
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygenic metabolism in nutritional obesity induced by olive oil. The influence of vitamin C
Eraci
Drehmer
 a,
Mari Ángeles
Navarro-Moreno
b,
Sandra
Carrera
c,
Vincent M.
Villar
d and
Mari Luz
Moreno
*b
a,
Mari Ángeles
Navarro-Moreno
b,
Sandra
Carrera
c,
Vincent M.
Villar
d and
Mari Luz
Moreno
*b
aDepartment of Health Sciences, Universidad Católica de Valencia San Vicente Mártir, Valencia, Spain
bDepartment of Basic Sciences, Universidad Católica de Valencia San Vicente Mártir. C/Ramiro de Maeztu, 14. 46900 Torrente, Valencia, Spain. E-mail: ml.moreno@ucv.es; Tel: +34 96 363 74 12 Ext 5538
cDepartment of Nutrition and Dietetics, Universidad Católica de Valencia San Vicente Mártir, Valencia, Spain
dDepartment of Biomedical Sciences, Universidad CEU Cardenal Herrera, Valencia, Spain
First published on 3rd June 2019
Abstract
Obesity is a medical and sociological problem of great importance due to the high percentage of people affected and the important health consequences that it involves. Most cases of obesity are related to an inadequate diet, rich in fats, which could lead to changes in the patient's oxygenic metabolism. That is why this study has been proposed to evaluate how some aspects of oxygenic metabolism are affected in a nutritional experimental model, with a controlled hyperlipidic liquid diet based on olive oil, and the effect of the antioxidant vitamin C on these conditions. Wistar rats were divided into four groups which received a control and hyperlipidic liquid diet for 30 days, with or without a vitamin C supplement (CO, COC, HO and HOC). First of all the body and fat tissue development was measured in the four groups. Our results showed that the excessive intake of nutritional and healthy fat such as olive oil did not prevent the appearance of obesity and the supplementation with vitamin C did not have a protective effect on body and fat development. The study of the antioxidant enzymes superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) in total liver, liver cytosol, abdominal white fat, brown fat and blood cells showed that vitamin C could have different selectivities and affinities for different enzymes and compartments/tissues of the body. Finally, the effect of vitamin C on various metabolic parameters (glucose, pyruvate, lactate, LDH, ATP, acetoacetate and beta-hydroxybutyrate) provided positive protection against oxidative stress especially under hyperlipidic conditions. All things considered, the present study concludes that vitamin C treatment could protect Wistar rats from the oxidative stress impairment induced by obesity generated by an excessive intake of fats.
1. Introduction
Obesity is considered one of the most important health issues in industrialized countries due to its increasing frequency and globalization.1–4 It presents as an epidemic worldwide.5–8 Multiple factors are implicated in its origin, such as social, psychological, metabolic, dietary and molecular ones, among others.9–12 Obesity is a chronic disease characterized by an accumulation of adipose tissue in the body, which results in an increase in body weight and eventually to health problems.13 It is a risk factor for the development of diseases such as diabetes, hypertension, coronary heart disease and some different types of cancer.14–25Obesity is related to mitochondrial metabolism.26–31 The excess of fat in the tissues and the overweight aspect of it enhance the production of reactive oxygen species (ROS), which contribute to the origin and establishment of oxidative stress.32–35
Although ROS are generated under normal physiological conditions, an excess in their production or lack of antioxidant defences can generate oxidative stress in organs and tissues leading to progressive and neurodegenerative diseases.36–39
Some studies have demonstrated that diets rich or enriched with antioxidants are useful in preventing functional impairment caused by oxidative stress.40–42 The antioxidant olive oil, which is present in the Mediterranean diet, produces a decrease in the expression of genes implicated in inflammatory and oxidative processes.43–45 Olive oil contains beneficial substances such as polyphenols and monounsaturated fatty acids that have a positive effect on oxygenic metabolism.46–49 Other substances, such as vitamin C in fruits and vegetables, have a positive effect on the protection against lipoperoxidation.50,51
The aim of the present study is to analyze the situation of oxygenic metabolism in obesity and the endogenous and exogenous cytotoxicity induced by ROS in Wistar rats fed with liquid diets based on olive oil and supplemented with vitamin C for 30 days.
2. Materials and methods
2.1. Materials
The reagents used in the experiments were analytical grade and were obtained from Sigma-Aldrich (USA), Boehringer Ingelheim (Germany), Merck (Germany), and Panreac (Spain).2.2. Animals
Male Wistar rats (150–180 g) (Charles River, Barcelona, Spain), housed one or two animals per cage (Panlab, Barcelona, Spain) under a 12 h light/dark cycle at 22 °C and 60% humidity and maintained on standard chow and water ad libitum, were used. This study was carried out in accordance with the guidelines established in the Spanish legislation (Royal Decree RD 53/2013). The protocol was approved by the Experimental Animal Ethics Committee of the Universidad Católica de Valencia “San Vicente Mártir” (Spain). Rats were divided into four groups which received a control and hyperlipidic liquid diet for 30 days, with or without a vitamin C supplement:Group 1: Control Olive Oil Diet (C.O.)
Group 2: Hyperlipidic Olive Oil Diet (H.O.)
Group 3: Control Olive Oil Diet + Vitamin C (C.O.C)
Group 4: Hyperlipidic Olive Oil Diet + Vitamin C (H.O.C)
The proportion of fats in the diet was provided by the inclusion of olive oil in the diet. The dietary intake of the animals was recorded daily and the body weight was measured weekly. After the 30 days of treatment, rats were anaesthetized with an overdose of isoflurane and sacrificed by cervical dislocation and all the samples (liver, white and brown fat and blood) were taken for the analytical determination.
2.3. Diets
The solid non-purified IPM R-20 diet commercialized by Letica (Barcelona, Spain) and commonly used in our university animal facility was taken as the reference for preparing the composition of our experimental liquid diets.The reagents used in the liquid diet production were obtained from Sigma-Aldrich (St Louis, Missouri, USA) (DL-methionine, choline, olive oil, dextrin, cellulose and xantham gum) and ICN-Biomedicals (Santa Ana, California, USA) (casein, vitamins and minerals). The liquid diets were produced following the directions of the American Institute of Nutrition.52 In order to obtain the overfeeding of the animals (H.O.), a supplement of lipids without changing the other components was given. A model of hyperlipidic and hyperenergetic diet (H.O.) was obtained with an increase of 45% of energy with respect to the control diet (C.O.).53Table 1 shows the final conditions of the dietary treatment which the rats received during the present study.
| Composition | Control diet (C.O.) | Hyperlipidic diet (H.O.) |
|---|---|---|
| C.O.: control olive oil diet; H.O.: hyperlipidic olive oil diet. | ||
| Casein | 52.0 g | 52.0 g |
| DL-Methionine | 0.8 g | 0.8 g |
| Choline, hydrochloride | 0.3 g | 0.3 g |
| Olive oil | 8.5 g | 57.7 g |
| Dextrin | 162.7 g | 162.7 g |
| Cellulose | 10.0 g | 10.0 g |
| Xantham gum | 2.0 g | 2.0 g |
| Vitamins AIN-76A | 2.6 g | 2.6 g |
| Minerals AIN-76 | 9.1 g | 9.1 g |
| Water | 1000 mL | 1000 mL |
A second model was formulated with the same dietary characteristics as the previous one but with a vitamin C supplement of 200 mg kg−1 day−1 (C.O.C and H.O.C).54 The two models of administered diets have the composition of macronutrients and energy shown in Table 2.
| Composition (per liter of diet) | C.O. diet | C.O.C diet | H.O. diet | H.O.C diet |
|---|---|---|---|---|
| C.O.: control olive oil diet; C.O.C: control olive oil diet supplemented with vitamin C; H.O.: hyperlipidic olive oil diet; H.O.C: hyperlipidic olive oil diet supplemented with vitamin C. | ||||
| Proteins | 52.8 g | 52.8 g | 52.8 g | 52.8 g |
| Fats | 8.5 g | 8.5 g | 57.7 g | 57.7 g |
| Carbohydrates | 166.3 g | 166.3 g | 166.3 g | 166.3 g |
| Energy | 960 kcal | 960 kcal | 1400 kcal | 1400 kcal |
| Intake | Isoenergetically limited compared to animals fed with a laboratory diet | Isoenergetically limited compared to animals fed with a laboratory diet | Isovolumetrically limited compared to animals fed with a laboratory diet | Isovolumetrically limited compared to animals fed with a laboratory diet |
| Treatment period | 30 days | 30 days | 30 days | 30 days |
| Vitamin C | 200 mg kg −1 day −1 | 200 mg kg −1 day −1 | ||
The composition of fatty acids from the olive oil supply of the diet was determined by gas chromatography at the “Instituto de la grasa”, Sevilla, Spain. The content of oleic acid was 81.3% and the content of antioxidants was namely alfa-tocoferol 20.9 ppm, gamma-tocoferol 2 ppm and delta-tocoferol 4 ppm.
All the diets given to the rats were stable not only during the administration to the animals, but also during the storage period. The highest quantity of lipids added to the hyperlipidic diet did not have the problems of dispersion stability of the mixture, not being necessary to increase the proportion of the thickening agent. Since the diets did not receive any conservative agent they were kept at 4 °C, in a closed environment protected from light and for a maximum period of 4 days.
The ingested quantity was obtained taking into account the administered quantity and the consumed amount of diet.
2.4. Sample processing
Immediately after the sacrifice of the rats 2 mL of blood samples were collected and 0.2 mL of heparine at 5% (Rovi, Madrid, Spain) was added and the liver was extracted and white adipose tissue from the abdominal area and all the brown fat from the dorsal part of the thorax were extracted as well.Each aliquot of the liver sample was homogenized in a Tris-HCl buffer (10 mM) with 0.25 M sucrose, 1 mM EDTA, pH 7.4, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (w:v). The homogenized mixture was filtered through a fine gauze and then centrifuged at 1900g for 20 minutes. After centrifugation, a millilitre sample of supernatant fluid was kept for the determination of total liver, and the remaining supernatant was once more centrifuged at 18000g for 60 minutes in order to obtain the cytosolic fraction for the determination of antioxidant enzymes.
:4 (w:v). The homogenized mixture was filtered through a fine gauze and then centrifuged at 1900g for 20 minutes. After centrifugation, a millilitre sample of supernatant fluid was kept for the determination of total liver, and the remaining supernatant was once more centrifuged at 18000g for 60 minutes in order to obtain the cytosolic fraction for the determination of antioxidant enzymes.
The white and brown fat were homogenized and centrifuged in the same manner as the liver sample. And the supernatant was used also for the determination of antioxidant enzymes.
