitosan, which is a polysaccharide derived from naturally occurring chitin extracted from the shells of e.g. shrimps and crabs,186 was functionalised to introduce quaternary ammonium moieties187,188 or imidazolium moieties189 and was subsequently evaluated as a bio-based and metal-free heterogeneous catalyst for the CO2/epoxide reaction, displaying similar but slightly lower activity compared to the best cellulose-based catalysts.
All the heterogeneous catalysts described above yield cyclic carbonate as the main product, because the nucleophile-to-Lewis acid ratio is >1, or because the activation of the epoxide occurs through mild hydrogen-bonding, or finally because the reaction temperature is relatively high (>100 °C). There exist also metal-based heterogeneous catalytic systems that allow the selective copolymerisation of CO2 with epoxides, generally with propylene oxide as substrate.192–195 The two main types of such heterogeneous catalysts are zinc carboxylates, with zinc glutarate (Table 6, entry 12) being the most active representative, and double metal cyanides.53 Zinc glutarate and other zinc carboxylates are industrially relevant, as they are easy to handle, non-toxic, air-stable and economically viable.2,6 Double metal cyanide catalysts [Zn3(M(CN)6)2, where the most commonly employed metals are M = Co(III) or Fe(III)]52,53 show high activity for polymerisation reactions with a variety of epoxides, including propylene oxide196–198 and cyclohexene oxide (Table 6, entry 13),191,196,198 but they exhibit low CO2 uptake and commonly yield poly(ether carbonates) rather than pure polycarbonates.53 In contrast to homogeneous catalytic systems, heterogeneous catalysts such as zinc carboxylates and double metal cyanides generally need more extreme reaction conditions for epoxide/CO2 polymerisations, namely a high CO2 pressure.53
2.3. Effect of CO2 source on catalyst performance
Although having received relatively little attention so far,199–204 the source of CO2 used for the reaction with epoxides is of importance, especially in the perspective of large scale production. When carbon dioxide is employed that has been captured from industrial sources such as the ones discussed in section 1 (Table 1), it often contains impurities, including H2S, CO, SOx, NOx and water.205 The effect of these impurities on the catalytic system is of crucial concern, as these compounds could potentially influence the activity of the catalytic system by poisoning the active sites or could even cause catalyst degradation. Few studies investigated the effect of using carbon dioxide with lower degree of purity and containing the contaminants commonly found in flue gases.50,199–204 In general, these studies showed that several catalytic systems active in the CO2/epoxide reaction (e.g. bimetallic aluminium salen/tetrabutylammonium bromide,199 immobilised bifunctional bimetallic aluminium salen,200,201 YCl3/tetrabutylammonium bromide,202 magnesium bimetallic macrocyclic phenolate203 and zirconium on silica/tetrabutylammonium bromide204) are quite insensitive towards common flue gas impurities. This supports the feasibility of this reaction as pathway for the utilisation of waste CO2.50 In the perspective of the industrial application of a novel catalyst in the reaction of CO2 with epoxide to produce either cyclic or polymeric carbonates, future research should increasingly include investigations of the influence of the CO2 purity on the catalyst performance.3. Applications of CO2-based cyclic and polymeric carbonates
The two products of the reaction between CO2 and epoxides, i.e. cyclic carbonates and polycarbonates, differ both in structure and in properties, thereby generally finding rather different kinds of applications.3.1. Properties of cyclic carbonates
The two cyclic carbonates that are most widely employed are ethylene carbonate (EC) and propylene carbonate (PC). Both are polar aprotic compounds – see Table 7 for their dielectric constant, dipole moment and other selected physicochemical properties.206 Due to their biodegradability, low toxicity, low vapour pressure and high flash point, these two cyclic carbonates are greener alternatives to traditional, more harmful polar aprotic solvents such as dimethylformamide (DMF), hexamethylphosphoramide (HMPA), N-methyl-2-pyrrolidone (NMP), dimethylacetamide and acetonitrile, with propylene carbonate being a more viable solvent as it is a liquid at room temperature whereas ethylene carbonate has a melting point of 36 °C.17,208–211 A drawback of propylene and ethylene carbonate is their relatively high boiling point, which implies a more difficult and costly separation and recycling.211 Their applications as solvents include cleaning,212 paint stripping213 and degreasing,214 and the use as electrolyte components in batteries (see section 3.5). Another relevant application of cyclic carbonates is as reactants for chemical syntheses, providing a greener alternative to toxic and/or volatile compounds as phosgene (see section 3.4.1), epoxides and cyanates. In the presence of a catalyst, typically an alkali compound, five-membered cyclic carbonates can be used to alkylate protic aromatics such as phenols, thiophenols, aniline and aromatic carboxylic acids (Fig. 5).207 Although the reaction can be carried out through a more direct path via reaction of the aromatic compounds with epoxides (e.g. ethylene oxide or propylene oxide), employing carbonates rather than epoxides has a few advantages: (i) cyclic carbonates are less toxic than the corresponding epoxides and, therefore, safer to handle; (ii) no need for high-pressure equipment, which would be required when working with volatile epoxides, as the alkylation reaction is typically carried out at relatively high temperatures (100–200 °C); (iii) no additional solvents are needed, as the cyclic carbonate can act as both solvent and reactant.207 Aromatic compounds that are typically used in this reaction include: hydroquinone, which is reacted with two molecules of EC to produce a compound employed as spacer in the synthesis of high-strength polyurethanes;215 cardanol (a compound derived from cashew nut oil), which is reacted with EC and PC to produce plasticisers;216 resorcinol, which upon reaction with EC and PC also produces compounds used as spacers in polyurethane synthesis;217 and cyanuric acid, which is reacted with EC to form a crosslinking agent for polyester resins used to coat underwater electric cables.207,218 In addition to the alkylation of aromatic compounds containing hydroxyl, thiol or amino groups, aromatics bearing carboxyl groups can be reacted with five-membered cyclic carbonates to introduce alkyl ester moieties.207 Moreover, the hydroxyl group of the product of this reaction (Fig. 5, X = COO) can react further with the carboxyl group of a second aromatic, thus forming a diester compound. Following this route, multifunctional carboxylic acids such as terephtalic acid have been reacted with PC or EC to synthesise polyester oligomers, which were subsequently used as prepolymers to produce polyester resins.207,219 In addition to the synthesis of building blocks for polymer production, the reaction of aromatic carboxylic acids with cyclic carbonates has also been employed in polymer modification, e.g. to lower the amount of free-acid end groups in poly(ethylene terephthalate) [PET] with the purpose of increasing the chemical resistance of the polymer220 or of increasing the rate of reaction with isocyanates to produce polyurethane foams;221 and to enhance the performance of superabsorbing polymers used in diapers and feminine hygiene products, by crosslinking the acid groups present in the employed polymer (sodium polyacrylate).207,222 Compared to the previously mentioned protic aromatic compounds containing hydroxyl or amino groups, the reaction of aliphatic alcohols and amines with five-membered cyclic carbonates proceeds differently (Fig. 6a). In the case of primary or secondary amines (and of ammonia), a urethane (carbamate) product is formed.207 Although similar compounds can be formed via the reaction of diols with urea, such route requires high temperatures (120–170 °C), while five-membered cyclic carbonates can react with primary amines at room temperature.207 In most cases a catalyst is not required, but strong bases can be used to increase the reaction rate.207 The use of cyclic carbonates and amines to produce urethane linkages provides a greener alternative to the common synthesis of polyurethanes, which is based on the reaction of toxic di- or polyisocyanates with polyols (see section 3.4.3 for further information on this).207,225 This type of reaction pathway also finds application in epoxy resins (see section 3.4.2).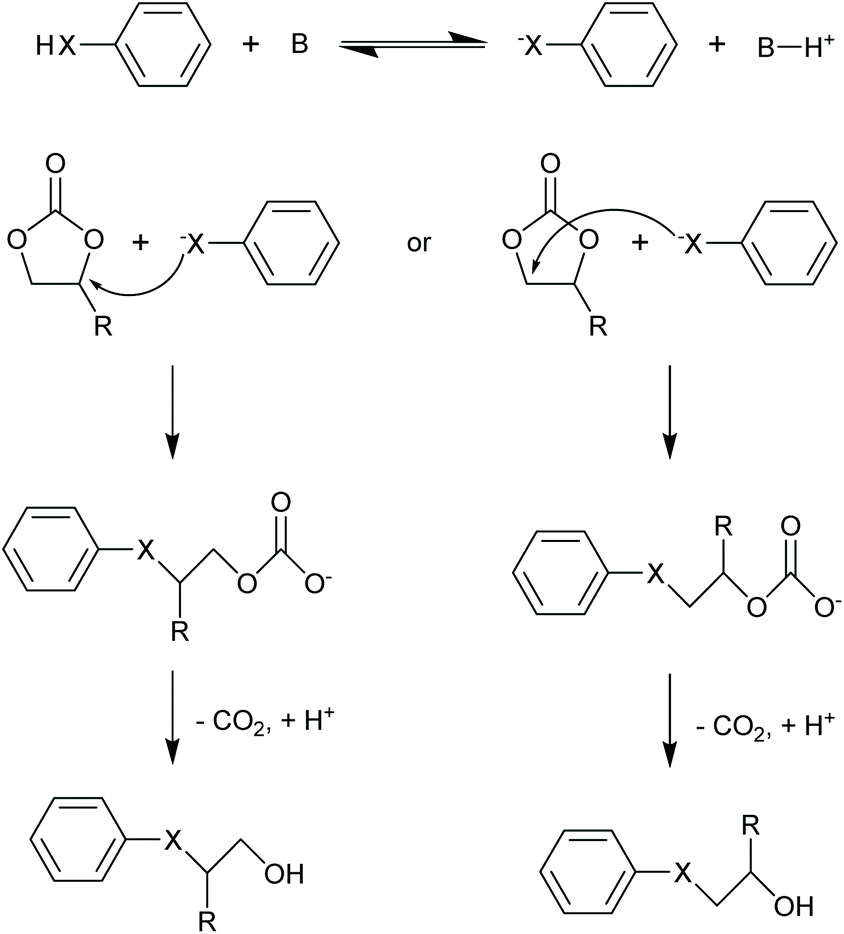 | ||
| Fig. 5 Alkylation of protic aromatic compounds with five-membered cyclic carbonates in the presence of an alkali catalyst. X = O, NH, S or COO; B is an alkali.207 | ||
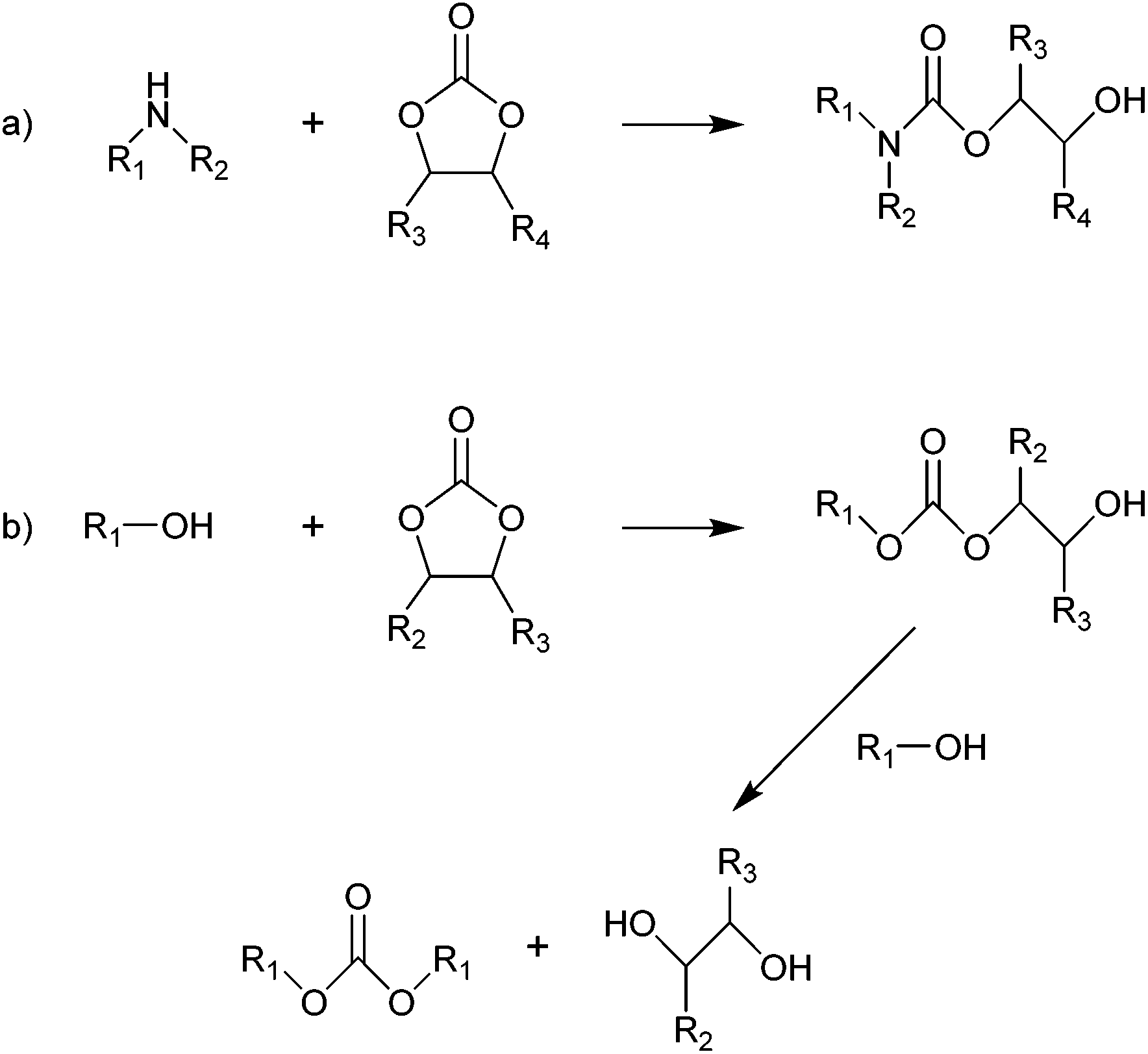 | ||
| Fig. 6 Reaction of five-membered cyclic carbonates with aliphatic amines (a) and alcohols (b). R1, R2, R3 and R4 = hydrogen, alkyl.207 | ||
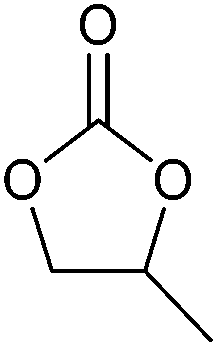
| Cyclic carbonate | Boiling point (°C) | Melting point (°C) | Flash point (°C) | Vapour pressure (kPa) | Dielectric constant | Dipole moment (D) | Density (g mL−1) | Hazards |
|---|---|---|---|---|---|---|---|---|
|
246 | 36 | 143 | 0.003 (25 °C) | 89.78 (40 °C) | 4.9 | 1.32 (39 °C) | Harmful if swallowed, causes serious eye irritation, may cause damage to organs (kidney) through prolonged or repeated exposure if swallowed |
|
242 | −49 | 135 | 0.004 (25 °C) | 66.14 (20 °C) | 4.9 | 1.20 (20 °C) | Causes serious eye irritation |
Aliphatic alcohols react with five-membered cyclic carbonates similarly to aliphatic amines, but a successive reaction with a second alcohol compound can also occur, thereby forming a linear carbonate and a diol (Fig. 6b).207 This additional step typically does not occur with aliphatic amines, except at high temperatures (≥150 °C).207 A well-known example of this reaction is the transcarbonation of cyclic carbonates (e.g. ethylene or propylene carbonate) with methanol to produce dimethyl carbonate (DMC), a non-toxic and biodegradable compound that can be used in methylation and carbonylation reactions as greener substitute for the highly toxic reactants conventionally used (dimethyl sulphate and phosgene, respectively).226 An important industrialised example of the application of a cyclic carbonate as alternative to the extremely toxic phosgene through transcarbonation reactions is the Asahi Kasei industrial process,227 which represents a greener route for the synthesis of aromatic bisphenol-A-based polycarbonates (see 3.4.1 for more detailed information). When, instead of simple alcohols, diols are employed in the reaction with five-membered cyclic carbonates, other five-membered cyclic carbonates, six-membered cyclic carbonates and polycarbonates can be formed.207 In this context, the reaction of ethylene carbonate and propylene carbonate with glycerol to form glycerol carbonate is a promising synthesis route as it allows converting a large-scale bio-based by-product from biodiesel manufacturing as glycerol into a valuable organic compound that can find application as low-volatility solvent in varnishes, glues, cosmetics and pharmaceuticals and as organic building block for preparing epichlorohydrin and polymers such as hyperbranched polyethers, polyesters, polycarbonates, polyurethanes and polyamides.23,35,36,228–232 This route to synthesise glycerol carbonate is more viable compared to the direct reaction of glycerol with CO2, which is thermodynamically unfavourable and thus results in low yields unless the formed water is removed from the reaction mixture.228,233 Among the products of the reaction of diols with five-membered cyclic carbonates, six-membered cyclic carbonates have been studied as starting materials for polymerisation reactions, as these compounds display lower thermodynamic stability and are thus more readily polymerised compared to their five-membered counterparts.43,207
3.2. Properties of polycarbonates prepared from CO2 and epoxides
Polycarbonates can be prepared by the polymerisation of carbon dioxide with a wide range of epoxides (Table 8), with propylene oxide and cyclohexene oxide being the most widely studied monomers. These polycarbonates are related to the bisphenol-A-based aromatic polycarbonates mentioned above, as they are all characterised by the carbonate group in their backbone, but differ markedly in terms of physicochemical properties and thus in potential applications due to the absence of aromaticity in the polycarbonate backbone. Bisphenol-A-based polycarbonates (BPA-PC) display excellent physicochemical properties such as relatively high Tg, high heat and impact resistance, transparency and dimensional stability, which make them suitable as engineering plastics for a wide variety of applications including automotive industry, construction, optics and electronics.17,227 On the other hand, the first generation of CO2/epoxide polycarbonates, often based on propylene oxide or cyclohexene oxide, have different thermal and mechanical properties as well as lower thermal stability compared to BPA-PC. As a result, these polycarbonates cannot be employed as engineering plastics.4,14,17 More specifically, poly(propylene carbonate)s (PPC) have significantly lower glass transition temperature (Tg) compared to bisphenol-A-based polycarbonate and thus display elastic behaviour (high elongation at break and impact strength) and lower tensile modulus and tensile strength (Table 8).17 Poly(cyclohexene carbonate)s (PCHC) have higher Tg and approach the mechanical properties of bisphenol-A-based polycarbonate in terms of tensile modulus and strength, but they display inferior impact properties and elongation at break and, thus, are brittle at room temperature (Table 8).17| Polycarbonate | M n (kg mol−1) [PDI] | T g (°C) | T d,initial (°C) | T d50% (°C) | Tensile modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) | Impact strength (J cm−1) | Transmission (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a T d5%. b T d,max. c T d10%. PDI: polydispersity index, Mw/Mn. | ||||||||||
| Poly(ethylene carbonate) | — | 10 | 217 | — | 3–8 | — | >600 | — | — | 242 |
| Poly(propylene carbonate) | 7.5 [3.8] | 28 | 235 | — | 212 | 9 | 8 | — | — | 242 |
| — | 38 | — | — | 1353 | 14.7 | 203.1 | 7.27 | — | 243 | |
| 69.5 [1.09] | 42 | — | 252 | — | — | — | — | — | 244 | |
| — | 25–45 | ±240 | — | 700–1400 | 7–30 | — | — | — | 245 | |
| Poly(butene carbonate) | 180 [1.15] | 9 | — | 241 | — | — | — | — | — | 246 |
| Poly(pentene carbonate) | 7.5 [5.1] | −4 | 246 | — | — | — | — | — | — | 242 |
| Poly(hexene carbonate) | 9.5 [4.8] | −10 | 250 | — | — | — | — | — | — | 242 |
| — | −15 | — | 253 | — | — | — | — | 246 | ||
| Poly(cyclohexene carbonate) | 8 [4.2] | 105 | 282 | — | 2460 | 11.8 | 0.5 | — | — | 242 |
| 63 [1.06] | 118 | — | 310 | — | — | — | — | — | 244 | |
| — | 125 | — | — | 2707 | 29.4 | 1.3 | 1.31 | — | 243 | |
| Poly(vinyl-cyclohexene carbonate) | — | 107 | — | — | 2110 | 36.7 | 1.3 | 1.02 | — | 243 |
| Poly(limonene carbonate) | 53.4 [1.10] | 130 | 250a | 265b | 950 | 55 | 15 | — | 94 | 238 |
| 3.69 [1.34] | 72 | 226c | — | — | — | — | — | — | 247 | |
| 10.6 [1.43] | 112 | — | — | — | — | — | — | — | 94 | |
| Poly(limonene oxide carbonate) | 11.4 [1.29] | 135 | — | — | — | — | — | — | — | 239 |
| Poly(indene carbonate) | 9.7 [1.12] | 138 | 239 | 257 | — | — | — | — | — | 237 |
| Bisphenol-A polycarbonate | — | 149 | — | 458 | 2000–2800 | 43–51 | 15–75 | 9 | 89 | 4, 234 and 235 |
Although the typical characteristics of BPA-PC (i.e. relatively high Tg, no crystallinity and outstanding impact behaviour),4,234,235 remain unreachable benchmarks for poly(propylene carbonate) and poly(cyclohexene carbonate), attempts have been reported to match the high Tg values (∼150 °C) by reacting CO2 with other epoxides. Examples include polycarbonates prepared from CO2 and indene oxide (with a Tg up to 138 °C vs. ∼150 °C for BPA-PC),236,237 limonene oxide (Tg of 130 °C, improved pencil hardness (B vs. 8B) and higher transparency (94% vs. 89%) compared to BPA-PC)234,238 and limonene dioxide (Tg up to 135 °C for unmodified CO2/limonene dioxide polycarbonates).239 Although the Tg of these CO2-based polycarbonates match that of BPA-PC, their impact behaviour was not studied, thereby not (yet) demonstrating their suitability as replacements for BPA-PC in rigid engineering plastic.
