
Geographical location specific composition of cultured microbiota and Lactobacillus occurrence in human breast milk in China

Abstract
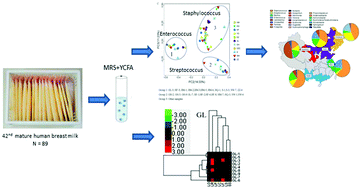
Breast milk bacteria play an important role in the early development of the gut microbiota and the immune system. Dominant living bacteria of 89 healthy Chinese women from 11 cities in five regions were analysed by broad-range yeast extract, casitone, and fatty acid and de Man, Rogosa, and Sharpe-based culturing coupled with 16S rRNA sequence and quantitative polymerase chain reaction. Principal coordinate analysis showed that human breast milk samples were classified into three groups, driven by Enterococcus (abundance in group 1, 63.13%), Streptococcus (abundance in group 2, 68.16%) and Staphylococcus (abundance in group 3, 55.17%). The microbiota profile was highly region-specific. Samples from the Northwest and North of China showed higher alpha diversity compared to other regions (p < 0.05). Staphylococcus, Streptococcus, and Enterococcus were the dominant genera in all samples. Lactobacillus had a high occurrence in samples from the Northwest and North, dominated by Lactobacillus reuteri and Lactobacillus gasseri. Samples of mothers with a high postpartum body mass index showed more Staphylococcus and less Lactobacillus and Streptococcus. Staphylococcus was negatively correlated with Lactobacillus and Streptococcus. The mode of delivery also affected the composition of microbiota, even after culture. These findings indicate differences between the North and South, provide effective information for collection of samples in which Lactobacillus is the predominant genus, and lower the detection limit for small amounts of bacteria.
 Please wait while we load your content...
Please wait while we load your content...