2 mL of blood samples were centrifuged at 1100g. The pellet was resuspended in the same amount of bidistilled water as the discarded supernatant and after vortexing the solution, it was left for 2 hours at 4 °C to obtain total hemolysis. Aliquots of 0.4 mL of samples were kept for the determination of glutathione peroxidase (GPx) and hemoglobin. In order to obtain the precipitation of hemoglobin, chloroform:ethanol (3:5, v/v) and 0.3 mL of bidistilled water were added. The samples were gently shaken and then centrifuged at 1100g for 10 minutes, and the supernatant fluid was kept in an Eppendorf tube to measure the activity of superoxide dismutase (SOD) and catalase (CAT).
2.5. Quantification of hemoglobin and antioxidant enzymatic systems
The measurement of the total hemoglobin quantity was performed through the use of a colorimetric commercial kit supplied by Boehringer Mannheim (Reference 124729). The spectrophotometric determination was done at 546 nm. This determination consists of reacting hemoglobin with a reagent that contains cyanide and ferrocyanide, which oxidizes hemoglobin to methaemoglobin which is converted into cyanomethaemoglobin. The color intensity of this compound is measured photocolorimetrically.SOD activity was measured at 450 nm by the inhibition of the xanthine/xanthine oxidase-mediated oxidation of cytochrome c using an assay kit manufactured by Biovision (Reference K335-100). The assay kit utilizes WST-1 that produces a water-soluble formazan dye upon reduction with superoxide anions. The rate of the reduction with a superoxide anion is linearly related to the xanthine oxidase activity and is inhibited by SOD. Therefore, the inhibition activity of SOD is determined by a colorimetric method. CAT activity was determined spectrophotometrically at 240 nm by the reduction of H2O2 at 25 °C.55 CAT catalyzes the decomposition of hydrogen peroxide into oxygen and water. The reduction of hydrogen peroxide per unit of time is proportional to the amount of catalase in the sample. GPx activity was assessed at 340 nm from the NADPH decrease at 37 °C.56 The quantification is based on the coupled reaction in which glutathione peroxidase catalyzes the oxidation reaction of glutathione (GSH) to glutathione disulfide (GSSG). Then, the enzyme glutathione reductase catalyzes the reduction of the disulfide glutathione (GSSG) in the presence of NADPH by forming glutathione (GSH). The protein content was measured by the Bradford assay (Bio-Rad reagent).
2.6. Study of hepatocytes isolated from rats
The cells were centrifuged at 200g for 3 min, the supernatant was discarded and the pellet was resuspended again with Krebs buffer supplemented with calcium at 37 °C. The samples were centrifuged once more at the same speed. The supernatant was discarded and the pellet was weighed and resuspended at the proportion of 10 mL g−1 of cells.
The metabolic studies used 2 mL of cell suspension and 2 mL of Krebs-Henseleit buffer was added to the samples. The hepatic suspension was distributed into 25 mL Erlenmeyer flasks.
The oxidants tert-butylhydroperoxide (t-BOOH) and dimethyl sulfoxide (DMSO) were added to the isolated hepatocytes. The samples were incubated at 37 °C for 60 minutes with gentle and constant shaking under an atmosphere of oxygen–carbon dioxide (95:5). After incubation, the reaction was stopped with perchloric acid at 20%. The samples were centrifuged at 1100g for 10 min, and the supernatant fluid was neutralized with KOH (20%).
A special treatment of the samples was required for the determination of LDH. After incubation and before cellular destruction with PCA at 20%, the samples were centrifuged at low speed (200g) for 5 min to avoid the breaking of the cell membranes in order to get the cytosolic lactate dehydrogenase (LDH) release. The supernatant fluid was kept for analysis.
2.7. Statistical analysis
Data are presented as the mean ± SEM of n experiments obtained from different animals. Statistical analysis was performed with SPSS software (19 version). Statistically significant differences in mean values were tested by analysis of variance. Then, the Scheffé test was carried out. The groups studied fitted into the normality that was analyzed by the Kolmogorov–Smirnov test and there was homogeneity of variances that was analyzed by the Levene test. Statistical differences were set at p < 0.05.3. Results
3.1. Nutrient intake and animal body development
The values of daily and total intakes of macronutrients and energy in the control and hyperlipidic diets as well as those supplemented with vitamin C are shown in Table 3. It can be observed that the animals of the groups supplemented with vitamin C ingested a slightly higher volume of diet (3%) from the beginning of the treatment compared with the groups without vitamin C supplementation.| Dietary treatment | ||||
|---|---|---|---|---|
| Nutrient intake | C.O. (n = 7) | C.O.C (n = 8) | H.O. (n = 8) | H.O.C (n = 9) |
| C.O.: control olive oil diet; C.O.C: control olive oil diet supplemented with vitamin C; H.O.: hyperlipidic olive oil diet; H.O.C: hyperlipidic olive oil diet supplemented with vitamin C. Values are expressed as mean ± SEM. | ||||
| Diet volume | ||||
| Daily (mL per 24 h) | 59.88 ± 0.65 | 64.43 ± 2.76 | 57.30 ± 2.10 | 63.03 ± 3.50 |
| Total (mL) | 1796.50 ± 19.50 | 1932.9 ± 88.57 | 1718 ± 60.50 | 1890 ± 106 |
| Proteins | ||||
| Daily (g per 24 h) | 3.12 ± 0.04 | 3.36 ± 0.14 | 2.98 ± 0.11 | 3.29 ± 0.18 |
| Total (g) | 93.47 ± 1.10 | 100.73 ± 4.31 | 89.51 ± 3.18 | 98.57 ± 5.50 |
| Fat | ||||
| Daily (g per 24 h) | 0.50 ± 0.01 | 0.54 ± 0.03 | 3.27 ± 0.12 | 3.59 ± 0.20 |
| Total (g) | 15.00 ± 0.23 | 16.16 ± 0.77 | 98.03 ± 3.48 | 107.80 ± 6.03 |
| Carbohydrates | ||||
| Daily (g per 24 h) | 9.82 ± 0.12 | 10.59 ± 0.45 | 9.41 ± 0.33 | 10.34 ± 0.59 |
| Total (g) | 294.56 ± 3.48 | 317.55 ± 13.64 | 282.23 ± 9.97 | 310.33 ± 17.70 |
| Energy | ||||
| Daily (kcal per 24 h) | 55.58 ± 0.67 | 59.90 ± 2.77 | 78.20 ± 2.77 | 86.02 ± 4.86 |
| Total (kcal) | 1667.42 ± 20 | 1797.14 ± 83.07 | 2346 ± 83.1 | 2580 ± 145.7 |
The body development of the experimental animals was assessed during all the dietary treatment and the results confirmed that the overfed animals increased their body weight by 75–100% compared to their controls (Table 4). In addition, this hyperlipidic group had a higher growth rate (68–75%) than the control group (33–43%).
| Weight (g) | C.O. (n = 8) | C.O.C (n = 10) | H.O. (n = 10) | H.O.C (n = 11) |
|---|---|---|---|---|
| C.O.: control olive oil diet; C.O.C: control olive oil diet supplemented with vitamin C; H.O.: hyperlipidic olive oil diet; H.O.C: hyperlipidic olive oil diet supplemented with vitamin C. Values are expressed as mean ± SEM. The statistical difference is indicated as follows: *p < 0.05, **p < 0.005 C.O. vs. C.O.C and H.O. vs. H.O.C; $p < 0.05, $$p < 0.005 C.O. vs. H.O. and C.O.C vs. H.O.C. | ||||
| Body development | ||||
| Day 1 | 181 ± 2.9 | 181 ± 4.3 | 178 ± 3.1 | 181 ± 3.7 |
| Day 7 | 199 ± 3.4 | 204 ± 4.9 | 212 ± 4.9 | 219 ± 3.8 |
| Day 14 | 212 ± 4 | 220 ± 5.8 | 242 ± 7.7 | 253 ± 5.8 |
| Day 21 | 230 ± 5 | 243 ± 8.1 | 272 ± 10 | 286 ± 8.5 |
| Day 30 | 241 ± 4.7 | 259 ± 9.8 | 300 ± 13 | 318 ± 13 |
| Total increase | 60.3 ± 4.6 | 78.4 ± 9** | 121.12 ± 2.9$$ | 137 ± 12$$* |
| Fat tissue development | ||||
| White Fat. Day 30 | 1.87 ± 0.43 | 1.71 ± 0.32 | 3.54 ± 0.82$$ | 4.15 ± 0.47$$ |
| Brown Fat. Day 30 | 0.38 ± 0.05 | 0.43 ± 0.07 | 0.45 ± 0.08$ | 0.54 ± 0.09$* |
Since the development of obesity is associated with an increase in lipid reserves, after animal sacrifice, the magnitude of two important fatty tissue masses susceptible to further development in the overfed animals was assessed: white adipose tissue and brown adipose tissue. The feeding with hyperlipidic diets caused a significant increase in the accumulation of adipose tissue in the animals. In addition, Table 4 demonstrates that the increase in adipose tissue is considerably higher than the general body development, with a greater rise in the animals of the hyperlipidic groups compared to their control groups.
3.2. Antioxidant enzymatic activities in different tissues and compartments
The activity of the antioxidant enzymes superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) was evaluated in total liver, in the liver cytosol, in abdominal white fat, in brown fat and in blood cells.In total liver, liver cytosol and white fat, our results show a significant decrease in the activity of SOD in the hyperlipidic groups, supplemented or not with vitamin C, compared to their controls (Fig. 1A, B and D). In addition, it can be observed that the administration of vitamin C did not modify the activity of the enzyme in these tissues except for in liver cytosol, where there was a significant increase in the activity of SOD in the hyperlipidic group supplemented with vitamin C (Fig. 1A, B and D).