An overview of selected thermal and mechanical properties of several polycarbonates is given in Table 8. It can be seen that the glass-transition temperature is strongly dependent on the molecular weight of the polymer (the Tg increases with the molecular weight up to a certain value of Mn, above which it remains approximately constant).17 The obtained molecular weights for CO2/epoxide copolymers are typically lower than the maximum theoretical values that would be calculated assuming a living anionic polymerisation mechanism. This occurs because the polymerisation is sensitive towards traces of protic compounds (i.e. water, alcohols or acids), that can cause chain transfer reactions (see Fig. 7) resulting in termination of the polymer chain and thus leading to relatively low molecular weight polymers.6,53 While the presence of adventitious water typically limits the molecular weight of polycarbonate chains (i.e. Mn < 20 kg mol−1), its role as chain transfer agent can also be exploited to generate low molecular weight polycarbonates with terminal hydroxyl groups,203 which can serve as polyols for polyurethane synthesis (see section 3.4.3). In addition to the influence of the polymer molecular weight on the glass transition temperature, the rigidity of the structure of the polycarbonate backbone affects the Tg, which decreases in the order of aromatic > alicyclic > linear aliphatic units in the backbone (i.e. Tg: BPA-PC > PCHC > PPC). Furthermore, a substantial decrease in Tg is observed upon an increase in the length of the alkyl side chains (Tg: poly(propylene carbonate) > poly(butane carbonate) > poly(pentene carbonate) > poly(hexane carbonate)). This can be ascribed to an increasing free volume of the polymeric chains, which results in less interaction between these chains, producing an increase in mobility.
 | ||
| Fig. 7 Chain transfer reaction with a protic compound (i.e. water, alcohols or acids) during the polymerisation of CO2 and epoxides catalysed by a Lewis acid (M) and a nucleophile (X). | ||
Although CO2-based polycarbonates are typically amorphous polymers, semi-crystalline stereoregular poly(propylene carbonate)s and poly(cyclohexene oxide)s were also synthesised, by employing a chiral catalyst, an enantiopure epoxide monomer, or both.17,240,241 These poly(cyclohexene oxide)s possess high melting temperature (Tm = 215–230 °C), which increases their potential to be used in products that are regularly subjected to high temperatures.240 The thermal properties of poly(propylene carbonate) also improved upon introduction of stereoregularity. More specifically, the initial degradation temperature of stereoblock poly(propylene carbonate) (Td5% = 253 °C) with isotactic (S)- and (R)-blocks is higher than that of atactic poly(propylene poly(propylene carbonate) (Td5% = 229 °C).241 Notably, the Tg of isotactic poly(propylene carbonate) was lower than that of atactic poly(propylene carbonate) with similar molecular weight (Tg = 24 °C vs. Tg = 34 °C, respectively).241 In general, CO2-epoxide copolymers are biodegradable.248–250 For example, it has been shown that poly(propylene carbonate) is biodegradable in air, water and soil, and that the process does not generate toxic substances.17
The properties of the polycarbonates prepared by the alternating CO2-epoxide polymerisation summarised above are promising for a variety of applications, which are discussed in detail in sections 3.4–3.7. The identification of these (potential) applications typically occurs by benchmarking the properties of these polycarbonates against those of other polymers. Additionally, other strategies can be considered for tuning the physicochemical properties of CO2/epoxide polycarbonates, including (i) terpolymerisation (i.e. reaction of CO2 with an epoxide and a third monomer that can be another epoxide, an anhydride or caprolactone);251–253 (ii) block-copolymerisation (i.e. sequential growth of a polymer consisting of different blocks of which at least one is based on CO2/epoxide copolymerisation)86,243,254 (iii) post-polymerisation modification of the functional groups that might be present along the backbone;252 (iv) end-group modification by using functional compounds as chain transfer agents (if the functional compound contains two or more groups that can give chain transfer, polymer extension or branching can be achieved17,255); (v) blending of polycarbonates with other polymers or with inorganic solids.17,256,257 Particularly, the post-polymerisation modification is a highly versatile approach: when epoxide substrates such as limonene oxide (LO), cyclohexadiene oxide (CHDO), vinyl-cyclohexene oxide (VCHO) or 2-vinyloxirane (VO) (Table 9) are used in the CO2/epoxide copolymerisation reaction, polycarbonates are produced that contain unsaturated moieties. These groups provide a synthetic toolbox to introduce a variety of functionalities, thereby making it possible to tune the chemical and mechanical properties of the polymer.234,247,258–265
| Epoxide | Structure | Boiling point (°C) | Hazards | Toxicity | Source (stage) |
|---|---|---|---|---|---|
| LC50 = lethal concentration of toxic agent sufficient to cause death of 50% of the subject population. LD50 = lethal dose of toxic agent sufficient to cause death of 50% of the subject population. n.a. = not available. | |||||
| Ethylene oxide (EO) |
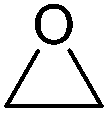
|
10.7 | Extremely flammable, toxic if inhaled, may cause genetic defects or cancer | LC50: 800 ppm (rat, 4 h, inhalation) | Bio-based (commercial),283 petroleum-based (commercial) |
| Propylene oxide (PO) |
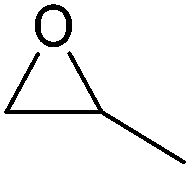
|
34 | Extremely flammable, toxic if inhaled, may cause genetic defects or cancer | LD50: 380 mg kg−1 (rat, oral) | Bio-based (proposed),283–285 petroleum-based (commercial) |
| Epichlorohydrin (ECH) |
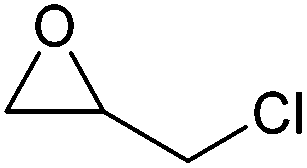
|
115–117 | Flammable, toxic, corrosive, may cause cancer | LD50: 90 mg kg−1 (rat, oral) | Bio-based (commercial),283 petroleum-based (commercial) |
| 2-Vinyloxirane (VO) |
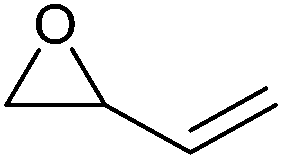
|
66 | Highly flammable, suspected of causing genetic defects | n.a. | Bio-based (partially commercial),286 petroleum-based (commercial) |
| Styrene oxide (SO) |
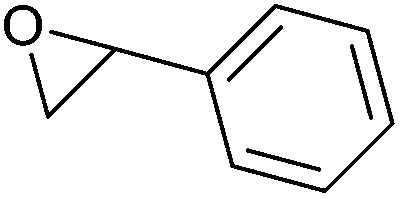
|
194 | Harmful, irritant, toxic if inhaled, may cause genetic defects or cancer | LD50: 1500 mg kg−1 (mouse, oral) | Petroleum-based (commercial) |
| Cyclohexene oxide (CHO) |
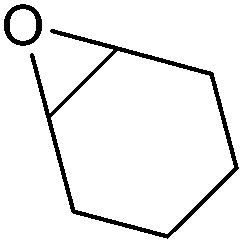
|
129–130 | Flammable, toxic, corrosive | LD50: 1058 mg kg−1 (rat, oral) | Bio-based (proposed),287 petroleum-based (commercial) |
| Cyclohexadiene oxide (CHDO) |
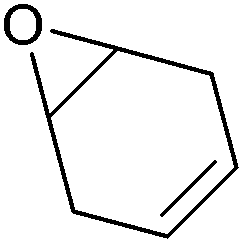
|
n.a. | n.a. | n.a. | Bio-based (proof of concept)264,287 |
| Vinyl-cyclohexene oxide (VCHO) |
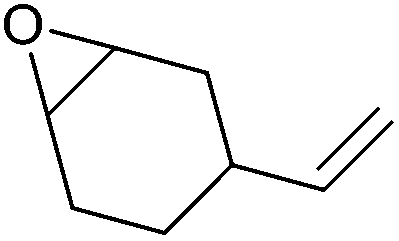
|
169 | Flammable, harmful, suspected of causing cancer | LD50: 1904 mg kg−1 (rat, oral) | Bio-based (precursors are commercially available),286 petroleum-based (commercial) |
| Limonene oxide (LO) |
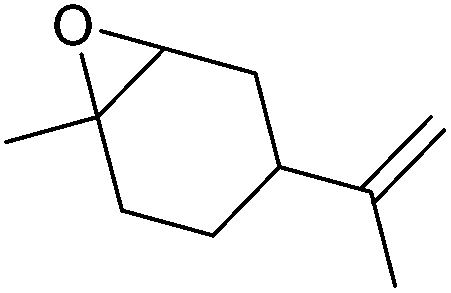
|
n.a. | Flammable, may cause eye, skin and respiratory system irritation | n.a. | Bio-based (commercial) |
The interest towards the industrial application of CO2-based polycarbonates is testified by the several patents describing these products and the processes for their manufacturing,266,267 filed by small, dedicated enterprises or large chemical companies including BASF,268,269 Covestro (former Bayer Material Science),270–273 Novomer274,275 (currently part of Saudi Aramco), Empower Materials,276 Cardia Bioplastics277 and SK Group.278,279 Additionally, some patents are owned by universities.280,281
3.3. Towards fully renewable CO2-based cyclic and polymeric carbonates
The two epoxides that are most commonly employed for the large-scale synthesis of CO2-based cyclic and polymeric carbonates, i.e. ethylene oxide and propylene oxide, are synthesised industrially from ethylene and propylene. Although commonly derived from petrochemical feedstock, interest has increased for developing bio-based routes for producing these two compounds. Ethylene can be produced via a bio-based route through the dehydration of ethanol282 or via the cracking of bio-naphtha.283 Several plants where bio-ethanol is converted into ethylene have been or are being established in Brazil, India and China.282,283 Additionally, various synthetic routes for the production of propylene from renewable resources have been suggested: (i) from methanol via methanol-to-olefins technology (MTO);283,285 (ii) by hydrodeoxygenation of glycerol, which is the main side product of biodiesel manufacturing;284 (iii) by dehydrogenation of propane obtained as a by-product of biodiesel manufacturing;283,285 (iv) by conversion of vegetable oil into propylene via catalytic cracking;283,285 (v) by dehydration of ethanol to ethylene, followed by dimerisation to form butene, and a final metathesis reaction step with ethylene;283,285 (vi) by dehydration of butanol (derived from the fermentation of carbohydrates or via a bio-based syngas-route) to yield butene, followed by a metathesis step with ethylene.283,285 In addition to the technological challenges in producing ethylene oxide and propylene oxide from bio-based sources, these routes should ideally also be cost-efficient in order to be able to compete with the low cost of their petroleum-based counterparts.Albeit the production of ethylene and propylene carbonate through the conversion of carbon dioxide with respectively ethylene oxide and propylene oxide is a well-established industrial process,129,288 the use of ethylene oxide and propylene oxide results in serious safety concerns due to the combination of high toxicity and volatility of both epoxides (especially ethylene oxide, which is a gas at room temperature). Cyclohexene oxide has a higher boiling point and is thus safer to use, but it is more expensive and is still usually derived from petroleum.238 Small amounts of cyclohexene have been observed during the hydrodeoxygenation of lignin-derivable phenols, although these quantities typically do not exceed more than a few percent.289–291 More recently, increasing attention has been given to the use of bio-based epoxides for the synthesis of cyclic and polymeric carbonates by reaction with CO2. Among these, limonene oxide and limonene dioxide87,234,238,239,247,260,292 can be produced through the well-studied epoxidation of (R)-(+)-limonene, a naturally occurring terpene that is extracted from e.g. citrus peels (generated as abundant waste of the juice extraction from orange and other citrus fruits).293 Unsaturated fatty acids and esters derived from triglycerides (e.g. those in vegetable oils) contain one or more double bonds that can be converted into epoxide groups and, subsequently, into cyclic or polymeric carbonates.294,295 Other bio-based epoxides can be prepared through multiple synthetic steps, and include 4-vinylcyclohexene oxide,258,263,296 which can be synthesised via Diels–Alder dimerisation of bio-butadiene297 followed by epoxidation of the double bond; 1,4-cyclohexadiene oxide,264,287 which can be synthesised via selective epoxidation of one of the two double bonds in 1,4-cyclohexadiene, a by-product of the self-metathesis of some polyunsaturated fatty acids derived from plant oils.264,287 Epichlorohydrin298,299 can be derived from bio-based glycerol in a two-step process, but the use of HCl as chloride source decreases the greenness of this option.300 Epichlorohydrin is also the starting compound used to prepare glycidyl ether-based epoxides, which have been employed as substrates in the CO2/epoxide polymerisation reaction.301–305 For all these bio-based resources, a challenge might be represented by their availability (and cost), which should match the desired market size of the targeted application of the cyclic or polymeric carbonate product.306
Although the utilisation of bio-based epoxides increases the renewable character of CO2-based cyclic and polymeric carbonates, the hazards originating from the high toxicity of epoxides are still a concern (Table 9). A solution would be to directly synthesise the CO2-based carbonates from the alkene from which the epoxides are generally prepared. This one-pot procedure would be advantageous both from the point of view of sustainability and of industrial applicability, as avoiding the purification and handling of epoxides would result in higher safety and lower process costs.14 This process requires a multifunctional catalytic system (either a one-component system or two separate non-interfering catalysts) that is able to promote both the oxidation of alkenes to epoxides and the subsequent reaction with CO2 to form carbonates. So far, only few studies have been conducted concerning this one-pot route, typically employing a combination of known oxidation catalysts with CO2/epoxide catalysts.14,307–312 The main issue encountered is the formation of side products during the epoxidation step, leading to low selectivity of the carbonate end-product. A further challenge is to develop a catalytic process operating at mild conditions, using a green oxidant such as H2O2 or O2 rather than organic peroxides.313
3.4. Application of cyclic and polymeric carbonates in the preparation of polymer products
Several strategies have been developed for the application of both cyclic and polymeric carbonates produced from CO2 in the preparation of polymer products. These strategies range from the use of cyclic carbonates as reactive intermediates in the formation of urethane linkages or as precursors for transcarbonation reactions to synthesise aromatic polycarbonates, to the application of CO2-based polycarbonates in functional coatings, as polyols for polyurethane production and in polymeric blends and composites. These strategies find a variety of applications, in some cases reaching industrial production, as reviewed in more detail in the following sub-sections (3.4.1–5).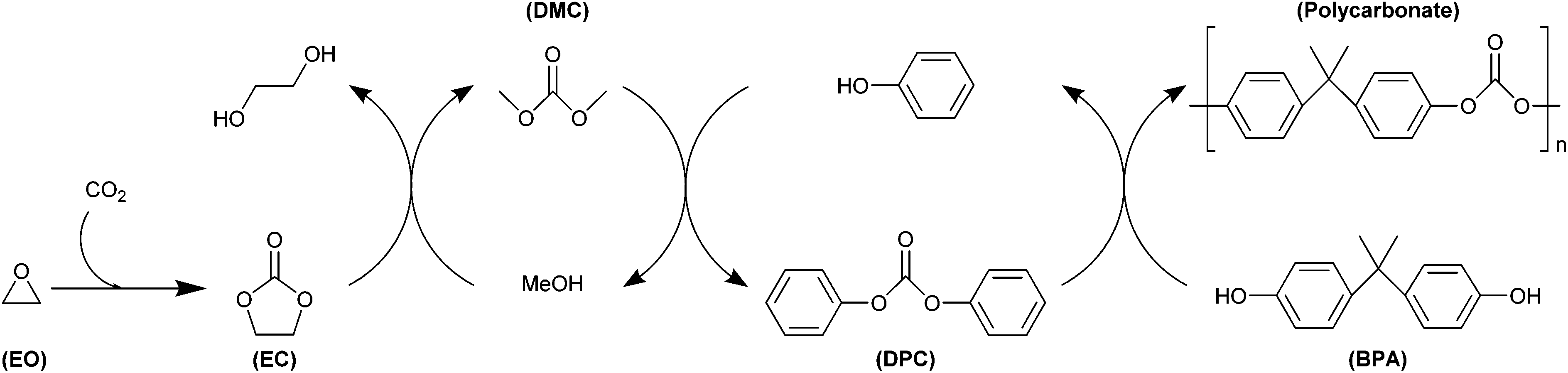 | ||
| Fig. 8 General overview of the Asahi Kasei non-phosgene polycarbonate production process. | ||
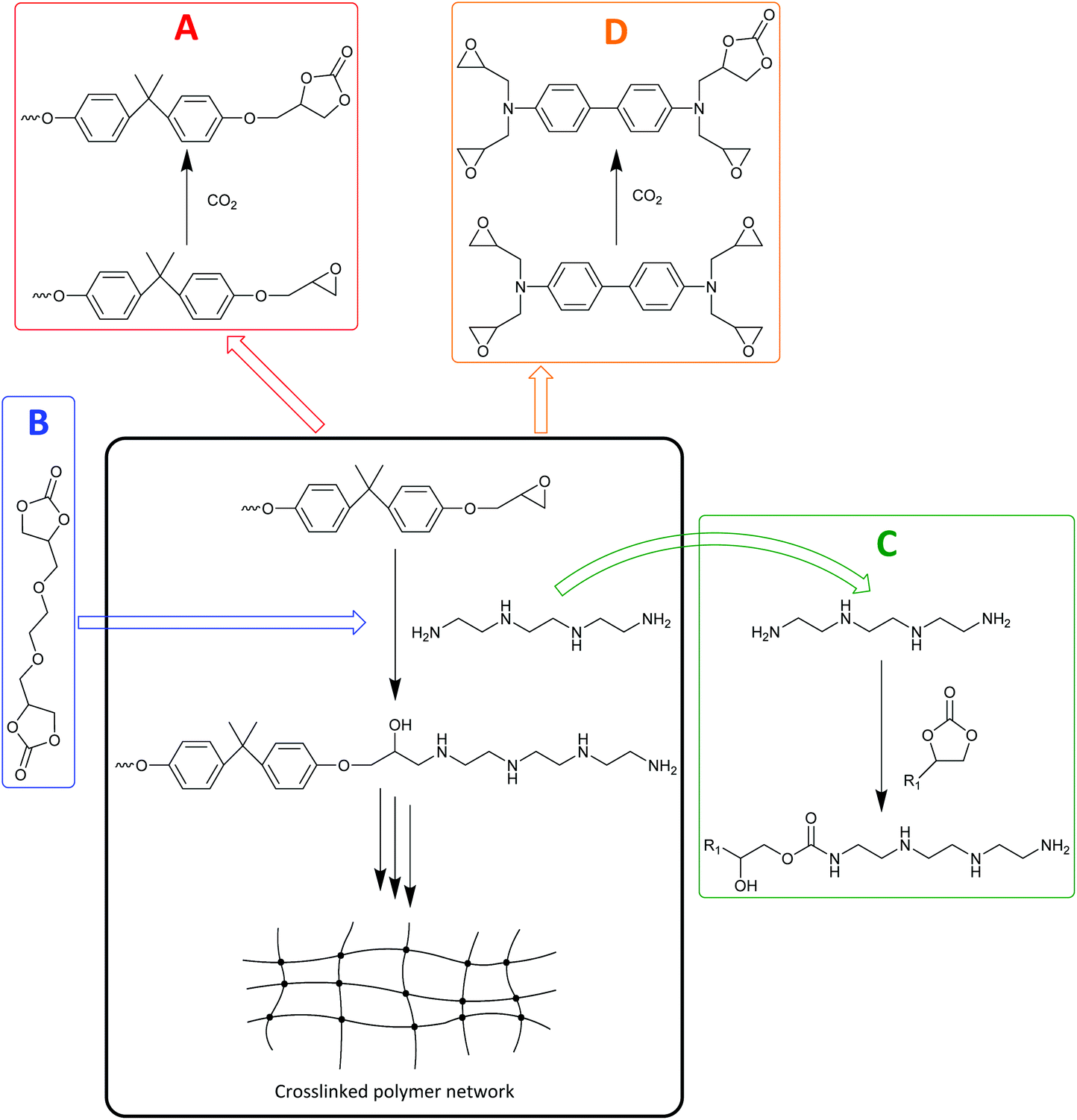 | ||
Fig. 9 Possible CO2-based modifications of the various components of epoxy resin formulations: (A) Conversion of terminal epoxide groups into cyclic carbonates. (B) Reactive cyclic carbonates as additives (note: only the cyclic carbonate analogue of triethylene glycol diglycidyl ether is shown as example). (C) Modification of triethylenetetramine with cyclic carbonates (note: multisubstituted products are not shown), R1 = e.g. –CH3, –C6H6, –CH2–O–CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2. (D) Modification of the tetrafunctional epoxy resin (4,4′-methylenebis(N,N-diglycidyl aniline). CH2. (D) Modification of the tetrafunctional epoxy resin (4,4′-methylenebis(N,N-diglycidyl aniline). | ||
| # | Composition | T g (°C) | T d,initial (°C) | Tensile strength (MPa) | Tensile modulus (MPa) | Yield stress (MPa) | Elongation at break (%) | Compressive properties | Lap shear strength (MPa) | Tensile-peel strength (kN m−1) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strength (MPa) | Strain (%) | |||||||||||
| 1 | Epoxy/triethylenetetramine | — | 208–210 | 29 | — | 11 | 4.0 | 118 | 13.5 | 6.3 | 0.60 | 324 |
| 2 | CO2-modified epoxy/triethylenetetramine | — | 208–210 | 23–29 | — | 8.5–12 | 3.2–3.8 | 196–235 | 42–50 | 7.5–8.7 | 1.47–2.10 | 324 |
| 3 | Epoxy/triethylenetetramine | — | — | 37 | — | 12 | 4.0 | 260 | 50 | 4.1 | 107 | 322 |
| 4 | Epoxy/TGDEC/triethylenetetramine | — | — | 47–69 | — | 13–15 | 5.4–8.0 | 235–275 | 45–55 | 5.1–5.7 | 108–127 | 322 |
| 5 | Epoxy/triethylenetetramine | — | — | 26 | 2010 | 7 | 1 | 223 | 63 | 6.4 | 0.059 | 325 |
| 6 | Epoxy/carbonate-modified triethylenetetramine | — | — | 17–72 | 1710–2110 | 5–12 | 1–8 | 88–261 | 36–58 | 7.6–14.4 | 0.11–0.22 | 325 |
| 7 | Tetrafunctional epoxide/diethylenetriamine | 186 | 325 | — | — | — | — | — | — | — | — | 318 |
| 8 | CO2-Modified tetrafunctional epoxide (48%)/diethylenetriamine | 149 | 309 | — | — | — | — | — | — | — | — | 318 |
| 9 | Poly(propylene carbonate-ether) polyurethane | 18.7 | — | 52 | 579 (100% elongation) | — | 410 | — | — | — | — | 326 |
| 10 | Poly(butylene adipate glycol) polyurethane | — | — | 68 | 57.8 (100% elongation) | — | 781 | — | — | — | — | 326 |
| 11 | Poly(propylene oxide glycol) polyurethane | — | — | 20 | 7.96 (100% elongation) | — | 930 | — | — | — | — | 326 |
| 12 | Polyadipate polyurethane | — | — | — | — | — | — | — | — | — | 21 | 327 |
| 13 | Poly(propylene carbonate)/polyadipate polyurethane | — | — | — | — | — | — | — | — | — | 14–22 | 327 |
| 14 | Polyhydroxyurethanes based on carbonated soybean oil/short amines | 15–18 | 227–239 (5 wt%) | — | 2.0 | — | 126–129 | — | — | — | — | 328 |
| 15 | Polyhydroxyurethanes based on carbonated soybean oil/oligoamide | −5–1 | 340–353 (5 wt%) | — | 169–224 | — | 286–406 | — | — | — | — | 328 |
| 16 | Limonene dicarbonate/polyhydroxyurethane networks | 55–70 | — | — | 2400–4100 | 7–21 | 1–2 | — | — | — | — | 329 |
A general drawback of polyurethane formulations for foams is that products prepared with polyesters as polyols exhibit excellent mechanical properties but poor hydrolysis resistance, while polyurethanes prepared using polyethers as polyols possess proper hydrolysis resistance but insufficient oxidation resistance and display poor mechanical properties.326,327 Since CO2-epoxide-based polycarbonates share similarities with both polyesters and polyethers with regard to the chemical structure but have lower tendency to hydrolyse compared to polyesters, polycarbonate polyols have been investigated as an alternative to conventional polyester and polyether polyols. Additionally, employing CO2-based poly(carbonate-ether) polyols is a greener option, as it allows increasing the renewable content of the polymer (up to 43 wt% CO2 in the case of poly(propylene carbonate)). This approach, commonly referred to as the “Dream Production”, has been pioneered by Bayer Material Science (currently Covestro),337,338 using a double metal cyanide (DMC) catalyst to produce poly(carbonate-ether) polyols from CO2, propylene oxide and an alcohol starter. For this application, poly(carbonate-ether) polyols are employed rather than polyols with only carbonate linkages, as the presence of ether bonds leads to polyols with lower Tg value [−60 °C for poly(propylene carbonate-ether) with a CO2-content of 7.1 wt%], which allows processing with the standard industrial equipment used in polyurethane foam production.337 It was estimated via life-cycle analysis that the production of these CO2-based poly(carbonate-ether) polyols requires 13–16% less fossil resources than the production of conventional petroleum-based polyols,339 indicating that substitution of petroleum-based polyols with CO2-based polyols is very promising, both from a sustainability point of view and with respect to cost reduction. Since 2016, Covestro has been commercially producing polyols with up to 20% CO2-content for use in flexible polyurethane foams, with an annual production capacity of 5000 tonnes.340 Another example of a commercial polyurethane product made from CO2-polycarbonate polyols is the foam prepared using poly(propylene carbonate) polyols by Saudi Aramco (Converge polyols, formerly part of Novomer).341 The advantages of CO2-based poly(carbonate-ether)s as polyols for polyurethane synthesis over their polyester and polyether counterparts was further demonstrated by synthesising a series of polyurethanes from poly(propylene carbonate-ether) polyols and comparing them with polyurethanes incorporating polyester (poly(butylene adipate glycol)) or polyether (poly(propylene oxide glycol)) segments.326 It was observed that the polycarbonate-based polyurethane (with 66% carbonate linkages and 34% ether linkages) exhibited better mechanical properties, although with a lower elongation at break compared to the other two polyurethanes, indicating a more rigid plastic behaviour (Table 10, entries 9–11). In addition, the poly(carbonate-ether)-based polyurethane exhibited better resistance against hydrolysis and oxidation compared to the polyester and polyether polyurethanes, which was ascribed to the presence of ether linkages with good hydrolysis resistance and to the relatively hydrophobic and rigid propylene carbonate repeating units, which might limit the permeation of water and, thus, increase the hydrolysis and oxidation resistance.