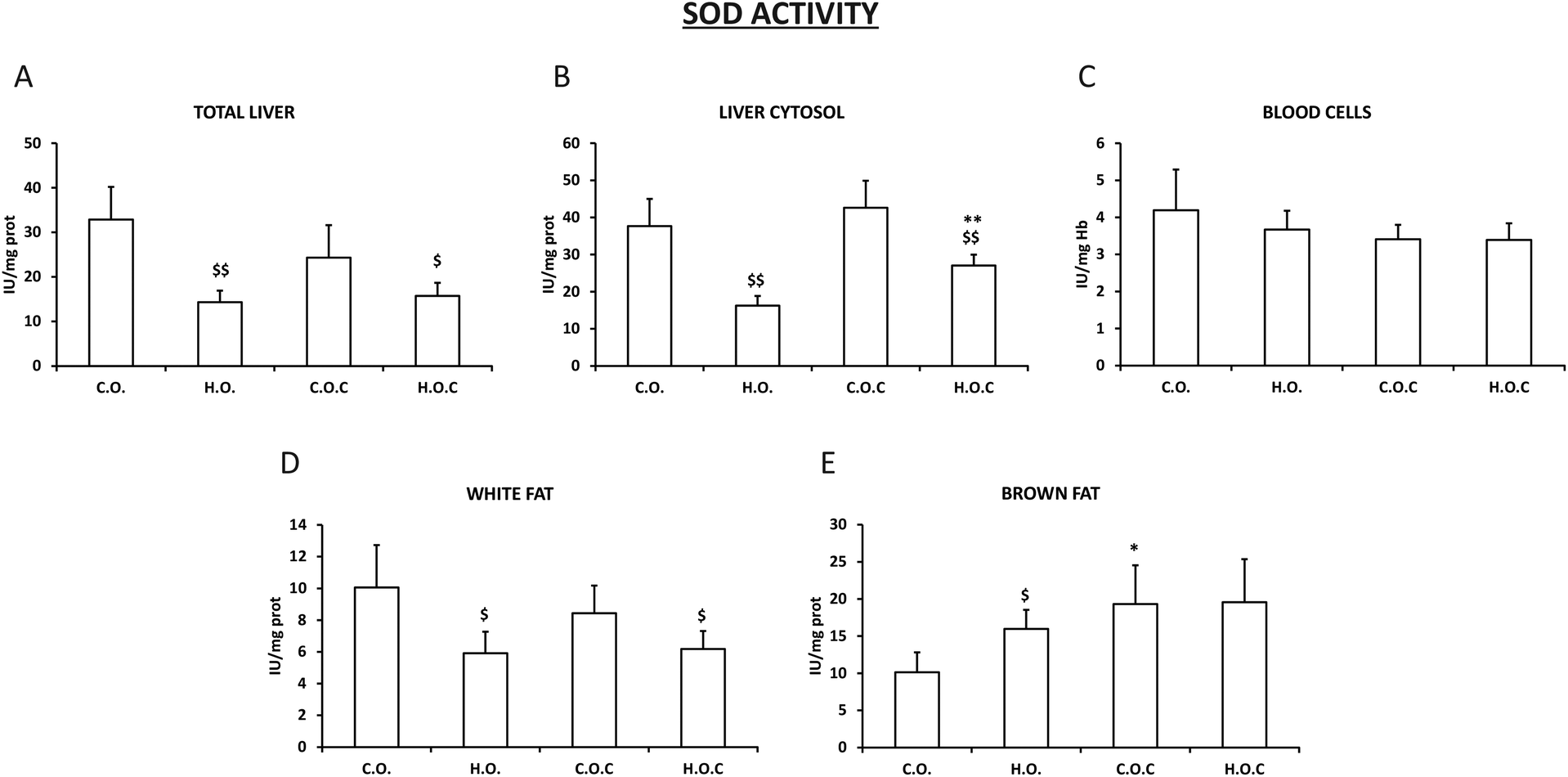 | ||
| Fig. 1 SOD activity in total liver (A), liver cytosol (B), blood cells (C), white fat (D) and brown fat (E). C.O.: control olive oil diet (n = 5); H.O.: hyperlipidic olive oil diet (n = 7); C.O.C: control olive oil diet supplemented with vitamin C (n = 8); H.O.C: hyperlipidic olive oil diet supplemented with vitamin C (n = 8). Values are expressed as mean ± SEM. The statistical difference is indicated as follows: *p < 0.05, **p < 0.005 C.O. vs. C.O.C and H.O. vs. H.O.C; $p < 0.05, $$p < 0.005 C.O. vs. H.O. and C.O.C vs. H.O.C. | ||
In brown fat, the SOD profile was more irregular than that observed in white fat. Thus, the enzymatic activity suffered an increase in the hyperlipidic groups. Only vitamin C supplementation had a positive effect in the control group (Fig. 1E). In the case of blood cells, there was no change in the activity of the enzyme in all the experimental groups studied (Fig. 1C).
The catalase activity observed in total liver and liver cytosol was very similar between them (Fig. 2A and B). In addition, this enzyme showed a very similar profile in total liver, liver cytosol, white fat and brown fat (Fig. 2A, B, D and E) to the previously described SOD (Fig. 1A, B and D) with the following exceptions: the administration of vitamin C produced a significant decrease in catalase activity in the control groups of total liver and liver cytosol (Fig. 2A and B). In white fat, only the hyperlipidic group supplemented with vitamin C showed a significant decrease in catalase activity (Fig. 2D). In the case of blood cells, the hyperlipidic group lost catalase activity which was recovered by the administration of vitamin C (Fig. 2C).
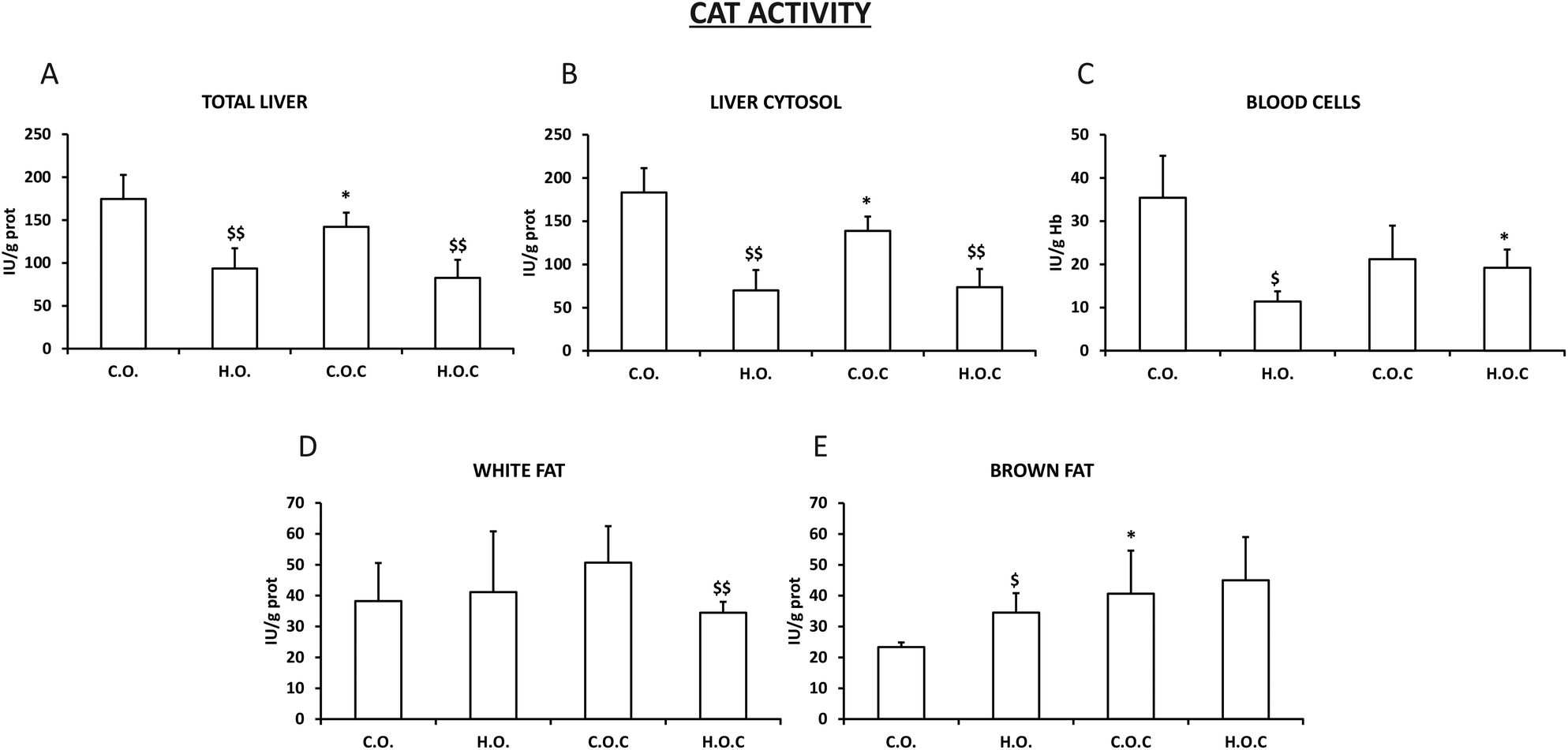 | ||
| Fig. 2 Catalase activity in total liver (A), liver cytosol (B), blood cells (C), white fat (D) and brown fat (E). C.O.: control olive oil diet (n = 5); H.O.: hyperlipidic olive oil diet (n = 7); C.O.C: control olive oil diet supplemented with vitamin C (n = 8); H.O.C: hyperlipidic olive oil diet supplemented with vitamin C (n = 8). Values are expressed as mean ± SEM. The statistical difference is indicated as follows: *p < 0.05 C.O. vs. C.O.C and H.O. vs. H.O.C; $p < 0.05, $$p < 0.005 C.O. vs. H.O. and C.O.C vs. H.O.C. | ||
The results obtained in the study of GPx activity in total liver and liver cytosol (Fig. 3A and B) were very similar to those observed in CAT activity in the same tissues (Fig. 2A and B). It is important to highlight that in the case of GPx, the administration of vitamin C to the hyperlipidic group mostly recovered the loss of enzymatic activity in total liver (Fig. 2A). As happened with the SOD activity in white fat (Fig. 1D), GPx in this tissue decreased in the hyperlipidic groups supplemented or not with vitamin C compared to their controls (Fig. 3D). Following the tendency observed in SOD and CAT activities in brown fat (Fig. 1E and 2E), the administration of vitamin C to the hyperlipidic groups enhanced the enzyme activity of the GPx (Fig. 3E). Finally, the hyperlipidic group supplemented with vitamin C showed a decrease in GPx activity in blood cells (Fig. 3C).
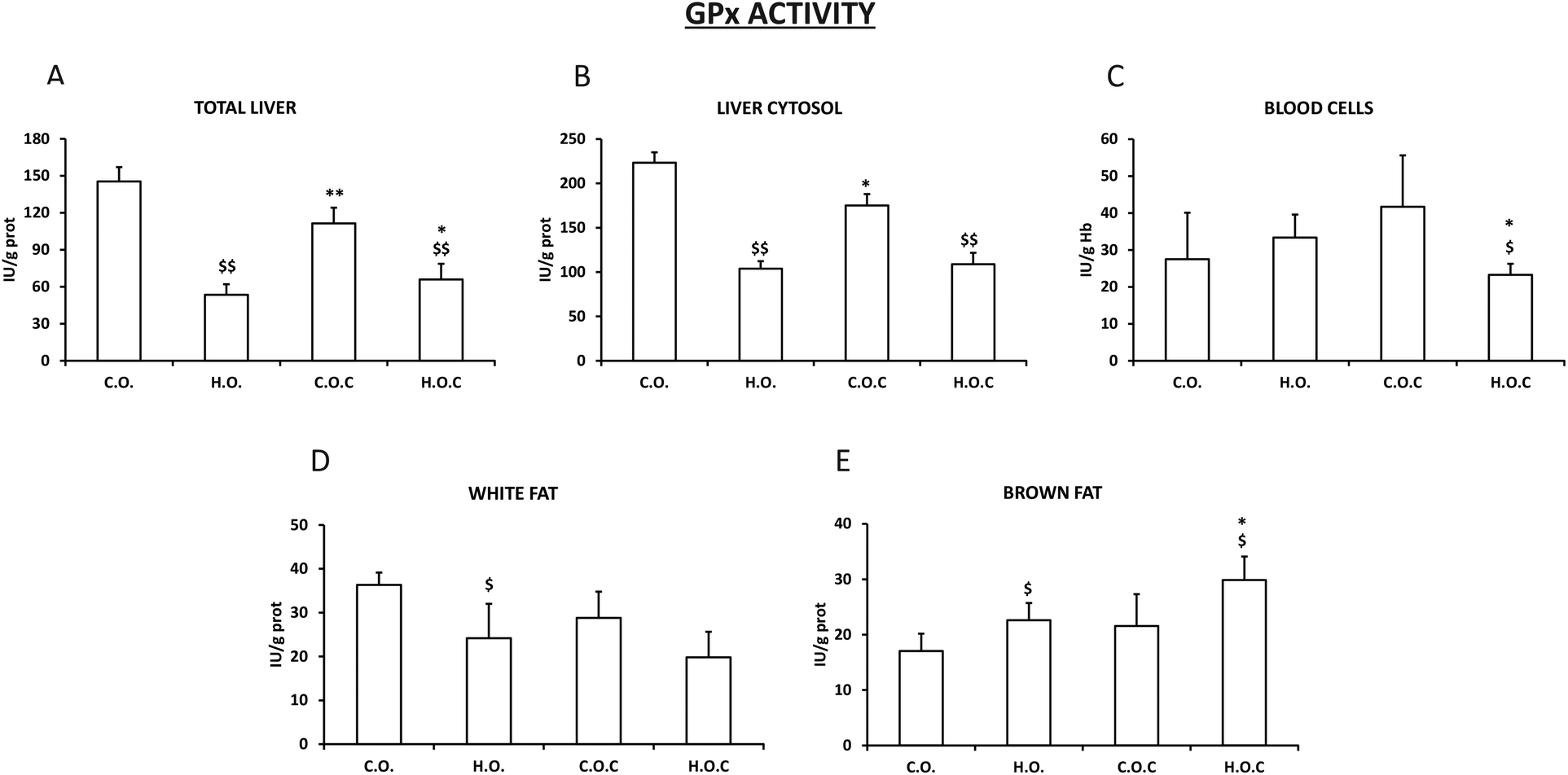 | ||
| Fig. 3 GPx activity in total liver (A), liver cytosol (B), blood cells (C), white fat (D) and brown fat (E). C.O.: control olive oil diet (n = 5); H.O.: hyperlipidic olive oil diet (n = 7); C.O.C: control olive oil diet supplemented with vitamin C (n = 8); H.O.C: hyperlipidic olive oil diet supplemented with vitamin C (n = 8). Values are expressed as mean ± SEM. The statistical difference is indicated as follows: *p < 0.05, **p < 0.005 C.O. vs. C.O.C and H.O. vs. H.O.C; $p < 0.05, $$p < 0.005 C.O. vs. H.O. and C.O.C vs. H.O.C. | ||
3.3. Hepatocytes
The next step in this study was to assess through various metabolic parameters (glucose, pyruvate, lactate, LDH, ATP, acetoacetate and beta-hydroxybutyrate) the antioxidant and cytoprotective effects of vitamin C in rat isolated hepatocytes that had been subjected to the 4 diets used in the present study (see the Materials and methods section). Oxidative stress was induced in hepatocytes by t-BOOH and DMSO.Glucose levels changed in all groups before and after the incubation period (Fig. 4A). The initial concentration of glucose in rat hepatocytes fed with a hyperlipidic diet was 65% lower than in the control diet. This decrease was reversed by the administration of vitamin C (Fig. 4A). After 60 min of incubation, an increase in glucose levels was observed in all the groups studied, an index of the glycogenolytic activity of the hepatocytes. t-BOOH incubation did not change this situation and the differences remained in the groups. However, a significant increase in glucose levels at 60 min of t-BOOH incubation was observed in the H.O.C group, compared to DMSO 60 min and control 60 min. DMSO did not alter the previously described results (Fig. 4A).
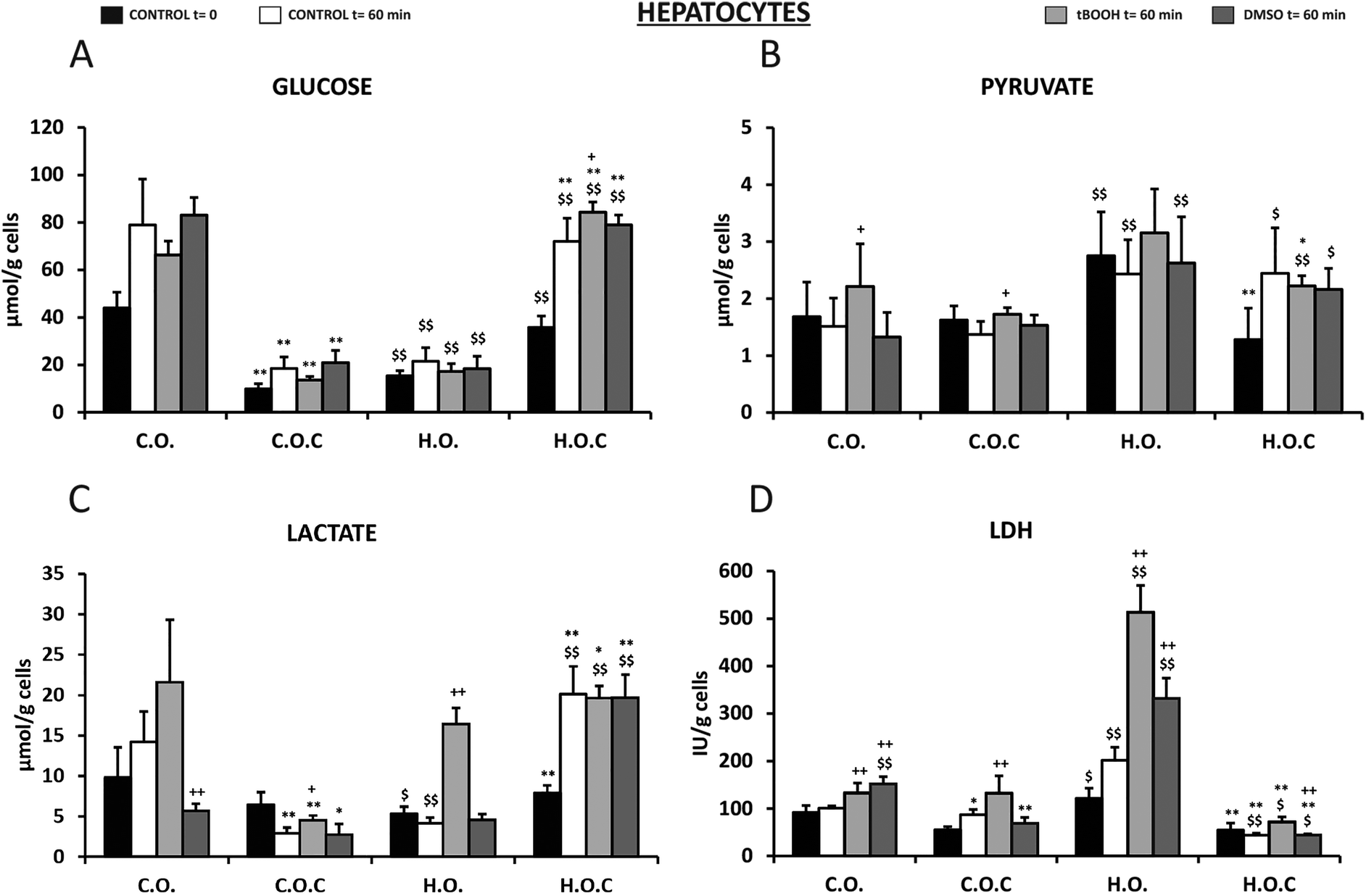 | ||
| Fig. 4 Effect of t-BOOH and DMSO on glucose levels (A), pyruvate levels (B), lactate levels (C) and LDH release (D) in rat isolated hepatocytes. C.O.: control olive oil diet; H.O.: hyperlipidic olive oil diet; C.O.C: control olive oil diet supplemented with vitamin C; H.O.C: hyperlipidic olive oil diet supplemented with vitamin C. Values are expressed as mean ± SEM. The number of rats per group was 5–8. The statistical difference is indicated as follows: *p < 0.05, **p < 0.005 C.O. vs. C.O.C and H.O. vs. H.O.C; $p < 0.05, $$p < 0.005 C.O. vs. H.O. and C.O.C vs. H.O.C; +p < 0.05, ++p < 0.005 t-BOOH 60 min or DMSO 60 min vs. control 60 min. | ||
In the case of pyruvate concentration (Fig. 4B), the levels of this metabolite under basal conditions were twice in the H.O. group than the C.O. group, which also occurred after incubation for 60 min. t-BOOH produced an increase compared to the 60 min control in the C.O. and C.O.C groups. DMSO did not trigger significant changes in reference to the respective controls of each group (Fig. 4B).
Contrary to what was observed in pyruvate, the initial lactate level (t = 0) was significantly lower in rat hepatocytes fed with a hyperlipidic diet and in this group vitamin C had an effect increasing the concentration of this metabolite (Fig. 4C). Unlike pyruvate, after 60 min incubation changes occurred depending on the nutritional group and the experimental condition studied. After this incubation period and in the absence of t-BOOH and DMSO, lactate levels increased in the control group (C.O.) and in the hyperlipidic group supplemented with vitamin C (H.O.C) (Fig. 4C). However, in the hyperlipidic group (H.O.) and in the control group supplemented with vitamin C (C.O.C) lactate values decreased. In the presence of t-BOOH, the concentration of lactate rose significantly in the H.O. group (Fig. 4C). Under these experimental conditions of oxidative stress, supplementation with vitamin C improved the lactate levels in the hyperlipidic group. DMSO provided lactate values much lower than those observed with t-BOOH. However, supplementation with vitamin C led to a significant increase in hyperlipidic rats (Fig. 4C).
Regarding the concentration of LDH released to the incubation medium by isolated hepatocytes of rats (Fig. 4D), we found a significant increase in LDH values in the H.O. group compared to the C.O. group during the incubation period. This would indicate cell damage. This effect was enhanced by the presence of t-BOOH. By supplementing with vitamin C a decrease in the release of LDH was observed, indicating a protective effect induced by vitamin C that is more pronounced in the hyperlipidic group. DMSO also induced a significant increase in the release of LDH to the medium in the control and hyperlipidic rats. In both cases, supplementation with vitamin C also exerted a protective effect by significantly decreasing LDH levels (Fig. 4D).