Numerous other accounts exist in the scientific literature on the usage of CO2-derived polycarbonates in polyurethane synthesis. An example thereof are the poly(urethane-urea)s where both the soft diol segments and the hard polyurea segments are derived from CO2, via the reaction of poly(propylene carbonate-ether) diols with isocyanates and oligoureas made from a diamine reacted with CO2.342 Another example are the poly(ethylene carbonate-ether)/MDI polyurethane elastomers displaying shape memory behaviour, which was attributed to the low Tg of the soft segments in the polyurethane (−4.5 °C).343 Other thermoplastic polyurethanes were prepared from various ratios of poly(propylene carbonate) polyols and polyether polyols reacted with an aliphatic diisocyanate and a diol as chain extender.344 Increasing polycarbonate content typically yielded, in addition to improved rigidity, enhanced corrosion resistance of coated metal surfaces compared to thermoplastic polyurethanes with higher polyether content. This was ascribed to the hydrophobic nature of the incorporated polycarbonate polyols, thereby shielding the metal surface from water and oxygen, as supported by the higher water contact angles displayed by the polyurethanes with higher polycarbonate content.
In addition to linear poly(carbonate-ether) polyols, star-shaped CO2-based polyols have also been prepared, via a two-step process starting with the synthesis of a hyper-branched poly(propylene oxide) copolymer with glycerol branching points, which was subsequently used as macro-initiator for the reaction with propylene oxide and CO2, generating a polyether core with poly(propylene carbonate) arms.345 These star-shaped structures possess low Tg values between −8 and 10 °C, due to the flexible polyether core. Other star-shaped CO2-based polyols were prepared via the reaction of CO2 and propylene oxide in the presence of trimesic acid as an initiation-transfer agent, resulting in oligo(carbonate-ether)s with three arms.346 These three-armed polyols were subsequently studied for their potential in shape-memory polyurethanes in biomedical applications (see section 3.6).347 A variety of compounds containing two or more –OH groups can be used as chain transfer agents (see section 3.2) from which polycarbonate branches can be grown. By employing organo-phosphorous compounds as chain transfer agents in CO2/epoxide polymerisation, flame-retarding poly(propylene carbonate) polyols can be prepared.255,348 Both the polycarbonate polyols, as well as the resulting thermoplastic polyurethane products were proved to be non-flammable. The flame-retarding properties of these thermoplastic polyurethanes might be exploited for applications in interior materials such as artificial leather, decorating sheets and hot-melt adhesives.255,348
The favourable properties of CO2-based carbonates as polyols in polyurethane formulations also promoted research focussed on specific applications, e.g. on polyurethane foams for automotive seating formulations.349 Polyurethane formulations were prepared using different ratios of CO2-based aliphatic polycarbonates, either linear or branched, mixed together with a fully petroleum-based polyether polyol, a blend of isocyanates and a mixture of additives. Wet compression set tests indicated that the fully petroleum-based foams retained their original shape better after deformation, while the higher compression modulus showed that the CO2-based foams are stiffer and deform less under load. This was explained by the smaller average pore size in polyurethane foams with increasing polycarbonate polyol content, caused by the higher viscosity of the CO2-based polyols compared to the polyether polyol, resulting in less pore expansion by gravity during the initial foaming stage. Another field of application where polyurethanes are widely employed is the footwear industry. Substitution of traditional polyester and polyether polyols was investigated by employing mixtures of poly(propylene carbonate) and poly(1,4-butylene adipate) polyols to produce polyurethane adhesive networks upon reaction with a diisocyanate (MDI).327 The thermal (i.e. melting viscosity and softening temperature) and adhesive properties (i.e. tensile-peel strength, Table 10 entries 12 and 13), of these polyurethanes were in suitable ranges for footwear applications. In practical applications where the degradation of the polyurethane network is not a drawback but rather a requirement, the biodegradable nature of CO2-based polycarbonates such as poly(propylene carbonate) can represent an asset. This is the case in marine anti-biofouling coatings, which aim at preventing the growth of marine organisms on ships by slow degradation accompanied by controlled release of anti-biofouling agents. In this context, polyurethane coatings with different molecular weights and incorporating an anti-biofouling agent were prepared from poly(propylene carbonate), hexamethylene diisocyanate and 1,4-butanediol.350 The polyurethane coating with the lowest molecular weight showed the best anti-biofouling performance, probably due to the highest degradation rate and, hence, the highest release rate of anti-biofouling agent. Based on the degradation data, the lifetime of the coating was estimated to be at least 6 months. These features are promising for marine anti-biofouling applications, although the mechanical properties of these coatings still have to be thoroughly investigated.
Although several CO2/epoxide polycarbonates have been shown to be suitable as precursors for polyurethanes, most epoxide building blocks are petroleum-based. In an effort to decrease the dependence from depleting fossil feedstocks, polycarbonates synthesised from bio-based epoxides such as limonene oxide have been investigated as greener polyols for polyurethane synthesis.351 For this purpose, poly(limonene carbonate) was synthesised and subjected to reaction with different compounds containing one or more hydroxyl groups (i.e. 1,3-propanediol (1,3-PD), 1,10-decanediol (1,10-DCD), isosorbide, trimethanolethane (TMP) and pentaerythritol (PE)) to produce hydroxyl end-capped, lower molecular weight polycarbonates suitable for polyurethane curing (Fig. 10A), with glass transition temperatures between 71 and 99 °C.351 Curing of the modified poly(limonene carbonate) polyols using a solvent-casting method with a curing agent with three isocyanate groups yielded crosslinked polyurethane coatings that displayed poor solvent resistance. This was ascribed to incomplete curing of the coating, which was related to the low reactivity of the secondary or tertiary hydroxyl moieties in the end-groups of the unfunctionalised poly(limonene carbonate)s (i.e. those with R1 = OH in Fig. 10). To overcome this limitation, poly(limonene carbonate) was functionalised with primary hydroxyl groups by reacting the pendant vinyl groups present in the polycarbonate backbone with a mercaptoalcohol (2-mercaptoethanol or 6-mercaptohexanol) through a thiol–ene reaction (Fig. 10B).259 Curing of the modified poly(limonene carbonate)s with polyisocyanate produced polyurethane coatings that were evaluated with respect to their chemical resistance, thermomechanical properties and mechanical performance (Table 11, entry 1). Acetone rubbing tests showed sufficient to good solvent resistance, whereas the measured König hardness of 167–182 s indicated that these polyurethane coatings can be classified as hard. On the other hand, the cured coatings lacked a good reverse impact resistance, which was ascribed to a low elongation at break caused by the brittle nature of the material. These results show that through multiple modification steps it is possible to functionalise fully renewable poly(limonene carbonate) in such a way that it can be used as a polyol in polyurethane coatings. However, the brittle character of poly(limonene carbonate) limits the applicability in polyurethane coatings that require high impact resistance. Although this system shows the potential of polyurethanes prepared from bio-based polyols derived from limonene oxide and CO2, it requires the use of di- or polyisocyanates, which is not preferred due to the health issues associated with these compounds.352 Furthermore, isocyanates are commercially produced using the toxic phosgene as a precursor, which is another reason to search for greener alternatives. In order to produce cyanate-free polyurethanes, different synthesis routes can be followed based on the reaction between carbonate moieties and amino groups (see section 3.1, Fig. 6a). A first route involves the reaction of two molecules of a cyclic carbonate with a diamine to produce a bis-hydroxyalkylcarbamate that can be subsequently polymerised through self-condensation to generate linear polyurethanes (Fig. 11A).353 A second pathway uses dialkyl carbonates (typically dimethyl carbonate) instead of cyclic carbonates, which can be reacted with a diamine to generate a bis-alkylcarbamate that can be polymerised to produce a polyurethane via reaction with a diol (Fig. 11B).353 The disadvantage of this route is that, in addition to the required second component (diol) for the polymerisation, an extra reaction step is necessary to synthesise the dialkyl carbonate from a CO2-based cyclic carbonate (typically ethylene carbonate or propylene carbonate) and two alcohols (see section 3.1, Fig. 6b). In a third approach, a bicyclic carbonate is reacted with a diamine, yielding polyhydroxyurethanes that slightly differ from typical cyclic carbonate/amine-based polyurethanes due to the presence of pendant hydroxyl groups in the polyurethane backbone (Fig. 11C).354 Polyhydroxyurethanes typically possess high degradation temperatures (up to 388 °C), improved chemical resistance and are hydrophilic due to the high density of hydroxyl groups and the resulting strong hydrogen bonds between these groups and the urethane moieties, rendering these polymers especially suitable for coating applications.354 The bicyclic carbonate can be a monomer (e.g. prepared from glycerol carbonate and a diacid/diacyl chloride such as terephthalic acid)335 or a polymer (e.g. poly(dimethylsiloxane)) bearing terminal cyclic carbonate groups.355 In order to increase the renewable nature of these isocyanate-free urethanes, a bio-based building block was developed by epoxidising the unsaturated bonds in soybean oil, followed by reaction with CO2 to introduce cyclic carbonate moieties.356 Subsequent reaction with di- and triamines resulted in crosslinked polyhydroxyurethane networks. A trend of increasing Tg and decreasing elongation at break was observed by decreasing the length of the diamines or by using triamines as curing agents. In another study on the synthesis of polyhydroxyurethanes from carbonated soybean oil, large diamines prepared via amidification of fatty acid dimers were used.328 Whereas the polyhydroxyuretanes produced from carbonated soybean oil reacted with short diamines (i.e. 1,4-butanediamine and 1,5-pentanediamine) were brittle, those prepared using the large diamines showed thermoplastic behaviour (Table 10, entries 14 and 15 respectively). This was ascribed to a higher intake of carbonated soybean oil in the former case, resulting in a higher crosslink density of the final polyhydroxyurethane network. In another study of bio-based carbonated precursors for polyurethanes, carbonated fatty acid diesters derived from sunflower oil were used as building blocks.357 Both fatty acid diesters with two internal epoxide moieties and with two terminal epoxide moieties were successfully converted into their cyclic carbonate analogues. Subsequent reaction of these bicyclic carbonates with diamines yielded linear polyurethanes with molecular weights up to 13500 g mol−1 and glass transition temperatures around −15 °C. Another type of bio-based bicyclic carbonate (Fig. 10C) can be obtained by the epoxidation of both double bonds of limonene (see section 3.3) followed by CO2-cycloadditon.329 This building block was used to synthesise both linear polyhydroxyurethanes and crosslinked thermoset polyhydroxyurethane networks by reaction with diamines or triamines, respectively. The mechanical properties of the obtained polyhydroxyurethane thermosets are shown in Table 10, entry 16. The relatively high Young's modulus and low elongation at break are an indication of strong and rigid structures, which might originate from the presence of hydroxyl groups and the resulting hydrogen bonding with the urethane moieties,354 but might also be related to the intrinsic rigidity of the limonene units.
 | ||
| Fig. 10 Limonene-based carbonates for polyurethane applications: (A) Poly(limonene carbonate) polyols (R1 = OH or functional group obtained by reaction with 1,3-PD, 1,10-DCD, isosorbide, TMP or PE). (B) Thiol-modified poly(limonene carbonate) polyols, incorporating primary hydroxyl groups as side groups (R2 = –(CH2)2– or –(CH2)6–). (C) Limonene dicarbonate as polyhydroxyurethane precursor. | ||
 | ||
| Fig. 11 Non-isocyanate polyurethanes based on the reaction of an organic carbonate with a diamine: (A) Reaction between two cyclic carbonates and a diamine producing a bis-hydroxyalkylcarbamate (R1 = alkyl). (B) Reaction between two alkyl carbonates and a diamine producing a bis-alkylcarbamate (R1, R2, R3 = alkyl). (C) Reaction between bicyclic carbonates and diamines producing polyhydroxyurethanes (R1 = alkyl, R2 = monomeric or polymeric unit). | ||
| # | Coating | T g (°C) | Acetone resistancea | Pencil hardness | König hardnessb (s) | Reverse impact resistance | Ref. |
|---|---|---|---|---|---|---|---|
| a 75 double rubs with a tissue soaked in acetone. b Standardised König pendulum hardness test, with the damping time reported in seconds. c Measured in accordance to ASTM D2794, 1 kg ball from 0.1 m height. d Measured in accordance to ASTM D2794, 1 kg ball from 0.5 m height. e Testing method not described. | |||||||
| 1 | Mercaptoalcohol-modified poly(limonene carbonate) polyurethane | — | Sufficient to good | 2H | 167–182 | Unsatisfactoryc | 259 |
| 2 | Poly(cyclohexene carbonate-co-4-vinylcyclohexene carbonate)/trithiol (solvent coated) | — | Good | — | — | Goodd | 251 |
| 3 | Poly(cyclohexene carbonate-co-4-vinylcyclohexene carbonate)/trithiol (powder coated) | 85–104 | Good | 6–8H | — | Unsatisfactoryd | 251 |
| 4 | Poly(limonene carbonate)/trithiol | — | Sufficient to good | H–2H | 103–114 | Unsatisfactorye | 358 |
| 5 | Poly(cyclohexadiene carbonate)/trithiol | — | Sufficient to good | H–2H | 123–159 | Unsatisfactorye | 358 |
| 6 | Poly(limonene-8,9-oxide carbonate)/trithiol | 150 | Good | 2H | — | — | 239 |
It should be noted that, although non-isocyanate polyurethanes prepared via the reaction between cyclic carbonates and amines are promising from an environmental and safety point of view, this reaction is quite slow compared to the reaction between isocyanates and alcohols,335 rendering these non-isocyanate polyurethanes unsuitable for application as rigid foams, which requires very rapid formation of the urethane network upon mixing of the precursor components.