Fig. 5A shows the ATP levels of the isolated rat hepatocytes of the different nutritional groups and under the different experimental conditions. In all the groups studied, both at the initial moment of incubation (t = 0) and after 60 min, ATP remained within normal values, but these values were lower when the cells were incubated in the absence of substrates as a consequence of its metabolic utilization. In all groups, this decrease ranged between 30 and 40% with respect to the corresponding initial value at t = 0 without any change in the ATP due to the dose of olive oil or vitamin C supplementation. When the cells were incubated in the presence of t-BOOH, the ATP concentration decreased even more, being significant in the case of the hyperlipidic diet (H.O.) group. However, the presence of vitamin C in the hyperlipidic diet protects against the decrease in ATP induced by t-BOOH. In the presence of DMSO, similar values were found to those obtained in the absence of substrates after 60 min of incubation. However, in the H.O.C group, ATP decreased significantly suggesting a possible cytotoxic interaction between these two compounds.
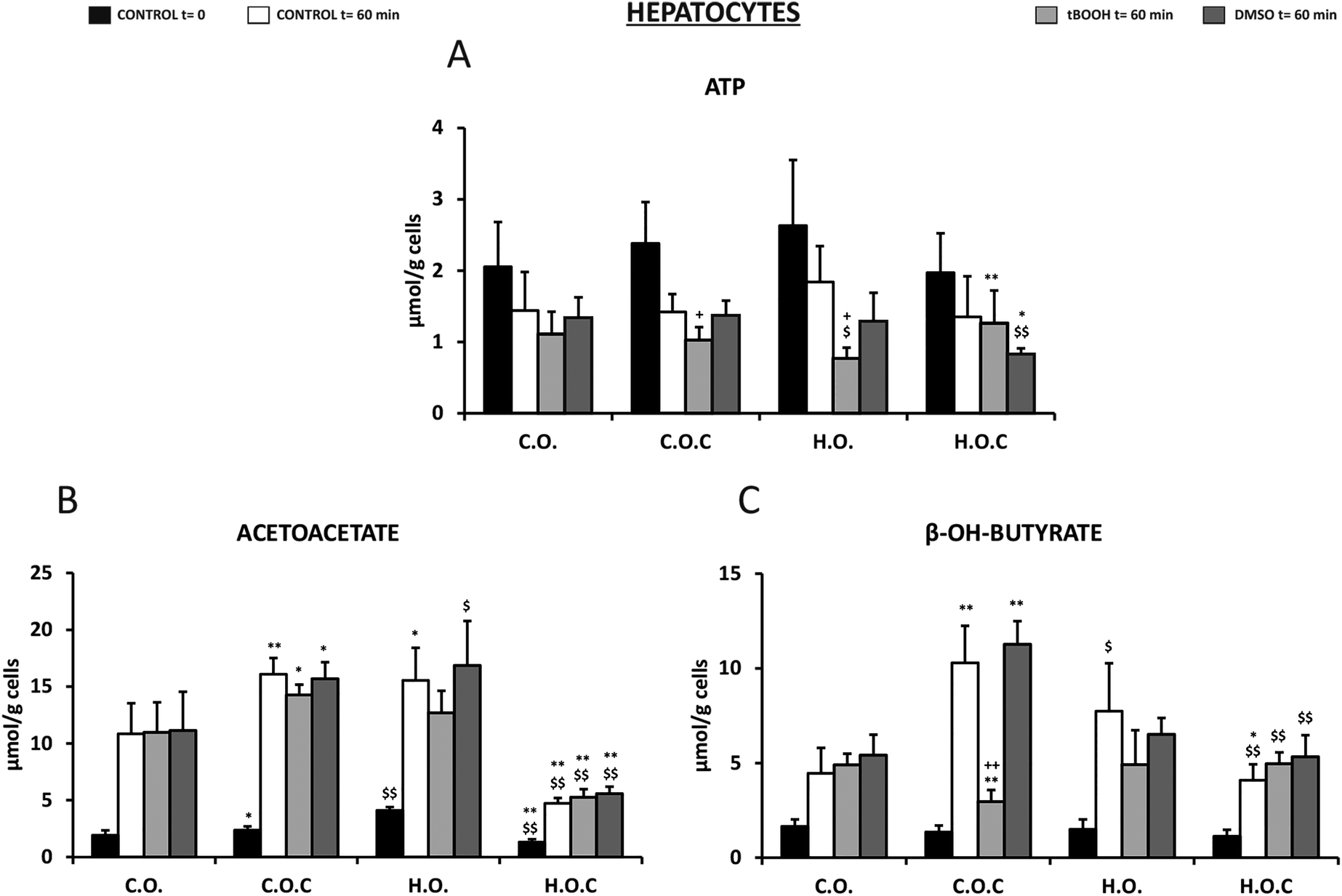 | ||
| Fig. 5 Effect of t-BOOH and DMSO on ATP (A), acetoacetate levels (B), and beta-hydroxybutyrate levels (C) in rat isolated hepatocytes. C.O.: control olive oil diet; H.O.: hyperlipidic olive oil diet; C.O.C: control olive oil diet supplemented with vitamin C; H.O.C: hyperlipidic olive oil diet supplemented with vitamin C. Values are expressed as mean ± SEM. The number of rats per group was 5–8. The statistical difference is indicated as follows: *p < 0.05, **p < 0.005 C.O. vs. C.O.C and H.O. vs. H.O.C; $p < 0.05, $$p < 0.005 C.O. vs. H.O. and C.O.C vs. H.O.C; +p < 0.05, ++p < 0.005 t-BOOH 60 min or DMSO 60 min vs. control 60 min. | ||
Rat hepatocytes of the C.O.C and H.O. groups showed significantly higher acetoacetate levels than the control (C.O.) (Fig. 5B). This was observed at t = 0 and at t = 60 min in the absence of substrates, with higher acetoacetate levels after the incubation period. Incubation with t-BOOH and DMSO produced a similar profile (Fig. 5B). Under all conditions, supplementation with vitamin C to the hyperlipidic group (H.O.C) significantly decreased the acetoacetate levels. Therefore, it could be suggested that vitamin C acted differently depending on the content of fatty acids in the diet (Fig. 5B).
Regarding the other ketone body studied, beta-hydroxybutyrate, values did not vary from one group to another in fresh isolated hepatocytes (t = 0) (Fig. 5C). At 60 min after incubation, a significant increase in the concentration of this metabolite was observed in the C.O.C and H.O. groups, being higher in the C.O.C group (Fig. 5C). In the presence of t-BOOH, supplementation with vitamin C in the control group significantly reduced the concentration of beta-hydroxybutyrate. In contrast, in the presence of DMSO the changes were in the opposite direction (Fig. 5C).
4. Discussion
4.1. Experimental model of nutritional obesity. Body and tissue development of animals
Studies in the field of biochemical changes that are associated with various nutritional alterations require a strict control of all the nutritional factors that affect the feeding of experimental animals; small modifications in the composition of the diet (which may affect the different types of macronutrients or the levels of micronutrients) can induce important metabolic changes and hide the modifications induced by the nutritional alteration under study.60,61 In this way, we must obtain a perfect control of the components of the diet supplied to the animals that will allow us to establish a more accurate cause–effect relationship and lead to a more correct interpretation of the results obtained.In our study, we developed a model of experimental nutritional obesity, in which the animals received a higher amount of daily energy, but this excess of energy was obtained exclusively from the ingestion of a higher quantity of lipids from a unique source. Investigations have demonstrated that the majority of human obesity cases with a nutritional etiology are produced by an excessive intake of lipids.62–67
This model of overfeeding was formulated based on the use of “liquid diets”, since this form of administration allows one to make dietary changes in a simple way and to know exactly the nutritional components of the diet. Thus, the diet administered to the control animals and that supplied to the overfed animals would only differ in the amount of energy ingested by the animals, with the difference provided only by the amount of lipids that they take in with the diet, maintaining the same proportion of the rest of the nutrients, vitamins and minerals and with the same nutritional quality. Therefore, the possible modifications of the oxygenic metabolism that we wanted to study would be exclusively due to a higher energy intake associated with a higher lipid intake and the type of fatty acids supplied to the experimental animals, without the introduction of other nutritional variables such as micronutrients, protein quality, the amount and type of carbohydrates, etc. All these factors could directly or indirectly affect the oxygenic metabolism.
The higher energy intake by the animals subjected to hyperenergetic and hyperlipidic liquid diets triggered a higher growth rate compared to the animals fed with liquid control diets. This increase was progressive and is related to the amount of energy ingested. An increase in white and brown fat occurs simultaneously in animals fed with hyperlipidic diets, as it happens in body development.68–71 Olive oil is one of the main components of the Mediterranean diet. It decreases body weight, body mass index and waist and hip circumferences.72 Some of the mechanisms that could be responsible for the decrease in body weight would be activating β-oxidation, inducing satiety, stimulating energy expenditure by inducing thermogenesis in brown adipose tissue, inhibiting adipocyte differentiation, promoting adipocyte apoptosis and increasing lipolysis. However, the effect of olive oil and the Mediterranean diet would be higher in association with an energy-restricted plan or physical activity improvements.73 This would explain our results in which we observed that, when an excess of fat is consumed, such as olive oil, which in normal quantities has beneficial effects on health, the positive effects on obesity disappear. The excessive intake of a nutritional and healthy fat such as olive oil did not prevent the appearance of obesity. Supplementation with vitamin C did not have a protective effect on body and fat development.
4.2. Oxygenic metabolism. Evolution of SOD, CAT and GPx
There is a complex defense system in organisms against ROS generated in response to different physiological or pathological situations. This system consists of an enzymatic antioxidant system formed by the studied enzymes SOD, catalase and GPx and an extensive non-enzymatic or chemical antioxidant system that the analyzed vitamin C is part of.74,75 The effectiveness of these three enzymes is due to a triple defensive action: they reduce ROS and they prevent the interaction of ROS to give rise to species of higher reactivity and finally they repair oxidized macromolecules. In the absence of oxidative stress, generated ROS are held to very low levels due to the coordinated action of these enzyme systems. If there is a reduction in the antioxidant capacity or unilateral deficiency of one of these enzyme systems, it will increase cell vulnerability against ROS.76 SOD is present in all cells that use oxygen in their metabolism. It catalyzes the conversion of radical superoxide into hydrogen peroxide and oxygen. Hydrogen peroxide is then transformed into water and oxygen by CAT and GPx.77,78Obesity is an important problem in modern society and is associated with oxidative stress.79 One of the main sources of oxidative stress in obesity is altered lipid homeostasis. In addition, ROS production is a consequence of free fatty acid oxidation.80 Obesity triggers changes in the concentration/activity of antioxidants, both non-enzymatic and enzymatic. Several studies indicate a consistent inverse relationship between body fat and total antioxidant capacity.81–84 Our findings are in agreement with previous results. In the present study the activities of the antioxidant enzymes SOD, CAT and GPx in total liver, liver cytosol, white fat, brown fat and blood cells were analyzed. After inducing obesity in rats, it was shown that the activity of SOD, CAT and GPx was significantly reduced in total liver and liver cytosol when compared with their respective control groups, supplemented or not with vitamin C (Fig. 1–3). White fat also had a similar pattern. It is postulated that changes in these three enzymes were associated with increased oxygen consumption and consequent production of superoxide radicals.85,86 Certainly, it is reasonable to think that increasing the amount of lipids in the diet will lead to the generation of a ROS cascade, where the activity of the antioxidant enzymes is affected by the concentration of superoxide radicals. In blood cells, the antioxidant capacity does not vary or decreases when obesity is induced. However, opposite to the pattern observed in total liver, liver cytosol and white fat occurs in brown fat. In brown adipose tissue, the excessive lipid intake induces an increase in the three enzymes (SOD, CAT and GPx) because of the compensatory adaptation to oxidative stress in obesity.87 This could be due to the fact that brown fat, unlike white fat, has a thermogenic function and has a greater amount of mitochondria,88 so its oxygenic metabolism is higher. Several studies have shown that obesity is accompanied by an increase in brown fat mitochondrial activity, inflammation and oxidative damage89,90 and that brown fat is more resistant to obesity than white fat and other body tissues90 which would explain our findings.