The fully bio-based polycarbonates synthesised from CO2 and limonene oxide already discussed in section 3.4.3 as components of polyurethane coatings can also be functionalised in a different way to enable the preparation of alternative polymeric coatings. Reaction of the pending vinyl groups of poly(limonene carbonate) (Fig. 10A) with a trithiol compound in the presence of a radical initiator and under UV irradiation produced transparent coatings (Table 11, entry 4).358 Using the same curing method, transparent coatings prepared from a different bio-based polycarbonate derived from cyclohexadiene oxide and CO2 were also tested with regard to their mechanical and adhesive properties (Table 11, entry 5).358 The coatings exhibited a hard and brittle character, typically observed for other CO2-polycarbonate based coatings (Table 11). The coatings prepared from poly(cyclohexadiene carbonate) typically possessed a higher König hardness than the poly(limonene carbonate)-based coatings, which was related to the higher intrinsic stiffness of the former polycarbonate due the absence of bulky isopropenyl groups present in poly(limonene carbonate), that possibly lead to enhanced free volume between the poly(limonene carbonate) chains. In addition to employing the vinyl groups of poly(limonene carbonate) for crosslinking purposes, these groups can also be converted into other functional groups via e.g. thiol–ene chemistry to produce functional coatings. For example, incorporation of quaternised ammonium moieties produced a coating with anti-bacterial properties.234 Another strategy consists in epoxidising both double bonds of limonene to prepare limonene dioxide and to polymerise this compound with CO2. The obtained polycarbonate displayed pendant epoxide groups, which were subsequently reacted with a trithiol compound to produce a thermoset coating with a very high Tg, and promising properties such as good acetone resistance and scratch resistance (Table 11, entry 6).239
3.5. Application of cyclic and polymeric carbonates in Li-ion batteries
Since half a century ago, batteries have become a crucial commodity in everyday life. Although the first electrochemical cell dates back to the end of the eighteenth century,363,364 it was during the late 1960s that innovation in the field of energy storage devices received an enormous boost due to widespread applications as those in consumer electronics, but also for more specific ones as medical implant devices.364 An important breakthrough that allowed an increase in energy density, and thus a decrease in the weight of batteries, was the development of rechargeable lithium-based batteries, which operate through transportation of Li+ cations between the two electrodes during charge and discharge (Fig. 12). Lithium has the highest electrochemical equivalent and is the lightest of all metals, which results in an exceptionally high energy density in Li-ion batteries.364–366 Nowadays, lithium-ion batteries are employed in a wide variety of applications, ranging from high-end (consumer) electronics and power tools to hybrid electric vehicles.367,368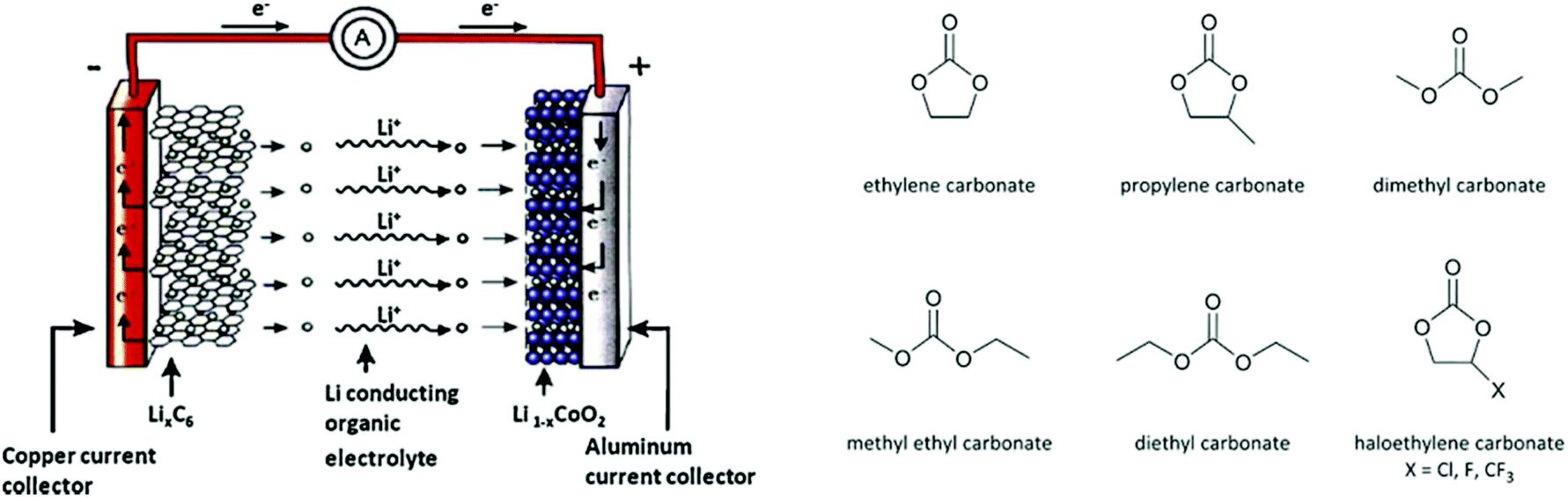 | ||
| Fig. 12 General scheme of a lithium-ion battery (left, reprinted from J. Power Sources, 2010, 195, 2419–2430, with permission from Elsevier) and the most common organic carbonates used in liquid electrolytes (right). | ||
Although liquid electrolytes have been employed in lithium-ion batteries for several decades and extensive research has been carried out to optimise these electrolytes with respect to performance and safety, obstacles still exist that are inherent to the use of conventional liquid electrolytes. One of the safety issues that arises from the use of conventional liquid electrolytes is the high flammability of the commonly used alkyl carbonates.368–370 Since various fluorinated organic compounds serve as flame-retardants, trifluoropropylene carbonate (flash point of 134 °C) was studied as a safer alternative to conventional alkyl carbonates, but its high viscosity limited the ionic conductivity of the electrolyte.369,385,386 Another safety issue is related to the typically employed LiPF6 salt, which becomes reactive towards the electrolyte components at temperatures above 55 °C, possibly leading to permanent damage and explosion hazards.369,370 As the seriousness of fire and explosion hazards becomes more pronounced with larger scale, these safety issues are particularly crucial for large-format batteries, and thereby limit the applicability of liquid electrolyte lithium-ion batteries in e.g. electrical vehicles and industrial operations.369,370
 | ||
| Fig. 13 Schematic drawing of different types of polymer electrolytes: (A) Solid polymer electrolyte. (B) Single lithium-ion conducting solid polymer electrolyte. (C) Gel polymer electrolyte. | ||
3.5.2.1. Polycarbonate-based solid polymer electrolytes. Poly(ethylene oxide) was the first polymer to be investigated for potential application in solid polymer electrolytes for lithium-based batteries.365,387 The first attempts to incorporate SPEs in lithium batteries were limited to large, stationary batteries, as these systems required relatively high operating temperatures (between 60 and 80 °C) and poly(ethylene oxide) tends to crystallise below 60 °C, which is accompanied by a significant drop in ionic conductivity of the electrolyte (between 10−6 and 10−5 S cm−1 at room temperature).366,388 This low ionic conductivity compared to the state-of-the-art organic liquid electrolyte solvents severely limits the application in lithium-ion batteries used at room temperature.365,369,371,389 To date, one of the few commercialised SPE-containing lithium-ion batteries is used as a power source for an electric car developed by Bolloré, wherein poly(ethylene oxide) is employed as SPE component.370 In recent years, the application of aliphatic polycarbonates, and specifically poly(ethylene carbonate), as solid polymeric electrolytes has gained increasing attention. Aliphatic polycarbonates generally display low glass transition temperature (see section 3.2, Table 8), accompanied by high chain mobility and ion transport required for proper battery operation.390 These features render aliphatic polycarbonates particularly attractive for electrolyte applications.391 Additionally, the polar nature of the carbonate group offers a high solvation power towards numerous salts.389 One of the first reports on CO2-derived polycarbonates as electrolytes was based on the use of various polycarbonates synthesised via the coupling of CO2 with a range of glycidyl ethers (Fig. 14A) using a zinc glutarate catalyst.304,392 Electrochemical analysis showed that these polycarbonate-based electrolytes (with ionic conductivities reaching 10−6 S cm−1 at 30 °C) did not represent an improvement compared to the benchmark poly(ethylene oxide) electrolytes. Increasing the amount of lithium salt in the electrolyte formulation and raising the operating temperature, drastically improved the ionic conductivity. Poly(n-butyl-glycidyl ether carbonate) with 65 wt% LiTFSI (lithium bis(trifluoromethane sulphonyl) imide) showed enhanced ionic conductivities in the order of 10−5 S cm−1 at 25 °C and 10−3 S cm−1 at 120 °C,305 the latter being close to the typical ionic conductivities for liquid electrolyte lithium-ion batteries at room temperature (≥10−3 S cm−1). For comparison, an electrolyte consisting of poly(ethylene oxide) with 65 wt% LiTFSI displayed ionic conductivity in the order of 10−5 S cm−1 at 20 °C and in the order of 10−3 S cm−1 at 80 °C.393 The presence of pendant ether groups is not a strict requirement, as proven by a poly(ethylene carbonate) electrolyte with 80 wt% loading of LiTFSI salt that displayed an ionic conductivity in the order of 10−5 S cm−1 at 30 °C. In general, the increase in the amount of lithium salt in poly(ethylene carbonate)-based electrolytes leads to an increase in ionic conductivity.394 However, the lithium salt concentration was also observed to impact the thermomechanical properties of the polymeric electrolyte, as the glass transition temperature and storage modulus initially increased for LiTFSI concentrations up to a molar ratio of 0.2 (approximately 39 wt% LiTFSI), while for higher lithium salt concentrations a decrease in these quantities was observed.394 The inclusion of several types of additives in the formulation of polymeric electrolytes based on polycarbonates has been studied. Ionic liquids have very low vapour pressures, are non-flammable and possess high ionic conductivities (in the order of 10−3 S cm−1 at room temperature),395 making them potentially valuable additives in (polymeric) electrolyte formulations.367 This was investigated by preparing a polycarbonate-based electrolyte consisting of poly(ethylene carbonate), a LiTFSI salt and a pyrrolidinium-based ionic liquid (Pyr14TFSI) as a plasticiser and an ionic conductivity enhancer.396 However, ionic conductivities ranging from 10−7 S cm−1 at 50 °C to 10−5 S cm−1 at 90 °C were obtained, which are too low for battery applications and are inferior compared to the benchmark poly(ethylene carbonate)/LiTFSI electrolytes.389 This drawback is partially counterbalanced by a rather high lithium transference number (0.66 at 80 °C).396 Inorganic fillers have been reported to increase the ionic conductivity of poly(ethylene oxide)-based electrolytes at ambient temperatures by preventing crystallisation at T < 60 °C, and to enhance their mechanical properties.388 In analogy to this approach, inorganic fillers have been included in polycarbonate-based electrolytes. For example, a submicron-sized silica fibre was added to the aforementioned poly(ethylene carbonate)/LiTFSI/Pyr14TFSI electrolyte formulation,397 leading to a slight enhancement of the mechanical properties (the Young's modulus increased from 0.35 to 0.39 MPa) and an increase in ionic conductivity (10−7 S cm−1 at 30 °C and 10−5 S cm−1 at 80 °C), which is nevertheless still far from the requirements for battery applications. The addition of TiO2 as inorganic filler to a poly(ethylene carbonate) electrolyte with LiFSI salt (with 0.53
:1 polymer-to-salt molar ratio) proved more effective,257 leading to an increase of the lithium transference number from 0.54 to 0.81 at 60 °C upon addition of 1 wt% TiO2, while also resulting in an increase in ionic conductivity, reaching values in the order of 10−5 S cm−1 at 30 °C and 10−3 S cm−1 at 80 °C. Another approach to improve the ionic conductivity of SPEs is to alter the chemical structure of the polymer chains, in order to decrease the glass transition temperature and, thus, to increase the chain mobility at lower temperatures. In the context of polycarbonate-based electrolytes, this pathway was explored by tuning the CO2/epoxide polymerisation reaction so that both carbonate and ether linkages are present in the polymer backbone.398 This allowed combining the high lithium transference number commonly associated with aliphatic polycarbonates with lower glass transition temperature bestowed by ether linkages, resulting in a poly(ethylene carbonate/ethylene oxide)/LiFSI electrolyte with an ionic conductivity in the order of 10−4 S cm−1 at 60 °C and a lithium transference number of 0.66 (under optimised salt concentrations).398 On the other hand, a low Tg can be a drawback because the practical application of polyelectrolytes also requires mechanical and dimensional stability. In the case of electrolytes based on poly(ethylene carbonate), glass transition temperatures as low as −47 °C have been reported,257 which limits the practical applicability of these polyelectrolytes as a self-standing membrane in battery applications.399 To overcome this obstacle, efforts have been made to combine a poly(ethylene carbonate)/LiFSI electrolyte with a three-dimensionally ordered macroporous polyimide matrix.399 Ionic conductivity in the order of 10−5 S cm−1 was reached at 30 °C, which was only slightly lower than the conductivity without the polyimide matrix. DSC measurements did not show any significant change in the glass transition temperature after combination of the electrolyte with the polyimide matrix, indicating that the matrix does not influence the ionic conductivity by altering the Tg. Although the authors did not perform any mechanical analysis, they were able to fabricate an all-solid-state lithium battery incorporating the poly(ethylene carbonate)/LiFSI/polyimide electrolyte, which showed a higher battery capacity than the same battery with a conventional ethylene oxide/propylene oxide copolymer electrolyte and did not suffer short circuit failures nor mechanical instability during 30 charge/discharge cycles.399
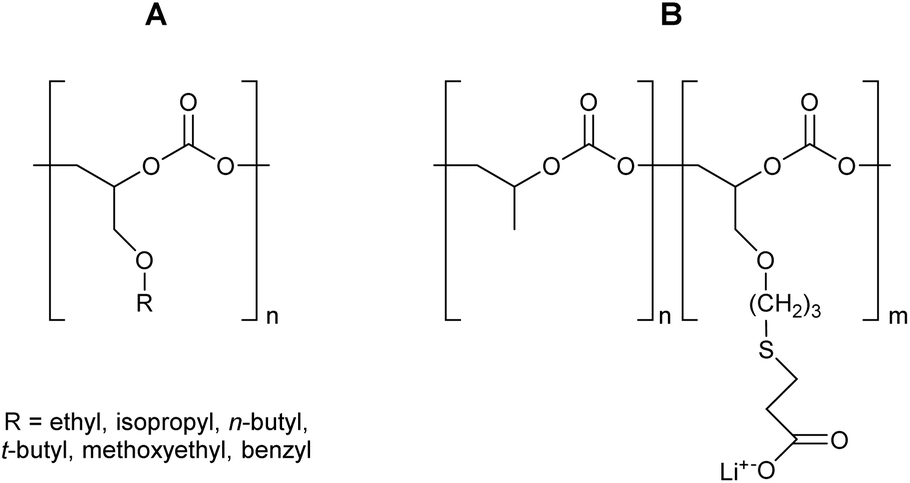 | ||
| Fig. 14 Glycidyl ether-based polycarbonates for application in polyelectrolytes: (A) Solid polymer electrolyte. (B) Single lithium-ion conducting solid polymer electrolyte. | ||
Next to the extensive research towards poly(ethylene carbonate)-based electrolytes, poly(propylene carbonate) has also been investigated as potential solid polymer electrolyte in energy storage devices.400 Various blends with different ratios of poly(propylene carbonate), poly(ethylene oxide) and a LiClO4 salt produced polymeric electrolytes that showed enhanced ionic conductivity compared to conventional pure poly(ethylene oxide).401 The highest measured ionic conductivity was in the order of 10−5 S cm−1 at room temperature for an electrolyte incorporating a 1:1 mass ratio of poly(propylene carbonate):poly(ethylene oxide) mixed with 10 wt% LiClO4. Polyelectrolytes consisting solely of poly(propylene carbonate) and LiClO4 possess an ionic conductivity in the order of 10−5 S cm−1 at room temperature and 10−3 S cm−1 at 80 °C for an electrolyte with a poly(propylene carbonate)-to-LiClO4 molar ratio of 10:1.402 It is worth noting that the ionic conductivity of poly(propylene carbonate)-based electrolytes is typically higher than that of poly(ethylene carbonate)-based electrolytes. This has been attributed to the presence of the pendant methyl groups on the poly(propylene carbonate) chains, imparting a lower degree of crystallinity, which was observed to positively influence the ionic conductivity.402 The addition of the 1-butyl-3-methylimidazolium tetrafluoroborate (BMIM+BF4−) ionic liquid to a poly(propylene carbonate) electrolyte containing LiClO4 led to an increase in ionic conductivity from 10−6 S cm−1 to 10−3 S cm−1 at 20 °C.403 This increase in conductivity is the opposite of what was observed for the addition of an ionic liquid to the above-mentioned poly(ethylene carbonate)-based electrolyte,396 where incorporation of a ionic liquid resulted in a decrease in conductivity (though it should be noted that the type of ionic liquid, the lithium salt and the relative amounts of these components differed in the two cases). The increase in conductivity of the poly(propylene carbonate)-based electrolyte observed in this work was explained by FTIR measurements, which showed that the presence of the ionic liquid cations caused a weakening of the complexation between poly(propylene carbonate) and Li-ions, resulting in a higher lithium-ion mobility. Thermal analysis indicated that addition of 3 mass eq. of BMIM+BF4− compared to poly(propylene carbonate) also caused a significant decrease in the Tg from 3 to −70 °C, while the thermal stability improved (Td5% increased from 166 to 231 °C). A drawback of this system is that at ionic liquid-to-polycarbonate weight ratios ≥1, the polyelectrolyte is not anymore a self-standing solid film, but rather a soft, sticky and non-fluidic gel. On the other hand, ionic conductivities at weight ratios <1 are in the range of 10−4 S cm−1 or less (20 °C), which is less attractive with respect to application in lithium batteries.
One of the disadvantages of using SPEs composed of a polymer matrix and a lithium salt is the generally low lithium transference number, caused by the fact that only a small amount of the ionic current is related to the motion of lithium-ions, while the rest of the ionic current is related to the motion of the mobile anions (e.g. TFSI−, FSI−, ClO4−).370,391 This leads to polarisation within the cell, giving rise to undesired effects such as voltage drop, limited power supply and, finally, leading to cell failure.370,391 A possible approach to overcome this limitation is the substitution of conventional polymer electrolyte systems with single lithium-ion conducting solid polymer electrolytes (SLIC-SPEs, Fig. 13B), which can be prepared by immobilising the anions in the polymeric backbone in the form of functional pendant groups.370,391 For this purpose, a single-ion polycarbonate was synthesised by reacting allyl glycidyl ether and propylene oxide with CO2, employing a zinc glutarate catalyst.391 The pendant vinyl groups on the polycarbonate backbone were functionalised with 3-mercaptopropionic acid and subsequently lithiated with lithium hydroxide to produce a single-ion-conducting polymer (Fig. 14B). This single-ion polycarbonate electrolyte displayed an ionic conductivity in the range of 10−4 S cm−1 at 80 °C and a lithium transference number of 0.86, which is considerably higher than most lithium transference numbers determined for analogous polycarbonate-based SPEs containing lithium salts.
3.5.2.2. Polycarbonate-based gel polymer electrolytes. Although solid polymer electrolytes generally impart better mechanical properties, flexible battery manufacturing and safer operation than liquid ones, they still suffer from moderate conductivities at operating conditions (vide supra). Gel polymer electrolyte (GPE)s contain both a polymeric matrix and a liquid in which a lithium salt is dissolved (Fig. 13C) and, in this sense, they can be considered hybrid systems between SPE and liquid electrolytes. Therefore, GPE systems may optimally combine the best of both worlds, although the risk is to combine the worst of both worlds as well. For example, GPEs typically display higher ionic conductivities than SPEs but the safety issues associated with liquid electrolytes (i.e. flammable components and electrochemical instability) are not completely avoided.368,370 GPEs are currently used in lithium-ion polymer batteries (LiPBs) employed in electrical vehicles and consumer goods such as mobile phones and notebooks, typically incorporating polymers such as poly(ethylene oxide), poly(acrylonitrile), poly(methyl methacrylate) and poly(vinylidene fluoride).368,369,373 CO2-based polycarbonates have also been studied as components of GPEs. Terpolymerisation of CO2, propylene oxide and maleic anhydride, followed by radical crosslinking with dicumyl peroxide as radical initiator, produced a polymer matrix that was used as a GPE in combination with a liquid LiClO4/ethylene carbonate/dimethyl carbonate electrolyte.404 This system showed rather high ionic conductivities (in the range of 10−3 S cm−1 at room temperature and 10−2 S cm−1 at 50 °C) and a lithium transference number of 0.42. A lithium cell incorporating this gel polyelectrolyte exhibited an initial battery capacity only slightly lower than that of commercial fully liquid electrolyte batteries.404,405 Blending of aliphatic polycarbonates with other polymers can also produce matrices with potential utilisation in GPE applications. A blended polybutadiene/poly(propylene carbonate) crosslinked matrix (70
:30 mass ratio) in which a liquid LiPF6/ethylene carbonate/dimethyl carbonate electrolyte was absorbed, displayed ionic conductivities in the range of 10−3 S cm−1 between room temperature and 80 °C.406 Thermal and mechanical analysis indicated that the polymeric electrolyte had proper thermal and mechanical stability due to a slightly higher initial degradation temperature than pure poly(propylene carbonate) (272 °C vs. 260 °C) and an achievable preparation of a robust, self-standing and flexible film with the absence of liquid flow. A lithium-ion battery cell containing this electrolyte maintained 84% of the original capacity value after 70 charge/discharge cycles. Similarly high ionic conductivities were measured for a different GPE system consisting of an electrospun poly(vinylidene fluoride)/poly(propylene carbonate) matrix swollen by a LiPF6/ethylene carbonate/dimethyl carbonate liquid electrolyte.407 Moreover, excellent cycling stability was observed with no apparent capacity decrease even after 100 charge/discharge cycles, whereas a commercial polymeric matrix based on polyethylene preserved 90% of the original capacity under the same conditions.
3.6. Polycarbonates in biomedical and pharmaceutical applications
Both natural as well as man-made polymers find a wide variety of uses in the field of medicine, where they are being employed in wound dressings, drug delivery applications, surgical implants and medical devices.408 For the specific utilisation of polymers inside the human body, the most important features are biodegradability and biocompatibility. Biodegradability should be carefully tuned because an unwanted degradation profile, e.g. slow degradation of short-term implants or fast degradation of long-term tissue scaffolds, and harmful degradation products can severely limit the range of applicability.409 Biocompatibility is also crucial because biomedical polymers that have to perform a task inside the human body (e.g. drug delivery systems and implants such as tissue scaffolds) should accomplish the desired function without causing any unwanted local or systemic effects in the patient.410 For the specific application of biomaterials as implants in the human body, the mechanical and chemical stability are also important, as the pH in different tissues ranges from 1 to 9 and daily activities exert stresses of approximately 4 MPa on bones and stresses in the range of 40–80 MPa on tendons and ligaments.411 Polyesters such as poly(lactic acid), poly(glycolic acid), poly(lactic-glycolic acid), polycaprolactone and polyhydroxyalkanoates are the most intensely studied polymers in the field of biomedical applications due to their biodegradability, low toxicity and the related good biocompatibility.408,409,412 However, one of the disadvantages of employing polyesters is that in vivo biodegradation of the majority of the used polyesters generates acidic products, thereby decreasing the local pH and triggering necrosis of host cells and inflammatory responses.409,413,414 Consequently, for implantation sites that exhibit low metabolic activity and, therefore, cannot sufficiently process degradation products, polyester-based materials are not an optimal choice.409 CO2-based aliphatic polycarbonates have been studied as a possible alternative to polyesters as materials for biomedical and pharmaceutical applications,248–250 owing to their biodegradability (see section 3.2) and good biocompatibility. Although typically not derived from CO2, a related aliphatic polycarbonate, poly(trimethylene carbonate), has already been approved for application in biological fields by the United States Food and Drug Administration (FDA).415,416 Poly(propylene carbonate) is the most widely studied CO2-based polymer for biomedical applications. Studies on both base-catalysed hydrolysis as well as thermal decomposition showed that propylene carbonate and 1,2-propanediol are the major initial degradation products of poly(propylene carbonate) decomposition.417,418 Apart from causing serious eye and slight skin irritation at high concentrations, propylene carbonate was reported not to display any significant toxicity during in vivo tests via oral insertion and inhalation (although the effects on the human body have not yet been thoroughly investigated),223,419 while 1,2-propanediol is commonly used in a variety of food applications and in the medical and pharmaceutical industry, it displays no toxicity and can be metabolised by the human body.420 Additionally, it has been suggested that poly(propylene carbonate) will eventually degrade into CO2 and water,413,414,421,422 which would trigger less inflammatory responses compared to the degradation products of polyesters, although no scientific study has been presented in which this degradation pathway is observed. Poly(propylene carbonate) typically displays an elastic behaviour and possesses an amorphous structure accompanied by a glass transition temperature in the range of body temperature (35–40 °C), which, in combination with its biodegradability, renders it potentially interesting for biomedical applications in soft tissue scaffolds or drug delivery. On the other hand, the low glass transition temperature and moderate mechanical strength (Table 12, entries 1a–d) make it unsuitable for application in rigid tissue scaffolds such as bone substituents.413,423 With the target of tuning the properties of poly(propylene carbonate) towards biomedical applications, this CO2-based polymer has been combined with other polymers (e.g. polyesters), either through terpolymerisation reactions or as blends. The thermal and mechanical properties of several researched poly(propylene carbonate)-based formulations for biomedical applications are summarised in Table 12. On a critical note, it can be seen that the characterisation of these properties is often incomplete. A more thorough analysis of the thermal and mechanical properties of these formulations would be advisable to be able to fully appraise the potential of these materials in biomedical applications such as tissue engineering. Polymer films of poly(carbonate-co-ester)s that were prepared by terpolymerisation of CO2, propylene oxide and caprolactone were compared with pure poly(propylene carbonate) and polycaprolactone with respect to the degradation behaviour employing various enzymes at either 37 or 60 °C.412,424 The degradation rates of poly(propylene carbonate-ε-caprolactone) were comparable to those of conventional polycaprolactone homopolymers. The study did not include an investigation of the structure and effects of the degradation products. This would have been desirable, since the products of polycaprolactone degradation have been reported to cause necrosis of host cells and inflammation.409,413,414 Another approach consisted in blending poly-3-hydroxybutyrate, which is characterised by a relatively stiff, rigid and brittle structure,425 with poly(propylene carbonate), which exhibits a more elastic behaviour with relatively low tensile strength.413In vivo radiolabelling studies showed that pure poly(propylene carbonate) degraded much faster than the poly(propylene carbonate)/poly-3-hydroxybutyrate blend. Both the pure polycarbonate and the blend exhibit good biocompatibility, as they did not show any toxicity in vivo and displayed proper biocompatibility with in vitro red blood cells. Although the mechanical properties of the poly(propylene carbonate)/poly-3-hydroxybutyrate blend still have to be investigated, the material shows potential to be used in biomedical applications that require longer degradation times than pure poly(propylene carbonate). Polymers incorporating glycerol building blocks (e.g. polyglycerol ethers, polyglycerol carbonates, polyether esters or polycarbonate esters containing glycerol units) have also received extensive attention in the field of biomedicine due to: (i) their biodegradability yielding non-toxic products such as glycerol and CO2; (ii) the presence of pendant hydroxyl groups that can be functionalised with chemotherapeutic agents, antibacterial compounds, anti-inflammatory agents, fluorescent tags or material-property modifiers; (iii) their suitability to be used in manufacturing techniques such as electrospinning.426 Within this group, CO2-based glycerol polycarbonates have been synthesised via polymerisation of benzyl glycidyl ether and carbon dioxide, followed by deprotection of the polymer to produce atactic and isotactic poly(1,2-glycerol carbonate)s.426 The degradation behaviour of these CO2-based poly(1,2-glycerol carbonate)s (Fig. 15A) was monitored (at 37 °C) and compared to the degradation of poly(1,3-glycerol carbonate) (Fig. 15B), a polymer that is synthesised via ring-opening polymerisation of six-membered cyclic carbonates and that has been investigated for use in biomedical applications such as drug-loaded buttressing films for the prevention of tumour recurrence after surgical resection,427 and particles for drug delivery.428 It was seen that the CO2-based poly(1,2-glycerol carbonate)s exhibited rather high degradation rates with a half-life time of 2–3 days, while poly(1,3-glycerol carbonate) did not show any degradation over a course of 4 days under the same conditions. This difference in degradation behaviour was attributed to a higher tendency of the primary hydroxyl group in the poly(1,2-glycerol carbonate) repeating unit to initiate an intramolecular attack compared to the secondary hydroxyl present in the poly(1,3-glycerol carbonate) repeating unit. This behaviour can prove advantageous for biomedical applications that require fast degradation rates. In a related study, poly(glyceric acid carbonate) (Fig. 15C) was synthesised as a CO2-based and biodegradable analogue of poly(acrylic acid) for application in e.g. drug delivery.429 Crosslinked hydrogels derived from poly(glyceric acid carbonate) readily degraded in basic water (pH = 8.4), while analogues crosslinked hydrogels based on poly(acrylic acid) did not. This was ascribed to the presence of biodegradable carbonate linkages in the poly(glyceric acid carbonate) chains. For the application of polymers in tissue scaffolds, the biocompatibility is also related to the attachment of living cells to the biomaterial and the subsequent spreading over the implant. In this context, the hydrophobicity of aliphatic polycarbonates such as poly(propylene carbonate) and poly(cyclohexene carbonate) is a limitation as it leads to poor cell adhesion and slow biodegradation.422,430 One way to overcome this issue is by incorporating hydrophilic moieties in the polycarbonate chains, e.g. via terpolymerisation of CO2, propylene oxide and a hydrophilic monomer such as ME3MO (2-((2-(2-(2-methoxyethoxy)ethoxy)ethoxy)methyl) oxirane (Fig. 15D).430 Polycarbonates containing at least 26 mol% of ME3MO showed reversible thermo-responsive solubility in water with lower critical solution temperatures between 7.0 and 35.2 °C. Another study investigating the hydrophilicity of CO2-based polycarbonates focussed on a series of poly(carbonate-ether)s made from CO2 and ethylene oxide, in which the carbonate content ranged from 1 to 43 mol%.434 It was observed that by varying the molecular weight of the polymer and the amount of carbonate units, water-soluble polycarbonates can be prepared with lower critical solution temperature between 21.5 and 84.1 °C. Specifically, poly(ethylene carbonate-ether) with 26 mol% carbonate content displayed a lower critical solution temperature of 36.1 °C (i.e. close to human body temperature). The possibility to tune poly(ethylene carbonate-ether) and the aforementioned polycarbonate derived from CO2/propylene oxide/Me3MO to possess lower critical solution temperature is promising with respect to application in biomedical materials, as thermo-responsive water solubility has potential in controlled drug delivery systems,435 construction of tissue engineering scaffolds,436 molecular gates and gene carriers.437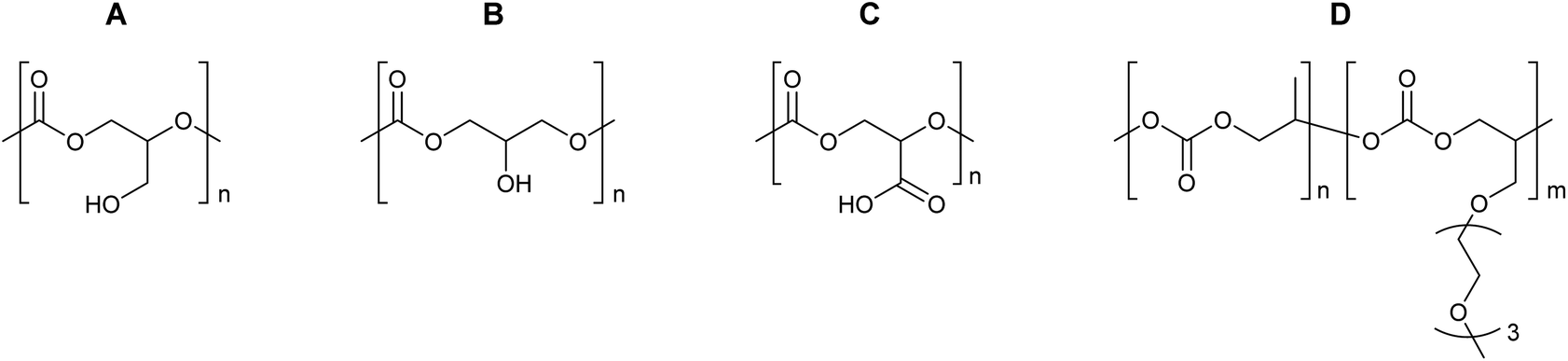 | ||
| Fig. 15 Examples of aliphatic polycarbonates that have been evaluated for their potential in biomedical applications: (A) Poly(1,2-glycerol carbonate). (B) Poly(1,3-glycerol carbonate). (C) Poly(glyceric acid carbonate). (D) Terpolymer synthesised from CO2, propylene oxide and ME3MO. | ||
| Polymer formulation | # | T g (°C) | T m (°C) | T d (°C) | Tensile modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) | Compressive modulus (MPa) | Compressive yield strength (MPa) | Storage modulus (MPa) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Poly(propylene carbonate) [PPC] | 1a | — | — | — | — | — | — | 0.38 | — | — | 431 |
| 1b | 25–45 | — | ±240 (initial) | 700–1400 | 7–30 | — | — | — | — | 245 | |
| 1c | 29 | — | — | — | — | — | — | 0.13 | 30 | 423 | |
| 1d | — | — | 245 (DSC) | — | — | — | 0.2 | — | — | 414 | |
| Electrospun PPC fibres | 2a | 38 | 103 | 210 (5 wt%) | 10190 |
22.11 | 10.71 | — | — | — | 432 |
| 2b | — | — | — | 429.63 | 7.37 | 154.93 | — | — | — | 433 | |
| Electrospun PPC/15 wt% gelatine/acetic acid | 3 | — | — | — | 295.72 | 7.72 | 95.26 | — | — | — | 433 |
| PPC/1 wt% graphite oxide | 4 | 39.5 | — | — | — | — | — | — | 1 | 1600 | 423 |
| PPC/50 wt% starch | 5 | — | — | 270 (DSC) | — | — | — | 33.9 | — | — | 414 |
| PPC/starch/plasticiser/bioglass | 6 | — | — | — | — | — | — | 80.0 | — | — | 421 |
In addition to terpolymerisation, surface modification of polycarbonates has also been explored to enhance the biocompatibility of aliphatic polycarbonates. A potential advantage of surface modifications is that only the polymeric surface is altered, while the bulk mechanical strength is maintained. Surface modification by aminolysis with polyethylenimine was used to introduce amino groups on the surface of a poly(propylene carbonate) membrane.422 These amino functionalities served as an initial layer for consecutive layer-by-layer assembly, in which multiple molecular layers of polyethylenimine/gelatine were deposited on the amino-functionalised poly(propylene carbonate) surface, based on the ionic interaction of the positively charged polyethyleneimine and negatively charged gelatine at pH = 7.4. A decrease in water contact angle after surface modification indicated the increased hydrophilicity of the modified poly(propylene carbonate). With respect to biocompatibility, the composite consisting of poly(propylene carbonate) with three bilayers of polyethylenimine/gelatine possessed enhanced compatibility with cells compared to both unmodified poly(propylene carbonate) as well as to a polycaprolactone benchmark, probably due to the presence of the outer gelatine layer that can suppress the cytotoxicity generally displayed by polyethyleneimine.422 The improved biocompatibility of poly(propylene carbonate)/polyethylenimine/gelatine with living cells indicates that the material could be a potential alternative to conventional polycaprolactone for tissue engineering applications. To investigate this further, foaming of poly(propylene carbonate) with supercritical CO2 and subsequent modification by aminolysis and layer-by-layer assembly techniques were employed to produce three-dimensional poly(propylene carbonate)/polyethyleneimine/gelatine foam structures.431 A maximum compressive modulus of 0.38 MPa was obtained for unmodified poly(propylene carbonate) (Table 12, entry 1a), while an average decrease in modulus of 13% was seen after modification with polyethyleneimine and gelatine. Despite this decrease, the values are still in the range required for meniscal tissue (0.22 MPa) and articular cartilage (0.51–15.3 MPa) repairs.431 However, for utilisation of the polymer in temporary tissue scaffolds that are meant to degrade in the human body, studies investigating the biodegradability of the polymer and the biocompatibility of the degradation products should still be conducted, especially since polyethyleneimine is known to exhibit cytotoxicity.
In the context of applied tissue engineering, the three-dimensional structure of biomedical scaffolds, though often not thoroughly investigated,438 can significantly influence the biodegradability and mechanical properties of the material. Ideally, the scaffold should display: (i) porosity that enables cell migration and the transport of nutrients and waste, (ii) a surface that promotes cell adhesion, growth, migration and differentiation, and (iii) a degradation profile that closely matches the regeneration profile of the desired natural tissue.433 Three-dimensional poly(propylene carbonate) architectures were prepared via electrospinning, which readily allows the production of interconnected flexible nanofibrous structures.432 These electrospun poly(propylene carbonate) structures displayed slightly lower thermal stability and slightly enhanced mechanical properties compared to conventional poly(propylene carbonate) (Table 12, compare entry 2a to 1b). Additionally, cell adhesion experiments indicated proper biocompatibility of the three-dimensional poly(propylene carbonate) structures, as the cells were seen to grow, migrate and differentiate. Electrospinning was also applied to blends of CO2-based polycarbonates with a hydrophilic polymer such as gelatine.433 Although cell culture experiments showed that these blends possessed enhanced biocompatibility compared to pure poly(propylene carbonate), high content of gelatine also resulted in a decline in mechanical strength compared to pure poly(propylene carbonate) fibres (Table 12, entry 2b), as a consequence of the poor mechanical properties of gelatine. This negative effect was partially countered by addition of a small amount of acetic acid (Table 12, entry 3), probably due to the increased miscibility of poly(propylene carbonate) and gelatine, which was related to the pH-dependent behaviour of gelatine. Whereas addition of gelatine led to a decrease in mechanical strength, the mechanical properties of poly(propylene carbonate) can be enhanced by means of other additives, which can be as diverse as graphite oxide423 or starch.414 Addition of as little as 1 wt% graphite oxide to poly(propylene carbonate) significantly increased the glass transition temperature, storage modulus and compressive yield strength of the obtained composite compared to the pure polymer (Table 12, compare entry 4 to 1c), without affecting negatively the biocompatibility.423 Also blending of poly(propylene carbonate) with starch remarkably affected the mechanical and thermal properties, as shown by the significant increase in the compressive modulus and decomposition temperature upon blending with 50 wt% starch (Table 12, compare entry 5 to 1d).414In vitro and in vivo biocompatibility studies combined with degradation measurements showed that the poly(propylene carbonate)/starch blends possessed excellent cytocompatibility, as shown by the formation of only a scar tissue as a mild inflammatory response after in vivo implantation of the composite. The long-term (2 months) in vivo degradation of the poly(propylene carbonate)/starch composite was compared with that of poly(lactic acid) and showed that, in contrast to poly(lactic acid), the polycarbonate/starch blend was properly tolerated, without the occurrence of inflammation and immune cell responses. In vitro biodegradation studies indicated that the degradation rate of the poly(propylene carbonate)/starch blend after 8 weeks (13 wt%) is comparable to that of poly(lactic acid) (8 wt%). Although the biocompatibility and biodegradation of the polycarbonate/starch blend seem promising, the relatively hydrophobic surface of the composite might limit its practical application as it can restrict or delay cell growth and ultimately hamper tissue regeneration.421 To enhance the interaction of the polycarbonate/starch composite with living cells, water and glycerol were added as plasticisers together with 10 wt% bioglass.421 The addition of plasticisers improved the compressive modulus considerably (Table 12, entries 5 and 6), while the addition of bioglass enhanced the cell growth on the polycarbonate/starch composite. The material was used to produce biomedical screws that were well tolerated in in vivo tests and exhibited higher degradation rates than the poly(lactic acid) analogues, accompanied by an enhanced bone regeneration profile. These features show that poly(propylene carbonate)/starch composites incorporating plasticisers and bioglass possess great potential for applications in musculoskeletal tissue repair.
In addition to rigid and strong mechanical structures useful as tissue scaffolds, flexible polymers also find use in biomedical applications, especially in cases in which it is difficult to insert biomedical implants in the human body due to obstruction by organs, bones and veins or in sensitive areas such as the eyes or arteries. In these cases, flexible shape-memory materials can offer advantages over rigid structures, as they can be folded, deformed and subsequently introduced, e.g. through small catheters, where they can recover their original shape once placed in the desired location and orientation.439 Within this group of materials, polyurethanes exhibiting shape-memory have been investigated for utilisation in endovascular intervention procedures.439 Shape-memory polyurethanes based on polycarbonate polyols (see also section 3.4.3) have also been reported.347 Star-type oligo(carbonate-ether)s with three arms were synthesised via the reaction of CO2 and propylene oxide in the presence of trimesic acid as an initiation-transfer agent.346 Consecutive reaction of these triol compounds with polyethylene glycol and 1,6-hexamethylene diisocyanate afforded crosslinked CO2-based polyurethane networks.347 Shape-memory tests showed that by tuning the molecular composition it was possible to obtain polyurethane networks that after deformation almost immediately recovered their original structure upon immersion in water at 70 °C. Furthermore, initial cytotoxicity tests indicated that the shape-memory polymer possesses good biocompatibility.
In addition to the utilisation of CO2-based polycarbonates in tissue engineering, research has also been devoted to the exploration of these polymers in pharmaceutical applications such as drug delivery440,441 and tumour imaging.415 A poly(carbonate-ester) synthesised via terpolymerisation of CO2, 1,2-butylene oxide and ε-caprolactone was examined as carrier for the antibiotic drug pazufloxacin mesilate.440 The drug release rate could be tuned by varying the carbonate/ester ratio: both faster hydrolytic degradation as well as higher drug release rates were observed for poly(carbonate-ester)s with higher ε-caprolactone content. Similar trends in drug release behaviour were observed for poly(carbonate-ester)s synthesised from CO2, propylene oxide and ε-caprolactone carrying the insecticide imidacloprid.441 This can be ascribed to the higher hydrolytic degradation rate of the ester blocks compared to the carbonate blocks. In recent work, an amphiphilic polycarbonate based on the triblock copolymer poly(allyl glycidyl ether carbonate)-poly(propylene carbonate)-poly(allyl glycidyl ether carbonate) was functionalised and labelled with gadolinium as MRI contrast agent.415 The resulting polymer micelles were evaluated for utilisation in tumour imaging. While in vitro investigations indicated that the polycarbonate/gadolinium micelles degraded into non-toxic products, in vivo experiments showed that the polymer micelles were cleared from the body within 72 h without any evidence of toxicity. Furthermore, the polycarbonate-based contrast agent showed superior MRI imaging capability compared to a commercial gadolinium contrast agent.