Olive oil is the principal source of fat in the Mediterranean diet. The antioxidant properties of olive oil have been widely studied in the literature.91,92 The main mechanism by which the components of olive oil have antioxidant effects involves inhibition and/or scavenging of ROS.93 Among the different components of olive oil include oleic acid which reduces intracellular ROS levels, h-sitosterol that enhances SOD activity and decreases radical superoxide levels, phenolic compounds which decreases lipid peroxidation and oleuropein that reduces oxidative DNA damage.93 However, our results show that when an excess of olive oil is ingested, the mechanism of action of all these components in the body changes and consequently their antioxidant properties disappear, which would explain the decrease in the activity of SOD, CAT and GPx when administering hyperlipidic diets. It would be necessary to carry out further studies to understand and find out the actual mechanism of action.
Vitamin C, also known as ascorbic acid, is a water-soluble vitamin that is abundant in vegetables and fruits.94 Vitamin C has become very popular for its antioxidant properties.95–97 Under oxidative stress situations vitamin C acts as a scavenger of endogenous and exogenous ROS becoming oxidized to its non-antioxidant and unstable form of dehydroascorbic acid.98 In this study, the antioxidant capacity of vitamin C through the measurement of SOD, CAT and GPx activities in different tissues of control rats and obese rats was analyzed. Supplementation with vitamin C in the control rats decreased the activity of CAT and GPx in total liver and liver cytosol, whereas in blood cells and white fat there were no changes (Fig. 1–3). Some authors sustain that “overdosing” with antioxidants is not effective in reducing oxidative stress due to suppression of normal physiological response systems following oxidative stress.99 This would support the findings observed in total liver and liver cytosol. When vitamin C was administered to obese rats a positive effect of the supplementation was observed because there was an increase in the activities of GPx in total liver and brown fat, SOD in liver cytosol and CAT in blood cells (Fig. 1–3). These findings support the fact that vitamin C could have different selectivities and affinities to diverse enzymes and compartments/tissues of the body. Vitamin C acts as a cofactor for a large group of enzymes100–102 and the present results would contribute to the different selectivities depending on the location and type of enzyme.
4.3. Metabolic study in rat isolated hepatocytes
In order to study the response of isolated hepatocytes to oxidative stress, t-BOOH and DMSO were used. These stressors were added to hepatocytes isolated from rats fed with olive oil and supplemented or not with an antioxidant and cytoprotective agent, namely vitamin C.To study the cell viability of these incubation groups, the levels of ATP and LDH were quantified. Lactate dehydrogenase is a cytoplasmic enzyme that catalyzes the conversion of pyruvate into lactate and NADH into NAD+ during glycolysis, and the reverse reaction during the Cori cycle.103 It is a very abundant enzyme in the liver among other tissues, and passes to the extracellular medium in response to cell damage, so its increase in serum is indicative of tissue destruction.103 With regard to the release of LDH in our study, it has a clear dependence on the nutritional status and the presence of antioxidants. Thus, it has been observed that the cytotoxicity induced by t-BOOH mainly and DMSO in terms of LDH release is higher in the hepatocytes of obese rats (Fig. 4D). The antioxidant effect of vitamin C is found and demonstrated by reverting these values to their normal numbers104,105 (Fig. 4D). ATP or adenosine triphosphate is the main energy source molecule for most cellular functions. ATP levels are also an indicator of cellular oxidative stress. Numerous studies have shown that oxidative stress increases mitochondrial electron transport, resulting in amplification of H2O2 production, ATP depletion and cell death.106,107 In addition, the decrease in ATP levels creates a “vicious circle” since it makes the cell more vulnerable to oxidative stress and favors the formation of more ROS and cell dysfunction.108 Thus, the significant decrease in ATP levels in hepatocytes isolated from rats fed with hyperlipidic diets in the presence of t-BOOH could be due to the oxidative stress generated in these conditions, because after the administration of the antioxidant vitamin C, the ATP formation is recovered. In the presence of DMSO the administration of vitamin C decreases the ATP levels significantly suggesting a possible cytotoxic interaction between these two compounds.
Glucose is the main molecule that provides energy and components for our body cells. Pyruvate and lactate are intermediate products formed during glucose metabolism. Scientists have known for many years that the normal breakdown of glucose is interrupted under oxidative stress, as can occur, for example, in inflammatory or toxic processes.109 Under these oxidative stress conditions, the normal functioning of numerous enzymes involved in glucose metabolism is impaired.109 Thus, variations in the cellular levels of glucose, pyruvate and lactate are an index of the cytosolic redox state.110,111 The present results show a decrease in glucose and lactate levels and an increase in pyruvate levels in the hepatocytes isolated from obese rats, which would indicate the oxidation of the cellular cytosolic state and therefore, an increase in oxidative stress.110,111 These results are reversed after supplementation with vitamin C. In this case, glucose and lactate levels significantly increase and pyruvate levels decrease, which suggest an increase in glycogenolysis and gluconeogenesis. Therefore, supplementation with vitamin C to the hepatocytes isolated from obese rats implies the reduction of the cytosolic redox state and cellular protection against oxidative stress.
Under conditions of long fasting (absence of glucose) or intake of diets with a high content of fats (as occurs in our experimental hyperlipidic group), an excess of fatty acids is produced in the body, which are converted into acetyl-CoA.112 The liver is able to convert the excess acetyl-CoA into the acetoacetate ketone body which is subsequently transformed into the beta-hydroxybutyrate ketone body.113 Under these conditions, acetoacetate and beta-hydroxybutyrate serve as fuel for obtaining energy by the tissues.113 Regarding the role of ketone bodies against oxidative stress, the results are conflicting. The experimental evidence is divided into studies that show that ketone bodies are antioxidants and other studies that show that they are prooxidants. This depends on whether the studies have been carried out in vitro or in vivo, the type of organ affected and the pathology.114,115 It has been shown that acetoacetate increases lipid peroxidation in human endothelial cells,116 hyperketonemia contributes to the oxidative stress observed in diabetes mellitus,117 and ketone bodies generate ROS in erythrocytes, monocytes and hepatic cell cultures in vitro.118,119 On the other hand, other studies have shown that ketone bodies are neuroprotective and perform antioxidant activity against a multitude of neurological diseases such as epilepsy and reduce oxidative stress in cardiac tissue.120 In the isolated rat hepatocytes of our study the levels of acetoacetate and beta-hydroxybutyrate increased in the group of obese rats (HO) compared to the control (CO). These levels decreased after the administration of vitamin C which would indicate that in our case, the intake of hyperlipidic diets generates an increase in ketone bodies that induce oxidative stress. However, when administering vitamin C to the control group (COC) the effect is opposite, so vitamin C acts differently depending on the fatty acid content of the diet.
5. Conclusions
In the present work, the results revealed that when an excess of fat is consumed, such as olive oil, which in normal quantities has beneficial effects on health, the positive effects disappear. The changes in the antioxidant activities of the enzymes SOD, CAT and GPx in different compartments/tissues of the body in obesity and after administering vitamin C suggest a distinct selectivity and affinity pattern for them. The antioxidant and protective effects of vitamin C against oxidative stress and obesity are evident when measuring some parameters of the carbohydrate and lipid metabolism such as glucose, pyruvate, lactate, LDH, ATP, acetoacetate and beta-hydroxybutyrate. However, future research is needed to elucidate the specific molecular mechanism behind the antioxidant capacity of vitamin C mainly under obesity situations.Conflicts of interest
The authors declare that there is no conflict of interest.Funding
This work was supported by the Universidad Católica de Valencia “San Vicente Mártir” (Grant Number: UCV257-001).Acknowledgements
We wish to thank Prof. Guillermo Sáez for his invaluable help in the study.References
- R. Sturm and A. Hattori, Int. J. Obes., 2013, 37, 889–891 CrossRef CAS PubMed.
- K. M. Flegal, D. Kruszon-Moran, M. D. Carroll, C. D. Fryar and C. L. Ogden, J. Am. Med. Assoc., 2016, 315, 2284–2291 CrossRef CAS PubMed.
- M. Bastien, P. Poirier, I. Leimeux and J. P. Després, Prog. Cardiovasc. Dis., 2014, 56, 369–381 CrossRef PubMed.
- R. J. Rosenthal, J. Morton, S. Brethauer, S. Mattar, E. De Maria, J. K. Benz, J. Titus and D. Sterrett, Surg. Obes. Relat. Dis., 2017, 13, 1643–1650 CrossRef PubMed.
- L. K. Twells, D. M. Gregory, J. Reddigan and W. K. Midodzi, CMAJ Open, 2014, 2, 18–26 CrossRef PubMed.
- S. Gallus, A. Lugo, B. Murisic, C. Bosetti, P. Boffetta and C. La Vecchia, Eur. J. Nutr., 2015, 54, 679–689 CrossRef PubMed.
- J. Wise, BMJ, 2014, 348, g3582, DOI:10.1136/bmj.g3582.
- C. S. Morgen and T. I. Sørensen, Nat. Rev. Endocrinol., 2014, 10, 513–514 CrossRef PubMed.
- O. A. Baothman, M. A. Zamzami, I. Taher, J. Abubaker and M. Abu-Farha, Lipids Health Dis., 2016, 15, 108 CrossRef PubMed.
- C. A. Roberto, B. Swinburn, C. Hawkes, T. T. Huang, S. A. Costa, M. Ashe, L. Zwicker, J. H. Cawley and K. D. Brownell, Lancet, 2015, 385, 2400–2409 CrossRef.
- A. G. Salici, P. Sisman, O. O. Gul, T. Karayel, S. Cander and C. Ersoy, Endocrine Abstracts, 2017, 49, 679, DOI:10.1530/endoabs.49.EP679.
- C. N. Markey, K. J. August, L. C. Bailey, P. M. Markey and C. S. Nave, Health Psychol. Open, 2016, 3, 1–4 Search PubMed.
- J. Korner and L. J. Aronne, J. Clin. Invest., 2003, 111, 565–570 CrossRef CAS PubMed.
- D. H. Sherling, P. Perumareddi and C. H. Hennekens, J. Cardiovasc. Pharmacol. Ther., 2017, 22, 365–367 CrossRef CAS PubMed.
- B. S. Karam, A. Chavez-Moreno, W. Koh, J. G. Akar and F. G. Akar, Cardiovasc. Diabetol., 2017, 16(1), 120 CrossRef PubMed.
- E. J. Gallagher and D. LeRoith, Physiol. Rev., 2015, 95, 727–748 CrossRef CAS PubMed.
- S. S. Doerstling, C. H. O'Flanagan and S. D. Hursting, Front. Oncol., 2017, 14, 216 CrossRef PubMed.
- R. Carreras-Torres, M. Johansson, V. Gaborieau, P. C. Haycock, K. H. Wade, C. L. Relton, R. M. Martin, G. Davey Smith and P. Brennan, J. Natl. Cancer Inst., 2017, 109, 1–9, DOI:10.1093/jnci/djx012.
- G. C. Kabat, M. Y. Kim, J. S. Lee, G. Y. Ho, S. B. Going, J. Beebe-Dimmer, J. E. Manson, R. T. Chlebowski and T. E. Rohan, Cancer Epidemiol. Biomarkers Prev., 2017, 26, 1730–1735 CrossRef PubMed.
- A. Al-Shorman, H. Al-Domi and M. Al-Atoum, Vascular, 2018, 26, 285–290, DOI:10.1177/1708538117735457.
- G. V. Halade and V. Kain, Compr. Physiol., 2017, 7, 1463–1477 Search PubMed.
- C. J. Lavie, P. A. McAuley, T. S. Church, R. V. Milani and S. N. Blair, J. Am. Coll. Cardiol., 2014, 63, 1345–1354 CrossRef PubMed.
- K. Rahmouni, Hypertension, 2014, 64, 215–221 CrossRef CAS PubMed.
- J. E. Hall, J. M. do Carmo, A. A. da Silva, Z. Wang and M. E. Hall, Circ. Res., 2015, 116, 991–1006 CrossRef CAS PubMed.
- W. S. Aronow, Ann. Transl. Med., 2017, 5, 350 CrossRef PubMed.
- A. Fernández-Sánchez, E. Madrigal-Santillán, M. Bautista, J. Esquivel-Soto, A. Morales-González, C. Esquivel-Chirino, I. Durante-Montiel, G. Sánchez-Rivera, C. Valadez-Vega and J. A. Morales-González, Int. J. Mol. Sci., 2011, 12, 3117–3132 CrossRef.
- S. Furukawa, T. Fujita, M. Shimabukuro, M. Iwaki, Y. Yamada, Y. Nakajima, O. Nakayama, M. Makishima, M. Matsuda and I. Shimomura, J. Clin. Invest., 2004, 114, 1752–1761 CrossRef CAS PubMed.
- A. B. Salmon, Antioxidants, 2016, 5, 24, DOI:10.3390/antiox5030024.
- L. Ramalingam, K. Menikdiwela, M. LeMieux, J. M. Dufour, G. Kaur, N. Kalupahana and N. Moustaid-Moussa, Biochim. Biophys. Acta, 2017, 1863, 1106–1114 CrossRef CAS PubMed.
- H. T. Hee-TaeRoh, S. Y. Cho and W. Y. So, J. Sport Health Sci., 2017, 6, 225–230 CrossRef PubMed.
- E. Ottobelli Chielle, W. M. de Souza, T. P. da Silva, R. N. Moresco and M. B. Moretto, Clin. Biochem., 2016, 49, 548–553 CrossRef CAS PubMed.
- V. Rani, G. Deep, R. K. Singh, K. Palle and U. C. Yadav, Life Sci., 2016, 148, 183–193 CrossRef CAS PubMed.
- G. Rowicka, H. Dyląg, J. Ambroszkiewicz, A. Riahi, H. Weker and M. Chełchowska, Oxid. Med. Cell. Longevity, 2017, 2017, 5621989, DOI:10.1155/2017/5621989.
- P. Newsholme, V. F. Cruzat, K. N. Keane, R. Carlessi and P. I. de Bittencourt Jr., Biochem. J., 2016, 473, 4527–4550 CrossRef CAS PubMed.
- A. Rimessi, M. Previati, F. Nigro, M. R. Wieckowski and P. Pinton, Int. J. Biochem. Cell Biol., 2016, 81, 281–293 CrossRef CAS.
- C. Y. Han, Diabetes Metab. J., 2016, 40, 272–279 CrossRef PubMed.
- M. Nita and A. Grzybowski, Oxid. Med. Cell. Longevity, 2016, 2016, 3164734, DOI:10.1155/2016/3164734.
- S. Manoharan, G. J. Guillemin, R. S. Abiramasundari, M. M. Essa, M. Akbar and M. D. Akbar, Oxid. Med. Cell. Longevity, 2016, 2016, 8590578, DOI:10.1155/2016/8590578.
- J. M. Van Raamsdonk, I. E. Vega and P. Brundin, Oncotarget, 2017, 8, 10777–10778 CrossRef PubMed.
- H. Zhang and R. Tsao, Curr. Opin. Food Sci., 2016, 8, 33–42 CrossRef.
- I. L. Chapple, H. R. Griffiths, M. R. Milward, M. R. Ling and M. M. Grant, Methods Mol. Biol., 2017, 1537, 61–77 CrossRef CAS PubMed.
- A. Bala, C. Mondal, P. K. Haldar and B. Khandelwal, Inflammopharmacology, 2017, 25, 595–607 CrossRef CAS PubMed.
- A. Sarkar and U. Ghosh, IJLTET, 2016, 6, 460–466 Search PubMed.
- A. Sureda, M. D. Bibiloni, M. Martorell, P. Buil-Cosiales, A. Marti, A. Pons, J. A. Tur and M. A. Martinez-Gonzalez, Mol. Nutr. Food Res., 2016, 60, 2654–2664 CrossRef CAS PubMed.
- M. A. Rincon-Cervera, R. Valenzuela, M. C. Hernandez-Rodas, M. Marambio, A. Espinosa, S. Mayer, N. Romero, C. Barrera, A. Valenzuela and L. A. Videla, Nutrition, 2016, 32, 1254–1267 CrossRef CAS PubMed.
- G. Presti, V. Guarrasi, E. Gulotta, F. Provenzano, A. Provenzano, S. Giuliano, M. Monfreda, M. R. Mangione, R. Passantino, P. L. San Biagio, M. A. Costa and D. Giacomazza, Biophys. Chem., 2017, 230, 109–116 CrossRef CAS PubMed.
- I. Kalaiselvan, M. Samuthirapandi, A. Govindaraju, D. Sheeja Malar and P. D. Kasi, Pharm. Biol., 2016, 54, 338–346 CrossRef CAS PubMed.
- M. Nadour, P. Michaud and F. Moulti-Mati, Appl. Biochem. Biotechnol., 2012, 167, 1802–1810 CrossRef CAS PubMed.
- B. Saibandith, J. P. E. Spencer, I. R. Rowland and D. M. Commane, Molecules, 2017, 22, 1082, DOI:10.3390/molecules22071082.
- C. R. Nogueira, F. Borges, E. Lameu, C. Franca and A. Ramalho, Nutr. Hosp., 2013, 28, 1666–1672 CAS.
- A. Kumar, A. Rahal, S. M. Zoheb, A. Prakash and R. Mandil, Toxicol. Environ. Chem., 2015, 96, 1248–1259 Search PubMed.
- P. G. Reeves, F. H. Nielsen and G. C. Fahey Jr., J. Nutr., 1993, 123, 1939–1951, DOI:10.1093/jn/123.11.1939.
- E. Estornell, T. Barber and J. Cabo, Br. J. Nutr., 1994, 72, 509–517 CrossRef CAS PubMed.
- R. Sakly, K. Zarrouk, A. Hedhili, A. Achour, Z. Kallel and A. Mbazzaa, Ann. Urol., 1991, 25, 242–245 CAS.
- J. F. Young, K. Rosenvold, J. Stagsted, J. H. Nielsen and H. J. Andersen, J. Agric. Food Chem., 2005, 53, 745–749 CrossRef CAS PubMed.
- A. G. Wolfgang and L. Flohe, Glutathione peroxidase. Handbook of Methods for Oxygen Radicals Research, CRC Press, USA, 1985 Search PubMed.
- P. O. Seglen, Biochim. Biophys. Acta, 1974, 338, 317–336 CrossRef CAS.
- E. Valero and F. García-Carmona, Anal. Biochem., 1996, 239, 47–52 CrossRef CAS PubMed.
- D. Williamson and J. Mellanby, Determination of d-(-)-3-Hydroxybutyrate. Methods of enzymatic analysis, Bergmeyer HB, USA, 1974 Search PubMed.
- Y. F. Djohan, E. Badia, B. Bonafos, G. Fouret, C. Lauret, A. M. Dupuy, E. Pinot, T. Sutra, S. Gaillet, K. Lambert, F. Raynaud, N. Gayrard, B. Jover, A. A. Monde, J. P. Cristol, C. Coudray and C. Feillet-Coudray, Eur. J. Nutr., 2018, 1–17, DOI:10.1007/s00394-018-1854-3.
- K. D. Hall, K. Y. Chen, J. Guo, Y. Y. Lam, R. L. Leibel, L. E. Mayer, M. L. Reitman, M. Rosenbaum, S. R. Smith, B. T. Walsh and E. Ravussin, Am. J. Clin. Nutr., 2016, 104, 324–333, DOI:10.3945/ajcn.116.133561.
- L. Schiavo, L. Busetto, M. Cesaretti, S. Zelber-Sagi, L. Deutsch and A. Lannelli, World J. Gastroenterol., 2018, 24, 3330–3346, DOI:10.3748/wjg.v24.i30.3330.
- L. T. F. Cheung, R. S. M. Chan, G. T. C. Ko, E. S. H. Lau, F. C. C. Chow and A. P. S. Kong, Nutr. J., 2018, 17, 63, DOI:10.1186/s12937-018-0374-6.
- X. F. Yang, Y. Q. Qiu, L. Wang, K. G. Gao and Z. Y. Jiang, J. Zhejiang Univ. Sci. B, 2018, 19, 884–894, DOI:10.1631/jzus.B1700507.
- A. Marina, A. D. von Frankenberg, S. Suvag, H. S. Callahan, M. Kratz, T. L. Richards and K. M. Utzschneider, Nutrients, 2014, 6, 4678–4690, DOI:10.3390/nu6114678.
- K. Beaulieu, M. Hopkins, J. Blundell and G. Finlayson, Int. J. Behav. Nutr. Phys. Act., 2017, 14, 14, DOI:10.1186/s12966-017-0473-3.
- S. Hu, L. Wang, D. Yang, L. Li, J. Togo, Y. Wu, Q. Liu, B. Li, M. Li, G. Wang, X. Zhang, C. Niu, J. Li, Y. Xu, E. Couper, A. Whittington-Davies, M. Mazidi, L. Luo, S. Wang, A. Douglas and J. R. Speakman, Cell Metab., 2018, 28, 415–431, DOI:10.1016/j.cmet.2018.06.010.