For the sake of completeness, it should be noted that not all polycarbonates that can be potentially employed in biomedical applications are synthesised via polymerisation of CO2 and epoxides, as aliphatic polymeric carbonates can also be produced via ring-opening polymerisation of six-membered cyclic carbonates.442–445Via this method several functional groups that are useful in biomedical applications can be incorporated in the polycarbonates,442–446 with potential applications including drug and gene delivery447–449 and antibacterial/antifouling coatings for medical devices.450 These six-membered cyclic carbonates can still be derived from CO2via two pathways: (1) synthesis of a five-membered cyclic carbonate through the reaction between CO2 and an epoxide, followed by transcarbonation with a 1,3-diol (see section 3.1). However, this route requires multiple reaction steps and will be hampered by the possible presence of other protic functionalities in the diol compound (i.e. hydroxyl or amino groups); (2) direct reaction of CO2 with an oxetane to form a six-membered cyclic carbonate, which is typically more difficult due to the lower accessibility and reactivity of oxetanes compared to epoxides.42
3.7. Other applications of CO2-based polycarbonates
In addition to the application of CO2-based polymeric carbonates in the major fields discussed in sections 3.4–3.6 (i.e. polymer products, battery applications and biomedical applications), these polycarbonates also find use in other applications. While some of these are yet of mere academic interest, others have already reached commercial status. Examples of the latter include the use of poly(propylene carbonate) and poly(cyclohexene carbonate) as sacrificial binder material in the production of ceramics and adhesives,15,242,451 owing to the clean and controllable thermal decomposition of these polycarbonates at temperatures below 300 °C. In recent years, Novomer investigated the end-chain modification of poly(propylene carbonate) and poly(ethylene carbonate) to incorporate functional groups and the use of these polycarbonates in block-copolymers with hydrophilic polymers.274,362,452 The obtained polymers were studied as drop-in alternatives to conventional polymers such as polypropylene and polystyrene.17 Another application for which poly(propylene carbonate) is considered of commercial interest is as packaging material, especially when the poly(propylene carbonate) chains have low ether linkage content and high head-to-tail ratio, which results in improved processability.453 Packaging material for food and medical devices is of particular interest, as it was observed that the mechanical properties (i.e. tensile modulus, ultimate strength and tear resistance) of poly(propylene carbonate) are comparable yet slightly better than those of low-density polyethylene, whereas the permeability to oxygen and water is remarkably lower.267,454 The excellent barrier properties for oxygen and water also creates potential for poly(propylene carbonate)s to be used as barrier adhesive in oxygen- and water-resistant materials.255,455 In contrast, poly(limonene carbonate) possesses very high gas permeability, which, combined with its relatively good mechanical properties, transparency and thermal insulation properties, renders it potentially suitable for application as “breathing glass” in energy-efficient buildings or in greenhouses, providing passive ventilation through these windows and thereby eliminating the need for additional ventilation systems.4564. Concluding remarks and perspectives
The synthesis of cyclic and polymeric carbonates via the reaction of CO2 with epoxides is a thriving pathway for the fixation of CO2 into valuable products, both from an academic as well as an industrial point of view. In the last decades, numerous homogeneous and heterogeneous catalytic systems (or combinations of the two) have been developed for this reaction, thus enabling the reaction of carbon dioxide with a wide array of epoxides, with high yields and selectivities towards either the cyclic carbonate or the polycarbonate products. Future advancements within CO2/epoxide catalysis will probably bring forth novel systems in the relatively new field of metal-free organocatalysis, as well as catalytic systems that are able to convert typically challenging substrates such as bio-based epoxides and functional epoxides. Both these advancements would further increase the green character of this reaction. In the perspective of commercial application, another challenge will be to develop robust and reusable catalysts that are able to perform under the often less-than-ideal conditions of industrial process (e.g. impurities in the CO2 feed), while still maintaining high activity and selectivity. Another challenge related to catalyst design will lie in developing multifunctional catalytic systems that are able to promote both the reaction between CO2 and epoxides and the prior epoxidation of the corresponding alkene with high activity and selectivity, in order to allow the one-pot synthesis of cyclic and polymeric carbonates from alkenes. This combined pathway would be advantageous from a green chemistry point of view since it would allow to avoid handling of toxic epoxides and would result in improved process efficiency.The products of the reaction between CO2 and epoxides, i.e. cyclic carbonates and polycarbonates, have been extensively studied with regard to their potential applications. One of the earliest and possibly largest applications of cyclic carbonates (mainly ethylene carbonate and propylene carbonate) is as electrolyte components in lithium-ion batteries, which was already commercialised almost three decades ago. On the other hand, the use of CO2-based polycarbonates in solid polymer electrolytes is still limited by unsatisfactory ionic conductivities and insufficient lithium transference numbers at ambient temperatures. The incorporation of additives such as inorganic fillers or ionic liquids may enhance these properties, though the influence of such additives still needs to be rationalised and controlled. Gel polymer electrolytes incorporating polycarbonates already possess electrochemical and mechanical properties that are suitable for practical application in lithium-ion batteries, but safety concerns related to the presence of flammable solvents still need to be tackled.
Another example of industrial application of cyclic carbonates is the Asahi Kasei process to produce bisphenol-A-based polycarbonates from ethylene carbonate as starting material,227 which received numerous rewards due to its several assets from the point of view of green chemistry. The industrial application of this process has been expanding in recent years, being estimated to soon reach an annual worldwide production capacity of 1 million tonnes of non-phosgene bisphenol-A-based polycarbonate. Also CO2-based polycarbonates have found industrial application in the field of polymer products, namely with their use as polyols in polyurethane production, which has been commercialised by Covestro with an annual capacity of 5000 tonnes of CO2-based polycarbonate polyols.340 The tuneable thermal, chemical, mechanical and biodegradable properties of CO2-based polycarbonates offer many opportunities for developing sustainable functional materials with potential in biomedical and pharmaceutical applications. However, on the road to commercialisation there is a need for more comprehensive, systematic studies in which both the functional properties of the employed polycarbonate systems (e.g. mechanical strength) and their biocompatibility and biodegradability are fully assessed.
When considering the current commercialised applications of CO2-based cyclic and polymeric carbonates, it can be seen that the epoxide starting materials employed in most cases (i.e. ethylene oxide and propylene oxide) are currently produced from fossil feedstock. A crucial future challenge will lie in replacing these petroleum-based epoxides with bio-based substitutes, while still maintaining the characteristics of the carbonate products that makes them suitable for their specific applications, e.g., high ionic conductivity in the case of cyclic or polymeric carbonates as components of battery electrolytes, appropriate thermal and mechanical properties in the case of polycarbonates in polymer products such as polyurethane foams, and proper biocompatibility in the case of polycarbonates as tissue scaffolds in biomedical applications. In order to attain these properties, various strategies discussed in this review can be followed, including reaction during or after formation of the carbonate products with other compounds (e.g. transcarbonation reactions of cyclic carbonates or block-copolymerisation of polycarbonates with other polymers), blending of the carbonate products with other materials and modification to introduce functional groups. The extent to which petroleum-based epoxides can be substituted by bio-based alternatives will also depend on the availability of the latter and, hence, the costs associated with the production of these compounds. In addition, although CO2-based polycarbonates are typically biodegradable, recycling of these materials is preferred from a circular economy point of view. In general, a life-cycle analysis using a cradle-to-grave approach (or even better a cradle-to-cradle approach) that takes into account the nature of the raw materials, how these are produced, how they are converted and whether the final products can be recycled will be essential to evaluate the potential commercialisation of new products based on CO2-based cyclic and polymeric carbonates. Although the path towards the substitution of fossil-based chemical products with renewable alternatives is still difficult and uncertain, this review highlighted the potential of CO2-based cyclic and polymeric carbonates to contribute to this crucial target for our planet.
Conflicts of interest
The authors state that there are no conflicts to declare.Notes and references
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- J. G. J. Olivier, G. Janssens-Maenhout, M. Muntean and J. A. H. W. Peters, Trends in global CO2 emissions: 2016 report, The Hague: PBL Netherlands Environmental Assessment Agency, Ispra: European Commission, Joint Research Centre, 2016 Search PubMed.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- Climate change 2007: the physical science basis: contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, ed. S. Solomon, Cambridge University Press, Cambridge, 2007 Search PubMed.
- IEA, CO2 emissions from fuel combustion 2016, OECD Publishing, Paris, 2016 Search PubMed.
- B. Diczfalusy and M. Wråke, Energy Technology Perspectives 2012: Pathways to a Clean Energy System, Organisation for Economic Cooperation and Development (OECD), International Energy Agency (IEA), Paris, 2012 Search PubMed.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- S. Anderson and R. Newell, Annu. Rev. Environ. Resour., 2004, 29, 109–142 CrossRef.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- R. Martín and A. W. Kleij, ChemSusChem, 2011, 4, 1259–1263 CrossRef PubMed.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169 RSC.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 41141 CrossRef.
- C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gómez, E. Martin, E. C. Escudero-Adán, C. Bo and A. W. Kleij, Chem. – Eur. J., 2014, 20, 2264–2275 CrossRef CAS PubMed.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083 RSC.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef CAS PubMed.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482 RSC.
- C. Schild, A. Wokaun, R. A. Koeppel and A. Baiker, J. Phys. Chem., 1991, 95, 6341–6346 CrossRef CAS.
- G. Garbarino, P. Riani, L. Magistri and G. Busca, Int. J. Hydrogen Energy, 2014, 39, 11557–11565 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- W.-H. Chen, M.-R. Lin, J.-J. Lu, Y. Chao and T.-S. Leu, Int. J. Hydrogen Energy, 2010, 35, 11787–11797 CrossRef CAS.
- T. L. LeValley, A. R. Richard and M. Fan, Int. J. Hydrogen Energy, 2014, 39, 16983–17000 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- S. Nahar, M. Zain, A. Kadhum, H. Hasan and M. Hasan, Materials, 2017, 10, 629 CrossRef PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, B. Maestro-Madurga, A. Pesquera-Rodríguez, C. Ramírez-López, L. Lorenzo-Ibarreta, J. Torrecilla-Soria and M. C. Villarán-Velasco, Appl. Catal., A, 2009, 366, 315–324 CrossRef.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part C: Polym. Lett., 1969, 7, 287–292 CAS.
- R. van Eldik and M. Aresta, CO2 Chemistry, Elsevier Science, Waltham, 2014 Search PubMed.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 121, 2990–2992 CrossRef.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- D.-H. Lan, N. Fan, Y. Wang, X. Gao, P. Zhang, L. Chen, C.-T. Au and S.-F. Yin, Chin. J. Catal., 2016, 37, 826–845 CrossRef CAS.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- Q. He, J. W. O'Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtis and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513–1528 RSC.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS PubMed.
- M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed.
- V. B. Saptal and B. M. Bhanage, Curr. Opin. Green Sustainable Chem., 2017, 3, 1–10 CrossRef.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- C. Romain, A. Thevenon, P. K. Saini and C. K. Williams, in Carbon Dioxide and Organometallics, ed. X.-B. Lu, Springer International Publishing, Cham, 2015, vol. 53, pp. 101–141 Search PubMed.
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- B.-H. Xu, J.-Q. Wang, J. Sun, Y. Huang, J.-P. Zhang, X.-P. Zhang and S.-J. Zhang, Green Chem., 2015, 17, 108–122 RSC.
- H.-Y. Ju, M.-D. Manju, K.-H. Kim, S.-W. Park and D.-W. Park, J. Ind. Eng. Chem., 2008, 14, 157–160 CrossRef CAS.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef.
- C. A. Montoya, A. B. Paninho, P. M. Felix, M. E. Zakrzewska, J. Vital, V. Najdanovic-Visak and A. V. M. Nunes, J. Supercrit. Fluids, 2015, 100, 155–159 CrossRef CAS.
- P. Mazo and L. Rios, J. Am. Oil Chem. Soc., 2013, 90, 725–730 CrossRef CAS.
- L. A. Bivona, O. Fichera, L. Fusaro, F. Giacalone, M. Buaki-Sogo, M. Gruttadauria and C. Aprile, Catal. Sci. Technol., 2015, 5, 5000–5007 RSC.
- W. N. Sit, S. M. Ng, K. Y. Kwong and C. P. Lau, J. Org. Chem., 2005, 70, 8583–8586 CrossRef CAS PubMed.
- R. A. Shiels and C. W. Jones, J. Mol. Catal. A: Chem., 2007, 261, 160–166 CrossRef CAS.
- K. R. Roshan, R. A. Palissery, A. C. Kathalikkattil, R. Babu, G. Mathai, H.-S. Lee and D.-W. Park, Catal. Sci. Technol., 2016, 6, 3997–4004 RSC.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304–1309 CrossRef CAS.
- T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 19, 8–13 CrossRef CAS.
- W. J. Kruper and D. D. Dellar, J. Org. Chem., 1995, 60, 725–727 CrossRef CAS.
- S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
- R. L. Paddock, Y. Hiyama, J. M. McKay and S. T. Nguyen, Tetrahedron Lett., 2004, 45, 2023–2026 CrossRef CAS.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS.
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489 RSC.
- T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498–11499 CrossRef CAS PubMed.
- D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335–6342 CrossRef CAS PubMed.
- X.-B. Lu, R. He and C.-X. Bai, J. Mol. Catal. A: Chem., 2002, 186, 1–11 CrossRef CAS.
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS PubMed.
- R. L. Paddock and S. T. Nguyen, Chem. Commun., 2004, 1622 RSC.
- A. Decortes, M. Martínez Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580 RSC.
- W. Xia, K. A. Salmeia, S. I. Vagin and B. Rieger, Chem. – Eur. J., 2015, 21, 4384–4390 CrossRef CAS PubMed.
- M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS.
- M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599–2602 CrossRef CAS PubMed.
- J. G. Kim, C. D. Cowman, A. M. LaPointe, U. Wiesner and G. W. Coates, Macromolecules, 2011, 44, 1110–1113 CrossRef CAS.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed.
- J. González-Fabra, F. Castro-Gómez, A. W. Kleij and C. Bo, ChemSusChem, 2017, 10, 1233–1240 CrossRef PubMed.
- L. Peña Carrodeguas, J. González-Fabra, F. Castro-Gómez, C. Bo and A. W. Kleij, Chem. – Eur. J., 2015, 21, 6115–6122 CrossRef PubMed.
- K. S. Smith and H. L. Huyck, Environ. Geochem. Miner. Depos., 1999, 6, 29–70 Search PubMed.
- H. Büttner, K. Lau, A. Spannenberg and T. Werner, ChemCatChem, 2015, 7, 459–467 CrossRef.
- C. Carvalho Rocha, T. Onfroy, J. Pilmé, A. Denicourt-Nowicki, A. Roucoux and F. Launay, J. Catal., 2016, 333, 29–39 CrossRef CAS.
- A. J. R. Amaral, J. F. J. Coelho and A. C. Serra, Tetrahedron Lett., 2013, 54, 5518–5522 CrossRef CAS.
- K. R. Roshan, T. Jose, D. Kim, K. A. Cherian and D. W. Park, Catal. Sci. Technol., 2014, 4, 963–970 RSC.
- H. Büttner, J. Steinbauer and T. Werner, ChemSusChem, 2015, 8, 2655–2669 CrossRef PubMed.
- T. Werner and H. Büttner, ChemSusChem, 2014, 7, 3268–3271 CrossRef CAS PubMed.
- J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- M. H. Anthofer, M. E. Wilhelm, M. Cokoja, M. Drees, W. A. Herrmann and F. E. Kühn, ChemCatChem, 2015, 7, 94–98 CrossRef CAS.
- M. Liu, L. Liang, X. Li, X. Gao and J. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- Z. Zhang, F. Fan, H. Xing, Q. Yang, Z. Bao and Q. Ren, ACS Sustainable Chem. Eng., 2017, 5, 2841–2846 CrossRef CAS.
- J. Ma, J. Liu, Z. Zhang and B. Han, Green Chem., 2012, 14, 2410–2420 RSC.
- M. Alves, B. Grignard, S. Gennen, R. Mereau, C. Detrembleur, C. Jerome and T. Tassaing, Catal. Sci. Technol., 2015, 5, 4636–4643 RSC.
- S. Sopeña, G. Fiorani, C. Martín and A. W. Kleij, ChemSusChem, 2015, 8, 3248–3254 CrossRef PubMed.
- J. Sun, J. Ren, S. Zhang and W. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS.
- Y.-M. Shen, W.-L. Duan and M. Shi, Adv. Synth. Catal., 2003, 345, 337–340 CrossRef CAS.
- J.-Q. Wang, J. Sun, W.-G. Cheng, K. Dong, X.-P. Zhang and S.-J. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 11021 RSC.
- A. M. Hardman-Baldwin and A. E. Mattson, ChemSusChem, 2014, 7, 3275–3278 CrossRef CAS PubMed.
- S. Gennen, M. Alves, R. Méreau, T. Tassaing, B. Gilbert, C. Detrembleur, C. Jerome and B. Grignard, ChemSusChem, 2015, 8, 1845–1849 CrossRef CAS PubMed.
- M. Alves, R. Méreau, B. Grignard, C. Detrembleur, C. Jerome and T. Tassaing, RSC Adv., 2016, 6, 36327–36335 RSC.
- J. Wang and Y. Zhang, ACS Catal., 2016, 6, 4871–4876 CrossRef CAS.
- X.-F. Liu, Q.-W. Song, S. Zhang and L.-N. He, Catal. Today, 2016, 263, 69–74 CrossRef CAS.
- N. Aoyagi, Y. Furusho and T. Endo, Tetrahedron Lett., 2013, 54, 7031–7034 CrossRef CAS.
- L. Martínez-Rodríguez, J. Otalora Garmilla and A. W. Kleij, ChemSusChem, 2016, 9, 749–755 CrossRef PubMed.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- P. Agrigento, S. M. Al-Amsyar, B. Sorée, M. Taherimehr, M. Gruttadauria, C. Aprile and P. P. Pescarmona, Catal. Sci. Technol., 2014, 4, 1598–1607 RSC.
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 119, 7393–7396 CrossRef.
- C. Aprile, F. Giacalone, P. Agrigento, L. F. Liotta, J. A. Martens, P. P. Pescarmona and M. Gruttadauria, ChemSusChem, 2011, 4, 1830–1837 CrossRef CAS PubMed.
- J.-Q. Wang, X.-D. Yue, F. Cai and L.-N. He, Catal. Commun., 2007, 8, 167–172 CrossRef CAS.
- L. Han, S.-W. Park and D.-W. Park, Energy Environ. Sci., 2009, 2, 1286 RSC.
- W. Cheng, X. Chen, J. Sun, J. Wang and S. Zhang, Catal. Today, 2013, 200, 117–124 CrossRef CAS.
- S. Udayakumar, M. Lee, H. Shim, S. Park and D. Park, Catal. Commun., 2009, 10, 659–664 CrossRef CAS.
- C. Calabrese, L. F. Liotta, E. Carbonell, F. Giacalone, M. Gruttadauria and C. Aprile, ChemSusChem, 2017, 10, 1202–1209 CrossRef CAS PubMed.
- S. Ghazali-Esfahani, H. Song, E. Păunescu, F. D. Bobbink, H. Liu, Z. Fei, G. Laurenczy, M. Bagherzadeh, N. Yan and P. J. Dyson, Green Chem., 2013, 15, 1584 RSC.
- Y. Du, F. Cai, D.-L. Kong and L.-N. He, Green Chem., 2005, 7, 518 RSC.
- J.-Q. Wang, D.-L. Kong, J.-Y. Chen, F. Cai and L.-N. He, J. Mol. Catal. A: Chem., 2006, 249, 143–148 CrossRef CAS.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef CAS.
- K. M. K. Yu, I. Curcic, J. Gabriel, H. Morganstewart and S. C. Tsang, J. Phys. Chem. A, 2010, 114, 3863–3872 CrossRef CAS PubMed.
- S. R. Jagtap, V. P. Raje, S. D. Samant and B. M. Bhanage, J. Mol. Catal. A: Chem., 2007, 266, 69–74 CrossRef CAS.
- A. Barbarini, R. Maggi, A. Mazzacani, G. Mori, G. Sartori and R. Sartorio, Tetrahedron Lett., 2003, 44, 2931–2934 CrossRef CAS.
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031 RSC.
- W.-Y. Gao, Y. Chen, Y. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. Cai, Y.-S. Chen and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 2615–2619 CrossRef CAS PubMed.
- M. North, P. Villuendas and C. Young, Chem. – Eur. J., 2009, 15, 11454–11457 CrossRef CAS PubMed.
- M. Liu, B. Liu, L. Liang, F. Wang, L. Shi and J. Sun, J. Mol. Catal. A: Chem., 2016, 418–419, 78–85 CrossRef CAS.
- J. Meléndez, M. North and P. Villuendas, Chem. Commun., 2009, 2577 RSC.
- H. He, J. A. Perman, G. Zhu and S. Ma, Small, 2016, 12, 6309–6324 CrossRef CAS PubMed.
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Front. Energy Res., 2015, 2, 63 Search PubMed.
- M. Taherimehr, B. Van de Voorde, L. H. Wee, J. A. Martens, D. E. De Vos and P. P. Pescarmona, ChemSusChem, 2017, 10, 1283–1291 CrossRef CAS PubMed.
- O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhinimaev and V. P. Fedin, J. Catal., 2013, 298, 179–185 CrossRef CAS.
- R. Babu, A. C. Kathalikkattil, R. Roshan, J. Tharun, D.-W. Kim and D.-W. Park, Green Chem., 2016, 18, 232–242 RSC.
- L. Liu, S.-M. Wang, Z.-B. Han, M. Ding, D.-Q. Yuan and H.-L. Jiang, Inorg. Chem., 2016, 55, 3558–3565 CrossRef CAS PubMed.
- Z.-R. Jiang, H. Wang, Y. Hu, J. Lu and H.-L. Jiang, ChemSusChem, 2015, 8, 878–885 CrossRef CAS PubMed.
- V. Guillerm, Ł. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, Ł. Wojtas, K. Adil and M. Eddaoudi, Nat. Chem., 2014, 6, 673–680 CrossRef CAS PubMed.
- D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180–183 CrossRef CAS.
- B. Mousavi, S. Chaemchuen, B. Moosavi, Z. Luo, N. Gholampour and F. Verpoort, New J. Chem., 2016, 40, 5170–5176 RSC.
- L. Yang, L. Yu, G. Diao, M. Sun, G. Cheng and S. Chen, J. Mol. Catal. A: Chem., 2014, 392, 278–283 CrossRef CAS.
- J. Tharun, G. Mathai, A. C. Kathalikkattil, R. Roshan, Y.-S. Won, S. J. Cho, J.-S. Chang and D.-W. Park, ChemPlusChem, 2015, 80, 715–721 CrossRef CAS.