- L. L. Tang, X. H. Tang, X. Li, H. B. Yu, Z. G. Xie, X. Y. Liu and Z. G. Zhou, Zhongguo Yixue Kexueyuan Xuebao, 2014, 36, 614–619, DOI:10.3881/j.issn.1000-503X.2014.06.010.
- W. Wang and P. Seale, Nat. Rev. Mol. Cell Biol., 2016, 17, 691–702, DOI:10.1038/nrm.2016.96.
- M. Okla, J. Kim, K. Koehler and S. Chung, Adv. Nutr., 2017, 8, 473–483, DOI:10.3945/an.116.014332.
- T. Montanari, N. Pošćić and M. Colitti, Obes. Rev., 2017, 18, 495–513, DOI:10.1111/obr.12520.
- S. W. Barth, T. C. L. Koch, B. Watzl, H. Dietrich, F. Will and A. Bub, Eur. J. Nutr., 2012, 51, 841–850, DOI:10.1007/s00394-011-0264-6.
- R. Huo, T. Du, Y. Xu, W. Xu, X. Chen, K. Sun and X. Yu, Eur. J. Clin. Nutr., 2015, 69, 1200–1208, DOI:10.1038/ejcn.2014.243.
- D. Wang, F. Li, Y. Chi and J. Xiang, Cell Stress Chaperones, 2012, 17, 423–433, DOI:10.1007/s12192-011-0317-z.
- W. Talbi, T. Ghazouani, D. Braconi, R. Ben Abdallah, F. Raboudi, A. Santucci and S. Fattouch, J. Appl. Microbiol., 2018, 126, 555–566, DOI:10.1111/jam.14150.
- J. R. Arthur, Cell. Mol. Life Sci., 2000, 57, 1825–1835 CrossRef CAS.
- J. Nordberg and E. S. Arnér, Free Radicals Biol. Med., 2001, 31, 1287–1312, DOI:10.1016/s0891-5849(01)00724-9.
- A. Holmgren, Antioxid. Redox Signaling, 2000, 4, 811–820 CrossRef PubMed.
- L. Lewandowski, M. Kepinska and H. Milnerowicz, Eur. J. Clin. Invest., 2018, 49, e13036, DOI:10.1111/eci.13036.
- M. Brownlee, Diabetes, 2005, 54, 1615–1625 CrossRef CAS.
- J. Bełtowski, G. Wójcicka, D. Górny and A. Marciniak, J. Physiol. Pharmacol., 2000, 51, 883–896 Search PubMed.
- H. H. Hermsdorff, B. Puchau, A. C. Volp, K. B. Barbosa, J. Bressan, M. Á. Zulet and J. A. Martínez, Nutr. Metab., 2011, 8, 59, DOI:10.1186/1743-7075-8-59.
- C. Chrysohoou, B. Demosthenes, B. Panagiotakos, C. Pitsavos, I. Skoumas, L. Papademetriou, M. Economou and C. Stefanadis, Nutr. Metab. Cardiovasc. Dis., 2007, 17, 590–597, DOI:10.1016/j.numecd.2006.05.007.
- F. Amirkhizi, F. Siassi, M. Djalali and S. Shahraki, Obes. Res. Clin. Pract., 2014, 8, 26–34, DOI:10.1016/j.orcp.2012.07.004.
- M. Togo, N. Konari, M. Tsukamoto, R. Kimoto, T. Yamaguchi, H. Takeda and I. Kambayashi, J. Int. Soc. Sports Nutr., 2018, 15, 13, DOI:10.1186/s12970-018-0217-z.
- S. Furukawa, T. Fujita, M. Shimabukuro, M. Iwaki, Y. Yamada, Y. Nakajima, O. Nakayama, M. Makishima, M. Matsuda and I. Shimomura, J. Clin. Invest., 2004, 114, 1752–1761 CrossRef CAS.
- C. Nakao, T. Ookawara, Y. Sato, T. Kizaki, N. Imazeki, O. Matsubara, S. Haga, K. Suzuki, N. Taniguchi and H. Ohno, Free Radical Res., 2000, 33, 229–241 CrossRef CAS PubMed.
- S. Enerbäck, N. Engl. J. Med., 2009, 360, 2021–2023, DOI:10.1056/NEJMcibr0809610.
- M. U. Din, T. Saari, J. Raiko, N. Kudomi, S. F. Maurer, M. Lahesmaa, T. Fromme, E. Z. Amri, M. Klingenspor, O. P. Nuutila and K. A. Virtanen, Cell Metab., 2018, 28, 207–216, DOI:10.1016/j.cmet.2018.05.020.
- M. Alcalá, M. Calderon-Dominguez, E. Bustos, P. Ramos, N. Casals, D. Serra, M. Viana and L. Herrero, Sci. Rep., 2017, 7, 16082, DOI:10.1038/s41598-017-16463-6.
- M. A. Carluccio, L. Siculella, M. A. Ancora, M. Massaro, E. Scoditti, C. Storelli, F. Visioli, A. Distante and R. de Caterina, Arterioscler., Thromb., Vasc. Biol., 2003, 23, 622–629, DOI:10.1161/01.ATV.0000062884.69432.A0.
- S. C. Edgecombe, G. L. Stretch and P. J. Hayball, J. Nutr., 2000, 130, 2996–3002, DOI:10.1093/jn/130.12.2996.
- S. Elias, S. Wisam, A. Luai, B. Massad and A. Nimer, Adv. Exp. Med. Biol., 2017, 960, 607–617, DOI:10.1007/978-3-319-48382-5_26.
- X. Y. Zhang, Z. P. Xu, W. Wang, J. B. Cao, Q. Fu, W. X. Zhao, Y. Li, X. L. Huo, L. M. Zhang, Y. F. Li and W. D. Mi, Int. Immunopharmacol., 2018, 65, 438–447, DOI:10.1016/j.intimp.2018.10.020.
- N. N. DePhillipo, Z. S. Aman, M. I. Kennedy, J. P. Begley, G. Moatshe and R. F. LaPrade, Orthop. J. Sports Med., 2018, 6, 2325967118804544, DOI:10.1177/2325967118804544.
- A. Kabirian, R. A. Batavani, S. Asri-Rezaei and A. Soleimanzadeh, Vet. Res. Forum, 2018, 9, 217–224, DOI:10.30466/vrf.2018.32085 . Epub 2018 Sep 15.
- Y. Min, T. Sun, Z. Niu and F. Liu, Anim. Reprod. Sci., 2016, 171, 1–6, DOI:10.1016/j.anireprosci.2016.04.005.
- A. Szarka, B. Tomasskovics and G. Bánhegyi, Int. J. Mol. Sci., 2012, 13, 4458–4483, DOI:10.3390/ijms13044458.
- M. Ristow, K. Zarse, A. Oberbach, N. Klöting, M. Birringer, M. Kiehntopf, M. Stumvoll, C. R. Kahn and M. Blüher, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8665–8670, DOI:10.1073/pnas.0903485106.
- M. G. Traber and J. F. Stevens, Free Radicals Biol. Med., 2011, 51, 1000–1013, DOI:10.1016/j.freeradbiomed.2011.05.017.
- V. Camarena and G. Wang, Cell. Mol. Life Sci., 2016, 73, 1645–1658, DOI:10.1007/s00018-016-2145-x.
- M. Rutkowski, T. Matuszewski, J. Kedziora, M. Paradowski, K. Kłos and A. Zakrzewski, Pol. Merkuriusz Lek., 2010, 29, 377–381 CAS.
- S. Kaja, A. J. Payne, Y. Naumchuk and P. Koulen, Curr. Protoc. Toxicol., 2017, 72, 1–10, DOI:10.1002/cptx.21.
- E. Maellaro, B. Del Bello, L. Sugherini, A. Pompella, A. F. Casini and M. Comporti, Xenobiotica, 1994, 24, 281–289, DOI:10.3109/00498259409043239.
- M. PSu, H. Chen, C. Wei, N. Chen and W. Wu, Int. Immunopharmacol., 2014, 22, 492–497, DOI:10.1016/j.intimp.2014.07.034.
- B. S. Tiwari, B. Belenghi and A. Levine, Plant Physiol., 2002, 128, 1271–1281, DOI:10.1104/pp.010999.
- A. Bakhtiarian, M. Abdollahi, S. M. Rezayat and H. Mohammadi, Pharm. Biomed. Res., 2015, 1, 10–19, DOI:10.18869/acadpub.pbr.1.2.10.
- F. Schütt, S. Aretz, G. U. Auffarth and J. Kopitz, Invest. Ophthalmol. Visual Sci., 2012, 53, 5354–5361, DOI:10.1167/iovs.12-9845.
- D. Peralta, A. K. Bronowska, B. Morgan, E. Dóka, K. Van Laer, P. Nagy, F. Gräter and T. P. Dick, Nat. Chem. Biol., 2015, 11, 156–163, DOI:10.1038/nchembio.1720.
- D. H. Williamson, P. Lund and H. A. Krebs, J. Biochem., 1967, 103, 514–527 CrossRef CAS PubMed.
- F. Q. Schafer and G. R. Buettner, Free Radicals Biol. Med., 2001, 30, 1191–1212, DOI:10.1016/S0891-5849(01)00480-4.
- L. Laffel, Diabetes/Metab. Res. Rev., 1999, 15, 412–426 CrossRef CAS.
- P. Puchalska and P. A. Crawford, Cell Metab., 2017, 25, 262–284, DOI:10.1016/j.cmet.2016.12.022.
- A. P. Beskow, C. G. Fernandes, G. Leipnitz, B. da Silva Lde, B. Seminotti, A. U. Amaral, A. T. Wyse, C. M. Wannmacher, C. R. Vargas, C. S. Dutra-Filho and M. Wajner, Metab. Brain Dis., 2008, 23, 411–425, DOI:10.1007/s11011-008-9108-3.
- P. A. McPherson and J. McEneny, J. Physiol. Biochem., 2012, 68, 141–151, DOI:10.1007/s13105-011-0112-4.
- S. K. Jain, K. Kannan and G. Lim, Free Radicals Biol. Med., 1998, 25, 1083–1088 CrossRef CAS.
- P. Kanikarla-Marie and S. K. Jain, Cell. Physiol. Biochem., 2015, 35, 364–373, DOI:10.1159/000369702.
- M. A. Abdelmegeed, S. K. Kim, K. J. Woodcroft and R. F. Novak, J. Pharmacol. Exp. Ther., 2004, 310, 728–736, DOI:10.1124/jpet.104.066522.
- S. K. Jain, R. McVie and J. A. Bocchini Jr., Pathophysiology, 2006, 13, 163–170, DOI:10.1016/j.pathophys.2006.05.005.
- R. L. Veech, B. Chance, Y. Kashiwaya, H. A. Lardy and G. F. Cahill Jr., IUBMB Life, 2001, 51, 241–247, DOI:10.1080/152165401753311780.
| This journal is © The Royal Society of Chemistry 2019 |