- H.-Y. Cho, D.-A. Yang, J. Kim, S.-Y. Jeong and W.-S. Ahn, Catal. Energy Environ. Technol. 13th Korea-Jpn. Symp. Catal., 2012, 185, 35–40 CAS.
- J. Tharun, K.-M. Bhin, R. Roshan, D. W. Kim, A. C. Kathalikkattil, R. Babu, H. Y. Ahn, Y. S. Won and D.-W. Park, Green Chem., 2016, 18, 2479–2487 RSC.
- T. Jose, Y. Hwang, D.-W. Kim, M.-I. Kim and D.-W. Park, Catal. Today, 2015, 245, 61–67 CrossRef CAS.
- X. Zhou, Y. Zhang, X. Yang, L. Zhao and G. Wang, J. Mol. Catal. A: Chem., 2012, 361–362, 12–16 CrossRef CAS.
- D. Ma, B. Li, K. Liu, X. Zhang, W. Zou, Y. Yang, G. Li, Z. Shi and S. Feng, J. Mater. Chem. A, 2015, 3, 23136–23142 RSC.
- A. C. Kathalikkattil, R. Babu, J. Tharun, R. Roshan and D.-W. Park, Catal. Surv. Asia, 2015, 19, 223–235 CrossRef CAS.
- W.-Y. Gao, C.-Y. Tsai, L. Wojtas, T. Thiounn, C.-C. Lin and S. Ma, Inorg. Chem., 2016, 55, 7291–7294 CrossRef CAS PubMed.
- P.-Z. Li, X.-J. Wang, J. Liu, J. S. Lim, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS PubMed.
- W. Wang, Y. Wang, C. Li, L. Yan, M. Jiang and Y. Ding, ACS Sustainable Chem. Eng., 2017, 5, 4523–4528 CrossRef CAS.
- Y. Chen, R. Luo, Q. Xu, J. Jiang, X. Zhou and H. Ji, ChemSusChem, 2017, 10, 2534–2541 CrossRef CAS PubMed.
- C. Li, W. Wang, L. Yan, Y. Wang, M. Jiang and Y. Ding, J. Mater. Chem. A, 2016, 4, 16017–16027 RSC.
- H. Li, C. Li, J. Chen, L. Liu and Q. Yang, Chem. – Asian J., 2017, 12, 1095–1103 CrossRef CAS.
- Z. Dai, Q. Sun, X. Liu, C. Bian, Q. Wu, S. Pan, L. Wang, X. Meng, F. Deng and F.-S. Xiao, J. Catal., 2016, 338, 202–209 CrossRef CAS.
- J. Chun, S. Kang, N. Kang, S. M. Lee, H. J. Kim and S. U. Son, J. Mater. Chem. A, 2013, 1, 5517 RSC.
- Y. Chen, R. Luo, Q. Xu, W. Zhang, X. Zhou and H. Ji, ChemCatChem, 2017, 9, 767–773 CrossRef CAS.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- T. Takahashi, T. Watahiki, S. Kitazume, H. Yasuda and T. Sakakura, Chem. Commun., 2006, 1664 RSC.
- J. Sun, W. Cheng, W. Fan, Y. Wang, Z. Meng and S. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS.
- R. A. Watile, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, Catal. Sci. Technol., 2012, 2, 1051 RSC.
- W.-L. Dai, L. Chen, S.-F. Yin, W.-H. Li, Y.-Y. Zhang, S.-L. Luo and C.-T. Au, Catal. Lett., 2010, 137, 74–80 CrossRef CAS.
- T.-Y. Shi, J.-Q. Wang, J. Sun, M.-H. Wang, W.-G. Cheng and S.-J. Zhang, RSC Adv., 2013, 3, 3726 RSC.
- C. J. Whiteoak, A. H. Henseler, C. Ayats, A. W. Kleij and M. A. Pericàs, Green Chem., 2014, 16, 1552–1559 RSC.
- L. Han, H.-J. Choi, S.-J. Choi, B. Liu and D.-W. Park, Green Chem., 2011, 13, 1023 RSC.
- A. Sainz Martinez, C. Hauzenberger, A. R. Sahoo, Z. Csendes, H. Hoffmann and K. Bica, ACS Sustainable Chem. Eng., 2018, 6, 13131–13139 CrossRef CAS.
- W. Zhang, Q. Wang, H. Wu, P. Wu and M. He, Green Chem., 2014, 16, 4767–4774 RSC.
- C. Kohrt and T. Werner, ChemSusChem, 2015, 8, 2031–2034 CrossRef CAS.
- X. Chen, J. Sun, J. Wang and W. Cheng, Tetrahedron Lett., 2012, 53, 2684–2688 CrossRef CAS.
- T. Jose, S. Cañellas, M. A. Pericàs and A. W. Kleij, Green Chem., 2017, 19, 5488–5493 RSC.
- Y. Zhi, P. Shao, X. Feng, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, J. Mater. Chem. A, 2018, 6, 374–382 RSC.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS PubMed.
- S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- J. Sun, W. Cheng, Z. Yang, J. Wang, T. Xu, J. Xin and S. Zhang, Green Chem., 2014, 16, 3071 RSC.
- K. R. Roshan, T. Jose, A. C. Kathalikkattil, D. W. Kim, B. Kim and D. W. Park, Appl. Catal., A, 2013, 467, 17–25 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- Y. Zhao, J.-S. Tian, X.-H. Qi, Z.-N. Han, Y.-Y. Zhuang and L.-N. He, J. Mol. Catal. A: Chem., 2007, 271, 284–289 CrossRef CAS.
- J. Tharun, Y. Hwang, R. Roshan, S. Ahn, A. C. Kathalikkattil and D.-W. Park, Catal. Sci. Technol., 2012, 2, 1674 RSC.
- J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654 RSC.
- S. Klaus, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, A. K. Ott and B. Rieger, J. Am. Chem. Soc., 2011, 133, 13151–13161 CrossRef CAS PubMed.
- S. Chen, G.-R. Qi, Z.-J. Hua and H.-Q. Yan, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5284–5291 CrossRef CAS.
- M. Ree, J. Y. Bae, J. H. Jung and T. J. Shin, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1863–1876 CrossRef CAS.
- Y. Z. Meng, L. C. Du, S. C. Tiong, Q. Zhu and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3579–3591 CrossRef CAS.
- M. Ree, J. Y. Bae, J. H. Jung, T. J. Shin, Y.-T. Hwang and T. Chang, Polym. Eng. Sci., 2000, 40, 1542–1552 CrossRef CAS.
- R. Eberhardt, M. Allmendinger, M. Zintl, C. Troll, G. A. Luinstra and B. Rieger, Macromol. Chem. Phys., 2004, 205, 42–47 CrossRef CAS.
- I. Kim, M. J. Yi, K. J. Lee, D.-W. Park, B. U. Kim and C.-S. Ha, Catal. Today, 2006, 111, 292–296 CrossRef CAS.
- S. Chen, Z. Hua, Z. Fang and G. Qi, Polymer, 2004, 45, 6519–6524 CrossRef CAS.
- I. Kim, M. J. Yi, S. H. Byun, D. W. Park, B. U. Kim and C. S. Ha, Macromol. Symp., 2005, 224, 181–192 CrossRef CAS.
- I. S. Metcalfe, M. North, R. Pasquale and A. Thursfield, Energy Environ. Sci., 2010, 3, 212–215 RSC.
- M. North, B. Wang and C. Young, Energy Environ. Sci., 2011, 4, 4163 RSC.
- J. Meléndez, M. North, P. Villuendas and C. Young, Dalton Trans., 2011, 40, 3885–3902 RSC.
- A. Barthel, Y. Saih, M. Gimenez, J. D. A. Pelletier, F. E. Kühn, V. D'Elia and J.-M. Basset, Green Chem., 2016, 18, 3116–3123 RSC.
- A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS.
- M. J. Kelly, A. Barthel, C. Maheu, O. Sodpiban, F.-B. Dega, S. V. C. Vummaleti, E. Abou-Hamad, J. D. A. Pelletier, L. Cavallo, V. D'Elia and J.-M. Basset, J. CO2 Util., 2017, 20, 243–252 CrossRef CAS.
- B. Wetenhall, J. M. Race and M. J. Downie, Int. J. Greenh. Gas Con., 2014, 30, 197–211 CrossRef CAS.
- J. Rumble, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 98th edn, 2017 Search PubMed.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- C. Beattie, M. North and P. Villuendas, Molecules, 2011, 16, 3420–3432 CrossRef CAS PubMed.
- H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 11, 1312 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- J. R. Machac and E. T. Marquis, WO Pat, 2000050524A1, 2001 Search PubMed.
- E. T. Marquis, R. E. Baldwin, J. R. Machac, K. Darragas and S. A. Woodrum, WO Pat, 2001042376A1, 2001 Search PubMed.
- J. A. Lucas and Z. E. Halar, US Pat, 5449474A, 1995 Search PubMed.
- M. Kawasaki, Y. Ohbuchi, Y. Maeda and S. Sato, US Pat, 5021534, 1991 Search PubMed.
- Z. Dai and M. J. Chen, US Pat, 6229054, 2001 Search PubMed.
- R. B. Durairaj, US Pat, 6303732, 2001 Search PubMed.
- F. A. Lavallee and F. A. Lavallee, US Pat, 6046326, 2000 Search PubMed.
- H. Nava, US Pat, 5714568, 1998 Search PubMed.
- S. D. Lazarus, K. Chakravarti, H. H. Rowan and J. G. Neal, US Pat, 4171422, 1982 Search PubMed.
- J. M. Trowell and S. F. Lange, US Pat, 4897429, 1990 Search PubMed.
- K. Dahmen and R. Mertens, US Pat, 5409771, 1995 Search PubMed.
- European Chemicals Agency (ECHA), Propylene carbonate substance information, https://echa.europa.eu/substance-information/-/substanceinfo/100.003.248, (accessed on December 11, 2018).
- European Chemicals Agency (ECHA), Ethylene carbonate substance information, https://echa.europa.eu/en/substance-information/-/substanceinfo/100.002.283, (accessed on December 11, 2018).
- H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Macromol. Rapid Commun., 2014, 35, 1238–1254 CrossRef CAS PubMed.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC.
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- G. Rokicki, P. Rakoczy, P. Parzuchowski and M. Sobiecki, Green Chem., 2005, 7, 529 RSC.
- J. Ma, J. Song, H. Liu, J. Liu, Z. Zhang, T. Jiang, H. Fan and B. Han, Green Chem., 2012, 14, 1743–1748 RSC.
- S. Kondawar and C. Rode, Energy Fuels, 2017, 31, 4361–4371 CrossRef CAS.
- J. George, Y. Patel, S. M. Pillai and P. Munshi, J. Mol. Catal. A: Chem., 2009, 304, 1–7 CrossRef CAS.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- H. A. Stretz, P. E. Cassidy and D. R. Paul, J. Appl. Polym. Sci., 1999, 74, 1508–1515 CrossRef CAS.
- D. J. Darensbourg and S. J. Wilson, J. Am. Chem. Soc., 2011, 133, 18610–18613 CrossRef CAS PubMed.
- D. J. Darensbourg and S. J. Wilson, Macromolecules, 2013, 46, 5929–5934 CrossRef CAS.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- C. Li, R. J. Sablong and C. E. Koning, Angew. Chem., Int. Ed., 2016, 55, 11572–11576 CrossRef CAS PubMed.
- G. Wu, S. Jiang, X. Lu, W. Ren and S. Yan, Chin. J. Polym. Sci., 2012, 30, 487–492 CrossRef CAS.
- K. Nakano, S. Hashimoto, M. Nakamura, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2011, 50, 4868–4871 CrossRef CAS PubMed.
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89, 1163–1176 CrossRef CAS.
- T.-J. Hsu and C.-S. Tan, Polymer, 2002, 43, 4535–4543 CrossRef CAS.
- W.-M. Ren, X. Zhang, Y. Liu, J.-F. Li, H. Wang and X.-B. Lu, Macromolecules, 2010, 43, 1396–1402 CrossRef CAS.
- G. Luinstra, Polym. Rev., 2008, 48, 192–219 CrossRef CAS.
- J. E. Seong, S. J. Na, A. Cyriac, B.-W. Kim and B. Y. Lee, Macromolecules, 2010, 43, 903–908 CrossRef CAS.
- C. Martín and A. W. Kleij, Macromolecules, 2016, 49, 6285–6295 CrossRef.
- J. H. Jung, M. Ree and H. Kim, Catal. Today, 2006, 115, 283–287 CrossRef CAS.
- L. C. Du, Y. Z. Meng, S. J. Wang and S. C. Tjong, J. Appl. Polym. Sci., 2004, 92, 1840–1846 CrossRef CAS.
- G. Kim, M. Ree, H. Kim, I. J. Kim, J. R. Kim and J. I. Lee, Macromol. Res., 2008, 16, 473–480 CrossRef CAS.
- C. E. Koning, R. J. Sablong, E. H. Nejad, R. Duchateau and P. Buijsen, Prog. Org. Coat., 2013, 76, 1704–1711 CrossRef CAS.
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- Y. Xu, L. Lin, M. Xiao, S. Wang, A. T. Smith, L. Sun and Y. Meng, Prog. Polym. Sci., 2018, 80, 163–182 CrossRef CAS.
- Y. Li, J. Hong, R. Wei, Y. Zhang, Z. Tong, X. Zhang, B. Du, J. Xu and Z. Fan, Chem. Sci., 2015, 6, 1530–1536 RSC.
- A. Cyriac, S. H. Lee, J. K. Varghese, J. H. Park, J. Y. Jeon, S. J. Kim and B. Y. Lee, Green Chem., 2011, 13, 3469 RSC.
- Z. Zhang, Z. Mo, H. Zhang, X. Wang and X. Zhao, Macromol. Chem. Phys., 2003, 204, 1557–1566 CrossRef CAS.
- Y. Tominaga and K. Yamazaki, Chem. Commun., 2014, 50, 4448 RSC.
- M. Taherimehr, J. P. Cardoso Costa Sertã, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8, 1034–1042 CrossRef CAS PubMed.
- C. Li, S. van Berkel, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2016, 85, 466–477 CrossRef CAS.
- N. Kindermann, À. Cristòfol and A. W. Kleij, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS.
- D. J. Darensbourg and F.-T. Tsai, Macromolecules, 2014, 47, 3806–3813 CrossRef CAS.
- J. Geschwind, F. Wurm and H. Frey, Macromol. Chem. Phys., 2013, 214, 892–901 CrossRef CAS.
- J.-F. Zhang, W.-M. Ren, X.-K. Sun, Y. Meng, B.-Y. Du and X.-H. Zhang, Macromolecules, 2011, 44, 9882–9886 CrossRef CAS.
- D. J. Darensbourg, W.-C. Chung, C. J. Arp, F.-T. Tsai and S. J. Kyran, Macromolecules, 2014, 47, 7347–7353 CrossRef CAS.
- D. J. Darensbourg and Y. Wang, Polym. Chem., 2015, 6, 1768–1776 RSC.
- G. Bleys, Technology Watch: Carbon Dioxide Conversion, Essenscia, 2016 Search PubMed.
- B. Bahramian, A. Fathi and F. Dehghani, Polym. Degrad. Stab., 2016, 133, 174–181 CrossRef CAS.
- A. K. Brym, P. Deglmann, J. Dengler, S. Klaus, M. Lehenmeier, P. Lutz-Kahler, B. Rieger and V. Warzelhan, WO Pat, 2012139994 A1, 2012 Search PubMed.
- P. Deglmann, A. K. Brym, B. Rieger, M. Lehenmeier and S. Klaus, WO Pat, 2013030317A1, 2013 Search PubMed.
- T. E. Müller, C. Gürtler, M. A. Subhani and W. Leitner, WO Pat, 2015128277A1, 2015 Search PubMed.
- J. Hofmann, S. Braun, K. Laemmer, A. Wolf and M. Traving, WO Pat, 2016001206A1, 2016 Search PubMed.
- T. E. Müller, C. Gürtler, M. Wohak, J. Hofmann and T. Carlson, WO Pat, 2016079065A1, 2016 Search PubMed.
- C. Wamprecht, W. Kaufhold and C. Gürtler, WO Pat, 2014060300A3, 2014 Search PubMed.
- S. D. Allen, A. E. Cherian, C. A. Simoneau and J. J. Farmer, WO Pat, 2011163133A1, 2011 Search PubMed.
- J. J. Farmer, WO Pat, 2012037282A3, 2012 Search PubMed.
- J. G. Sant'Angelo, X. Chen and V. D. McGinnis, US Pat, 6248860B1, 2001 Search PubMed.
- C. Chen and J. Scheirs, WO Pat, 2011026171A1, 2012 Search PubMed.
- J. Jeong, S. J. Na, S. Sudevan, M. Ok, Y. Han, K. J. Chung, B. Y. Lee, A. Cyriac and S. Lee, WO Pat, 2012108696A3, 2016 Search PubMed.
- K. J. Chung, Y. G. Han, J. S. Jeong, V. J. Kodiyan, B. Y. Lee, S. J. Na, M. A. Ok and S. Sudevan, WO Pat, 2011105846A3, 2012 Search PubMed.
- G. W. Coates, S. Lee, Z. Qin and N. J. Robertson, WO Pat, 2008024363A3, 2008 Search PubMed.
- K. Nozaki and K. Nakano, WO Pat, 2008150033A1, 2008 Search PubMed.
- A. Mohsenzadeh, A. Zamani and M. J. Taherzadeh, ChemBioEng Rev., 2017, 4, 75–91 CrossRef CAS.
- E. de Jong, A. Higson, P. Walsh and M. Wellisch, Bio-based Chemicals: Value Added Products From Biorefineries, IEA Bioenergy, 2012 Search PubMed.
- V. Zacharopoulou, E. S. Vasiliadou and A. A. Lemonidou, Green Chem., 2015, 17, 903–912 RSC.
- J. S. Plotkin, Green Propylene, Nexant, 2009 Search PubMed.
- Genomatica, https://www.genomatica.com/, (accessed on March 16, 2018).
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- B. Elvers, Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2003 Search PubMed.
- M. M. Ahmad, M. F. R. Nordin and M. T. Azizan, Am. J. Appl. Sci., 2010, 7, 746–755 CrossRef CAS.
- F. E. Massoth, P. Politzer, M. C. Concha, J. S. Murray, J. Jakowski and J. Simons, J. Phys. Chem. B, 2006, 110, 14283–14291 CrossRef CAS PubMed.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen and A. D. Jensen, ACS Catal., 2013, 3, 1774–1785 CrossRef CAS.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 127, 1231–1234 CrossRef.
- V. Negro, G. Mancini, B. Ruggeri and D. Fino, Bioresour. Technol., 2016, 214, 806–815 CrossRef CAS PubMed.
- N. Tenhumberg, H. Büttner, B. Schäffner, D. Kruse, M. Blumenstein and T. Werner, Green Chem., 2016, 18, 3775–3788 RSC.
- F.-T. Tsai, Y. Wang and D. J. Darensbourg, J. Am. Chem. Soc., 2016, 138, 4626–4633 CrossRef CAS PubMed.
- G. Si, L. Zhang, B. Han, H. Zhang, X. Li and B. Liu, RSC Adv., 2016, 6, 22821–22826 RSC.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917–7953 RSC.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, T.-Q. Xu and D. J. Darensbourg, J. Am. Chem. Soc., 2011, 133, 15191–15199 CrossRef CAS PubMed.
- G.-P. Wu, P.-X. Xu, X.-B. Lu, Y.-P. Zu, S.-H. Wei, W.-M. Ren and D. J. Darensbourg, Macromolecules, 2013, 46, 2128–2133 CrossRef CAS.
- B. M. Bell, J. R. Briggs, R. M. Campbell, S. M. Chambers, P. D. Gaarenstroom, J. G. Hippler, B. D. Hook, K. Kearns, J. M. Kenney, W. J. Kruper, D. J. Schreck, C. N. Theriault and C. P. Wolfe, Clean: Soil, Air, Water, 2008, 36, 657–661 CAS.
- Y. Hu, L. Qiao, Y. Qin, X. Zhao, X. Chen, X. Wang and F. Wang, Macromolecules, 2009, 42, 9251–9254 CrossRef CAS.
- J. Hilf, M. Scharfenberg, J. Poon, C. Moers and H. Frey, Macromol. Rapid Commun., 2015, 36, 174–179 CrossRef CAS PubMed.
- J. Geschwind and H. Frey, Macromolecules, 2013, 46, 3280–3287 CrossRef CAS.
- Y. Tominaga, T. Shimomura and M. Nakamura, Polymer, 2010, 51, 4295–4298 CrossRef CAS.
- M. D. Konieczynska, X. Lin, H. Zhang and M. W. Grinstaff, ACS Macro Lett., 2015, 4, 533–537 CrossRef CAS.
- F. Parrino, A. Fidalgo, L. Palmisano, L. M. Ilharco, M. Pagliaro and R. Ciriminna, ACS Omega, 2018, 3, 4884–4890 CrossRef CAS.
- J. Sun, L. Liang, J. Sun, Y. Jiang, K. Lin, X. Xu and R. Wang, Catal. Surv. Asia, 2011, 15, 49–54 CrossRef CAS.
- N. V. Maksimchuk, I. D. Ivanchikova, A. B. Ayupov and O. A. Kholdeeva, Appl. Catal., B, 2016, 181, 363–370 CrossRef CAS.
- F. D. Bobbink and P. J. Dyson, J. Catal., 2016, 343, 52–61 CrossRef CAS.
- F. Chen, T. Dong, T. Xu, X. Li and C. Hu, Green Chem., 2011, 13, 2518–2524 RSC.
- J. Wu, J. A. Kozak, F. Simeon, T. A. Hatton and T. F. Jamison, Chem. Sci., 2014, 5, 1227–1231 RSC.
- X. Yang, J. Wu, X. Mao, T. F. Jamison and T. A. Hatton, Chem. Commun., 2014, 50, 3245–3248 RSC.
- D. Zhang, E. A. del Rio-Chanona, J. L. Wagner and N. Shah, Sustainable Prod. Consumption, 2018, 14, 152–160 CrossRef.
- IHS Markit: Polycarbonate Market Overview, May 2018, http://www.media-tech.net/2018/wp-content/uploads/2018/06/Mediatech-2018-Polycarbonate-Market_Wiesweg.pdf, (accessed on August 15, 2018).
- O. Haba, I. Itakura, M. Ueda and S. Kuze, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2087–2093 CrossRef CAS.
- R. Huang, Z. Liu, H. Yin, Z. Dang, P. Wu, N. Zhu and Z. Lin, Sci. Total Environ., 2018, 626, 971–981 CrossRef CAS PubMed.
- K. Horie, B. Máximo, R. B. Fox, J. He, M. Hess, J. Kahovec, T. Kitayama, P. Kubisa, E. Maréchal, W. Mormann, R. F. T. Stepto, D. Tabak, J. Vohlídal, E. S. Wilks and W. J. Work, Pure Appl. Chem., 2004, 76, 889 CAS.
- M. Khoshkish, H. Bouhendi and M. Vafayan, J. Mater. Sci., 2014, 49, 6813–6819 CrossRef CAS.
- F.-L. Jin, X. Li and S.-J. Park, J. Ind. Eng. Chem., 2015, 29, 1–11 CrossRef CAS.
- J. V. Alemán, A. V. Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvíl, I. Meisel, I. Mita, G. Moad, S. Penczek and R. F. T. Stepto, Pure Appl. Chem., 2007, 79, 1801 Search PubMed.
- H. Gu, C. Ma, J. Gu, J. Guo, X. Yan, J. Huang, Q. Zhang and Z. Guo, J. Mater. Chem. C, 2016, 4, 5890–5906 RSC.
- G. Rokicki and C. Wojciechowski, J. Appl. Polym. Sci., 1990, 41, 647–659 CrossRef CAS.
- J. H. Hodgkin, G. P. Simon and R. J. Varley, Polym. Adv. Technol., 1998, 9, 3–10 CrossRef CAS.
- G. Rokicki and M. Lewandowski, Macromol. Mater. Eng., 1987, 148, 53–66 CAS.
- G. Rokicki and R. Laziński, Macromol. Mater. Eng., 1989, 170, 211–225 CAS.
- J. Wang, H. Zhang, Y. Miao, L. Qiao, X. Wang and F. Wang, Green Chem., 2016, 18, 524–530 RSC.
- E. Orgilés-Calpena, F. Arán-Aís, A. M. Torró-Palau and C. Orgilés-Barceló, Int. J. Adhes. Adhes., 2016, 70, 218–224 CrossRef.
- L. Poussard, J. Mariage, B. Grignard, C. Detrembleur, C. Jérôme, C. Calberg, B. Heinrichs, J. De Winter, P. Gerbaux, J.-M. Raquez, L. Bonnaud and P. Dubois, Macromolecules, 2016, 49, 2162–2171 CrossRef CAS.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447 RSC.
- Statista, Market size forecast of polyurethane worldwide from 2016 to 2021, https://www.statista.com/statistics/720341/global-polyurethane-market-size-forecast/, (accessed on 27 August 2018).
- N. Mahmood, Z. Yuan, J. Schmidt and C. Xu, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef CAS.
- J.-M. Raquez, M. Deléglise, M.-F. Lacrampe and P. Krawczak, Prog. Polym. Sci., 2010, 35, 487–509 CrossRef CAS.
- A. Kuta, Z. Hrdlička, A. Strachota and M. Špírková, Mater. Manuf. Processes, 2009, 24, 1214–1216 CrossRef CAS.
- P. Cinelli, I. Anguillesi and A. Lazzeri, Eur. Polym. J., 2013, 49, 1174–1184 CrossRef CAS.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- B.-H. Lee and H.-J. Kim, Polym. Degrad. Stab., 2006, 91, 1025–1035 CrossRef CAS.
- J. Langanke, A. Wolf, J. Hofmann, K. Böhm, M. A. Subhani, T. E. Müller, W. Leitner and C. Gürtler, Green Chem., 2014, 16, 1865–1870 RSC.
- J. Langanke and A. Wolf, Org. Process Res. Dev., 2015, 19, 735–739 CrossRef CAS.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- Covestro, CO2 Dreams, https://www.co2-dreams.covestro.com, (accessed on August 16, 2018).
- Converge polyols, http://www.saudiaramco.com/en/home/our-business/downstream/converge.html, (accessed on August 10, 2018).
- Z. Ying, C. Wu, S. Jiang, R. Shi, B. Zhang, C. Zhang and F. Zhao, Green Chem., 2016, 18, 3614–3619 RSC.
- S. Xu and M. Zhang, J. Appl. Polym. Sci., 2007, 104, 3818–3826 CrossRef CAS.
- P. Alagi, R. Ghorpade, Y. J. Choi, U. Patil, I. Kim, J. H. Baik and S. C. Hong, ACS Sustainable Chem. Eng., 2017, 5, 3871–3881 CrossRef CAS.
- J. Hilf, P. Schulze, J. Seiwert and H. Frey, Macromol. Rapid Commun., 2014, 35, 198–203 CrossRef CAS PubMed.
- S. Liu, Y. Qin, X. Chen, X. Wang and F. Wang, Polym. Chem., 2014, 5, 6171–6179 RSC.
- S. Liu, Y. Qin, X. Wang and F. Wang, J. Renewable Mater., 2015, 3, 101–112 CrossRef CAS.
- S. H. Lee, A. Cyriac, J. Y. Jeon and B. Y. Lee, Polym. Chem., 2012, 3, 1215 RSC.
- M. DeBolt, A. Kiziltas, D. Mielewski, S. Waddington and M. J. Nagridge, J. Appl. Polym. Sci., 2016, 133, 44086 CrossRef.
- Y. Chen, Z. Liu, S. Han, J. Han and D. Jiang, J. Appl. Polym. Sci., 2016, 133, 43667 Search PubMed.
- C. Li, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2015, 67, 449–458 CrossRef CAS.
- D. Bello, C. A. Herrick, T. J. Smith, S. R. Woskie, R. P. Streicher, M. R. Cullen, Y. Liu and C. A. Redlich, Environ. Health Perspect., 2006, 115, 328–335 CrossRef PubMed.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- B. Nohra, L. Candy, J.-F. Blanco, C. Guerin, Y. Raoul and Z. Mouloungui, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- K. R. Aguiar, V. G. Santos, M. N. Eberlin, K. Rischka, M. Noeske, G. Tremiliosi-Filho and U. P. Rodrigues-Filho, RSC Adv., 2014, 4, 24334–24343 RSC.
- B. Tamami, S. Sohn and G. L. Wilkes, J. Appl. Polym. Sci., 2004, 92, 883–891 CrossRef CAS.
- A. Boyer, E. Cloutet, T. Tassaing, B. Gadenne, C. Alfos and H. Cramail, Green Chem., 2010, 12, 2205 RSC.
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Polym. Chem., 2017, 8, 6099–6105 RSC.
- J. Y. Jeon, J. J. Lee, J. K. Varghese, S. J. Na, S. Sujith, M. J. Go, J. Lee, M.-A. Ok and B. Y. Lee, Dalton Trans., 2013, 42, 9245–9254 RSC.
- G. A. Luinstra and E. Borchardt, in Synthetic biodegradable polymers, Springer, Berlin, 2012, pp. 29–48 Search PubMed.
- X. L. Lu, F. G. Du, X. C. Ge, M. Xiao and Y. Z. Meng, J. Biomed. Mater. Res., Part A, 2006, 77A, 653–658 CrossRef CAS PubMed.
- S. D. Allen, J. J. Farmer and C. A. Simoneau, WO Pat2012154849A1, 2012 Search PubMed.
- V. S. Bagotsky, J. Solid State Electrochem., 2011, 15, 1559–1562 CrossRef CAS.
- B. Scrosati, J. Solid State Electrochem., 2011, 15, 1623–1630 CrossRef CAS.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- T. B. Reddy and S. Hossain, in Handbook of batteries, McGraw-Hill, New York, 2002, vol. 3, pp. 1–62 Search PubMed.
- D. Fauteux, A. Massucco, M. McLin, M. Van Buren and J. Shi, Electrochim. Acta, 1995, 40, 2185–2190 CrossRef CAS.
- C. A. Vincent, Solid State Ionics, 2000, 134, 159–167 CrossRef CAS.
- Y. Nishi, H. Azuma and A. Omaru, US Pat4959281, 1990 Search PubMed.
- Lithium Ion Rechargeable Batteries, ed. K. Ozawa, Wiley-VCH Verlag GmbH & Co. KGaA, Tokyo, 2009 Search PubMed.
- S.-K. Jeong, M. Inaba, R. Mogi, Y. Iriyama, T. Abe and Z. Ogumi, Langmuir, 2001, 17, 8281–8286 CrossRef CAS.
- J.-T. Lee, Y.-W. Lin and Y.-S. Jan, J. Power Sources, 2004, 132, 244–248 CrossRef CAS.
- M. Winter, R. Imhof, F. Joho and P. Novák, J. Power Sources, 1999, 81–82, 818–823 CrossRef CAS.
- M. C. Smart, B. V. Ratnakumar, V. S. Ryan-Mowrey, S. Surampudi, G. K. S. Prakash, J. Hu and I. Cheung, J. Power Sources, 2003, 119–121, 359–367 CrossRef CAS.
- Z. X. Shu, R. S. McMillan, J. J. Murray and I. J. Davidson, J. Electrochem. Soc., 1995, 142, L161–L162 CrossRef CAS.
- Z. X. Shu, R. S. McMillan, J. J. Murray and I. J. Davidson, J. Electrochem. Soc., 1996, 143, 2230–2235 CrossRef CAS.
- M. Winter and P. Novák, J. Electrochem. Soc., 1998, 145, L27–L30 CrossRef CAS.
- R. McMillan, H. Slegr, Z. X. Shu and W. Wang, J. Power Sources, 1999, 81, 20–26 CrossRef.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- M. Inaba, Y. Kawatate, A. Funabiki, S.-K. Jeong, T. Abe and Z. Ogumi, Electrochim. Acta, 1999, 45, 99–105 CrossRef CAS.
- J. Arai, H. Katayama and H. Akahoshi, J. Electrochem. Soc., 2002, 149, A217 CrossRef CAS.
- G. S. MacGlashan, Y. G. Andreev and P. G. Bruce, Nature, 1999, 398, 792–794 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 3 CrossRef.
- Y. Tominaga, V. Nanthana and D. Tohyama, Polym. J., 2012, 44, 1155 CrossRef CAS.
- M. A. Ratner and D. F. Shriver, Chem. Rev., 1988, 88, 109–124 CrossRef CAS.
- K. Deng, S. Wang, S. Ren, D. Han, M. Xiao and Y. Meng, ACS Appl. Mater. Interfaces, 2016, 8, 33642–33648 CrossRef CAS PubMed.
- M. Nakamura and Y. Tominaga, Electrochim. Acta, 2011, 57, 36–39 CrossRef CAS.
- M. Marzantowicz, J. R. Dygas, F. Krok, J. L. Nowiński, A. Tomaszewska, Z. Florjańczyk and E. Zygadło-Monikowska, J. Power Sources, 2006, 159, 420–430 CrossRef CAS.
- T. Okumura and S. Nishimura, Solid State Ionics, 2014, 267, 68–73 CrossRef CAS.
- M. Galiński, A. Lewandowski and I. Stępniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- K. Kimura, J. Hassoun, S. Panero, B. Scrosati and Y. Tominaga, Ionics, 2015, 21, 895–900 CrossRef CAS.
- K. Kimura, H. Matsumoto, J. Hassoun, S. Panero, B. Scrosati and Y. Tominaga, Electrochim. Acta, 2015, 175, 134–140 CrossRef CAS.
- T. Morioka, K. Nakano and Y. Tominaga, Macromol. Rapid Commun., 2017, 38, 1600652 CrossRef PubMed.
- K. Kimura, M. Yajima and Y. Tominaga, Electrochem. Commun., 2016, 66, 46–48 CrossRef CAS.
- H. Yue, J. Li, Q. Wang, C. Li, J. Zhang, Q. Li, X. Li, H. Zhang and S. Yang, ACS Sustainable Chem. Eng., 2018, 6, 268–274 CrossRef CAS.
- X.-Y. Yu, M. Xiao, S.-J. Wang, Q.-Q. Zhao and Y.-Z. Meng, J. Appl. Polym. Sci., 2010, 115, 2718–2722 CrossRef CAS.
- G. P. Dukhanin, A. N. Gaidadin and I. A. Novakov, Russ. J. Appl. Chem., 2014, 87, 1868–1871 CrossRef CAS.
- D. Zhou, R. Zhou, C. Chen, W.-A. Yee, J. Kong, G. Ding and X. Lu, J. Phys. Chem. B, 2013, 117, 7783–7789 CrossRef CAS PubMed.
- X. Yu, M. Xiao, S. Wang, D. Han and Y. Meng, J. Appl. Polym. Sci., 2010, 118, 2078–2083 CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- X. Huang, J. Huang, J. Wu, X. Yu, Q. Gao, Y. Luo and H. Hu, RSC Adv., 2015, 5, 52978–52984 RSC.
- X. Huang, S. Zeng, J. Liu, T. He, L. Sun, D. Xu, X. Yu, Y. Luo, W. Zhou and J. Wu, J. Phys. Chem. C, 2015, 119, 27882–27891 CrossRef CAS.
- W. Amass, A. Amass and B. Tighe, Polym. Int., 1998, 47, 89–144 CrossRef CAS.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- D. F. Williams, Biomaterials, 2008, 29, 2941–2953 CrossRef CAS PubMed.
- S. Ramakrishna, J. Mayer, E. Wintermantel and K. W. Leong, Compos. Sci. Technol., 2001, 61, 1189–1224 CrossRef CAS.
- Y. Hwang, H. Kim and M. Ree, Macromol. Symp., 2005, 224, 227–238 CrossRef CAS.
- J. Ma, Z. Liu, F. Wang, Q. Zhou, C. Feng and F. Li, J. Bionic Eng., 2013, 10, 514–521 CrossRef.
- I. Manavitehrani, A. Fathi, Y. Wang, P. K. Maitz and F. Dehghani, ACS Appl. Mater. Interfaces, 2015, 7, 22421–22430 CrossRef CAS PubMed.
- Y. Li, S. Liu, X. Zhao, Y. Wang, J. Liu, X. Wang and L. Lu, Theranostics, 2017, 7, 4689–4698 CrossRef CAS PubMed.
- X. Jiang, H. Xin, J. Gu, X. Xu, W. Xia, S. Chen, Y. Xie, L. Chen, Y. Chen, X. Sha and X. Fang, Biomaterials, 2013, 34, 1739–1746 CrossRef CAS PubMed.
- M. Qingwei, Q. Yusheng, Z. Xiaojiang, W. Xianhong and W. Fosong, Acta Polym. Sin., 2010, 217–221 Search PubMed.
- X. H. Li, Y. Z. Meng, Q. Zhu and S. C. Tjong, Polym. Degrad. Stab., 2003, 81, 157–165 CrossRef CAS.
- K. R. Stone, Environmental Profile for Propylene Carbonate, United States Environmental Protection Agency, Washington DC, 1998 Search PubMed.
- J. A. Ruddick, Toxicol. Appl. Pharmacol., 1972, 21, 102–111 CrossRef CAS PubMed.
- I. Manavitehrani, A. Fathi, Y. Wang, P. K. Maitz, F. Mirmohseni, T. L. Cheng, L. Peacock, D. G. Little, A. Schindeler and F. Dehghani, Biomacromolecules, 2017, 18, 1736–1746 CrossRef CAS PubMed.
- X. Zhong, Z. Lu, P. Valtchev, H. Wei, H. Zreiqat and F. Dehghani, Colloids Surf., B, 2012, 93, 75–84 CrossRef CAS PubMed.
- G. Yang, J. Su, J. Gao, X. Hu, C. Geng and Q. Fu, J. Supercrit. Fluids, 2013, 73, 1–9 CrossRef CAS.
- Y. Hwang, M. Ree and H. Kim, Catal. Today, 2006, 115, 288–294 CrossRef CAS.
- S. P. Valappil, S. K. Misra, A. R. Boccaccini and I. Roy, Expert Rev. Med. Devices, 2006, 3, 853–868 CrossRef CAS PubMed.
- H. Zhang and M. W. Grinstaff, J. Am. Chem. Soc., 2013, 135, 6806–6809 CrossRef CAS PubMed.
- R. Liu, J. B. Wolinsky, J. Walpole, E. Southard, L. R. Chirieac, M. W. Grinstaff and Y. L. Colson, Ann. Surg. Oncol., 2010, 17, 1203–1213 CrossRef PubMed.
- P. Kallinteri, S. Higgins, G. A. Hutcheon, C. B. St. Pourçain and M. C. Garnett, Biomacromolecules, 2005, 6, 1885–1894 CrossRef CAS PubMed.
- H. Zhang, X. Lin, S. Chin and M. W. Grinstaff, J. Am. Chem. Soc., 2015, 137, 12660–12666 CrossRef CAS PubMed.
- L. Gu, Y. Qin, Y. Gao, X. Wang and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2834–2840 CrossRef CAS.
- X. Zhong and F. Dehghani, Green Chem., 2012, 14, 2523 RSC.
- N. Nagiah, U. T. Sivagnanam, R. Mohan, N. T. Srinivasan and P. K. Sehgal, Adv. Eng. Mater., 2012, 14, B138–B148 CrossRef.
- X. Jing, M. R. Salick, T. Cordie, H.-Y. Mi, X.-F. Peng and L.-S. Turng, Ind. Eng. Chem. Res., 2014, 53, 9391–9400 CrossRef CAS.
- L. Gu, Y. Gao, Y. Qin, X. Chen, X. Wang and F. Wang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 282–289 CrossRef CAS.
- J. Kost and R. Langer, Adv. Drug Delivery Rev., 2012, 64, 327–341 CrossRef.
- N. M. Alves, I. Pashkuleva, R. L. Reis and J. F. Mano, Small, 2010, 6, 2208–2220 CrossRef CAS PubMed.
- I. Roy and M. N. Gupta, Chem. Biol., 2003, 10, 1161–1171 CrossRef CAS PubMed.
- A. Welle, M. Kröger, M. Döring, K. Niederer, E. Pindel and I. S. Chronakis, Biomaterials, 2007, 28, 2211–2219 CrossRef CAS PubMed.
- A. Metcalfe, A.-C. Desfaits, I. Salazkin, L. Yahia, W. M. Sokolowski and J. Raymond, Biomaterials, 2003, 24, 491–497 CrossRef CAS PubMed.
- Y. Liu, K. Huang, D. Peng, S. Liu and H. Wu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2152–2160 CrossRef CAS.
- H. Li and Y. Niu, Int. J. Polym. Mater. Polym. Biomater., 2018, 67, 192–198 CrossRef CAS.
- N. H. Park, M. Fevre, Z. X. Voo, R. J. Ono, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2016, 5, 1247–1252 CrossRef CAS.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- J. Feng, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2012, 37, 211–236 CrossRef CAS.
- F.-Y. Qiu, M. Zhang, F.-S. Du and Z.-C. Li, Macromolecules, 2017, 50, 23–34 CrossRef CAS.
- K. Fukushima, Biomater. Sci., 2016, 4, 9–24 RSC.
- W. Chen, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2014, 190, 398–414 CrossRef CAS PubMed.
- Z. Y. Ong, K. Fukushima, D. J. Coady, Y.-Y. Yang, P. L. R. Ee and J. L. Hedrick, J. Controlled Release, 2011, 152, 120–126 CrossRef CAS PubMed.
- X. Zhang, Z. Zhang, X. Su, M. Cai, R. Zhuo and Z. Zhong, Biomaterials, 2013, 34, 10296–10304 CrossRef CAS PubMed.
- C. Yang, X. Ding, R. J. Ono, H. Lee, L. Y. Hsu, Y. W. Tong, J. Hedrick and Y. Y. Yang, Adv. Mater., 2014, 26, 7346–7351 CrossRef CAS PubMed.
- Empower Materials, http://www.empowermaterials.com, (accessed on August 1, 2017).
- S. D. Allen, C. A. Simoneau, J. M. Salladay, D. M. Hatfield and J. W. Stevens, WO Pat, 2010062703A1, 2011 Search PubMed.
- S. D. Allen, EP Pat, 2451860A2, 2012 Search PubMed.
- F. Gao, Q. Zhou, Y. Dong, Y. Qin, X. Wang, Z. Xiaojiang and W. Fosong, J. Polym. Res., 2012, 19, 9877 CrossRef.
- D. D. Dixon and M. E. Ford, US Pat, 4142021A, 1979 Search PubMed.
- O. Hauenstein, M. M. Rahman, M. Elsayed, R. Krause-Rehberg, S. Agarwal, V. Abetz and A. Greiner, Adv. Mater. Technol., 2017, 2, 1700026 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |