 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of tetrafluoropropenes by rhodium(I) germyl and silyl complexes†
Maria
Talavera
 a,
Robert
Müller
b,
Theresia
Ahrens
a,
Cortney N.
von Hahmann
a,
Beatrice
Braun-Cula
a,
Martin
Kaupp
*b and
Thomas
Braun
*a
a,
Robert
Müller
b,
Theresia
Ahrens
a,
Cortney N.
von Hahmann
a,
Beatrice
Braun-Cula
a,
Martin
Kaupp
*b and
Thomas
Braun
*a
aHumboldt-Universität zu Berlin, Department of Chemistry, Brook-Taylor-Straße 2, 12489 Berlin, Germany. E-mail: thomas.braun@cms.hu-berlin.de; Tel: +49 30 2093 3913
bInstitut für Chemie, Theoretische Chemie/Quantenchemie, Technische Universität Berlin, Sekr. C7, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: martin.kaupp@tu-berlin.de
First published on 20th September 2019
Abstract
The reaction of the rhodium(I) complexes [Rh(E)(PEt3)3] (E = GePh3 (1), Si(OEt)3 (5)) with HFO-1234yf (2,3,3,3-tetrafluoropropene) afforded [Rh(F)(PEt3)3] (2) and the functionalized olefins Z-CF3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(E) (E = GePh3 (4a), Si(OEt)3 (7)). Conceivable reaction pathways were assessed using DFT calculations. Reactions of [Rh(E)(PEt3)3] with HFO-1234ze (E-1,3,3,3-tetrafluoropropene) yielded the rhodium fluorido complex 2 and [Rh{(E)-CHCH(CF3)}(PEt3)3] (9) via two different reaction pathways. Using complexes 1 and 5 as catalysts, functionalized building blocks were obtained.
CH(E) (E = GePh3 (4a), Si(OEt)3 (7)). Conceivable reaction pathways were assessed using DFT calculations. Reactions of [Rh(E)(PEt3)3] with HFO-1234ze (E-1,3,3,3-tetrafluoropropene) yielded the rhodium fluorido complex 2 and [Rh{(E)-CHCH(CF3)}(PEt3)3] (9) via two different reaction pathways. Using complexes 1 and 5 as catalysts, functionalized building blocks were obtained.
Introduction
The chemistry of fluorinated compounds has grown in recent decades due to their broad applications as building blocks in various fields, such as for agrochemicals or pharmaceuticals or in material science.1 The fluorinated tetrafluoropropenes HFO-1234yf (2,3,3,3-tetrafluoropropene) and HFO-1234ze (E-1,3,3,3-tetrafluoropropene) show good environmental properties which qualify them to be useful as refrigerants, but they might also be interesting substrates for obtaining new building blocks.2 Reactivity studies are rather rare, but involve some organometallic transformations such as C–F bond activation reactions.3 Thus, Ogoshi and co-workers have described monodefluoroborylation and monodefluorosilylation reactions of both HFO-1234yf and HFO-1234ze using a copper catalyst and Bpin (pin = pinacolate) derivatives.3b,d In addition, Crimmin et al. used an aluminum(I) complex for an oxidative addition of the fluoroolefins resulting in a C–F bond activation.3a We recently reported exceptional C–H bond activation at HFO-1234yf initiated by the rhodium(I) fluorido complex 2 in the presence of fluorosilanes to yield [Rh{(E)-C(CF3)CHF}(PEt3)3] (3).4 The conversion also involves a 1,2-fluoride shift (Scheme 1). Catalytic hydrodefluorination reactions on using the rhodium hydrido complex [Rh(H)(PEt3)3] (8) as catalyst were also observed.4
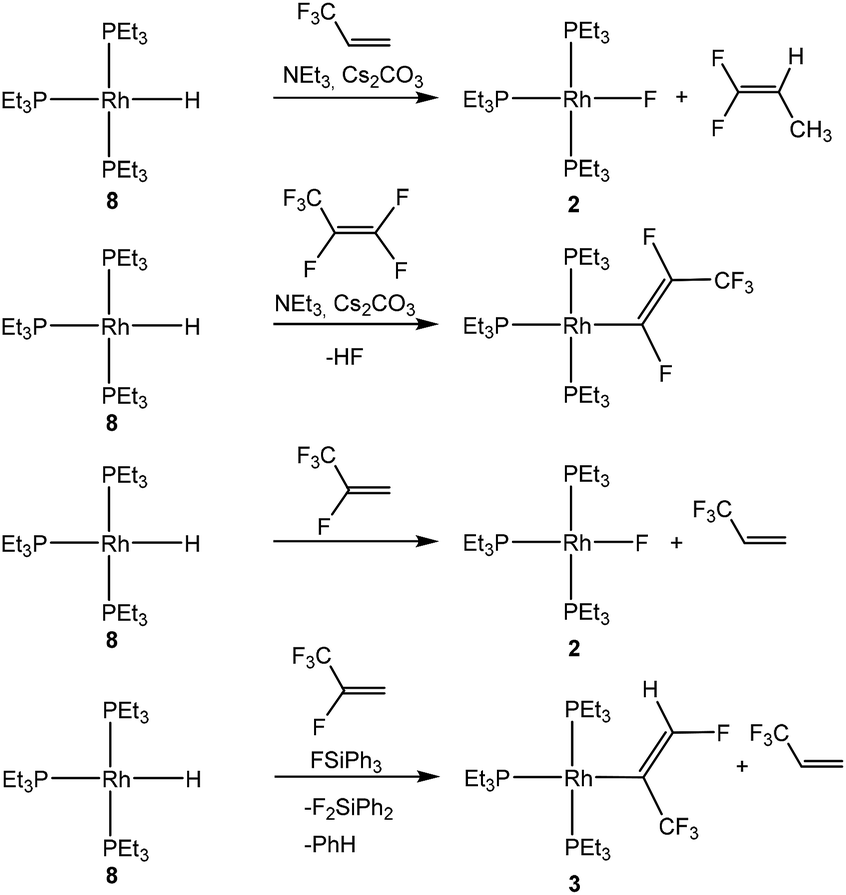 | ||
| Scheme 1 Known reactivity pathways of [Rh(H)(PEt3)3] (8) towards fluoroolefins. | ||
C–F bond activation reactions of fluoroorganics at transition metal complexes are well-known,5 and a useful strategy to achieve this activation consists of the formation of a thermodynamically stable B–F, Si–F, Ge–F, H–F or Al–F bond.5s,6 Interesting rhodium complexes to activate fluoroolefins are, in addition to [Rh(H)(PEt3)3] (8),7 the rhodium(I) germyl complex [Rh(GePh3)(PEt3)3] (1)8 and the rhodium(I) silyl complex [Rh(Si(OEt)3)(PEt3)3] (5).9 Reactivity studies revealed that they exhibit different reaction pathways towards fluorinated olefins, such as olefin coordination to complex 5. Alternatively, C–F bond activation steps lead either to the formation of a rhodium–carbon bond to form a fluorinated ligand or to the formation of a rhodium–fluorine bond and a derived fluorinated olefin. Examples are shown in Schemes 1 and 2 of the reactivity of complexes 1, 5 or 8 with hexafluoropropene8,10 and 3,3,3-trifluoropropene.7d
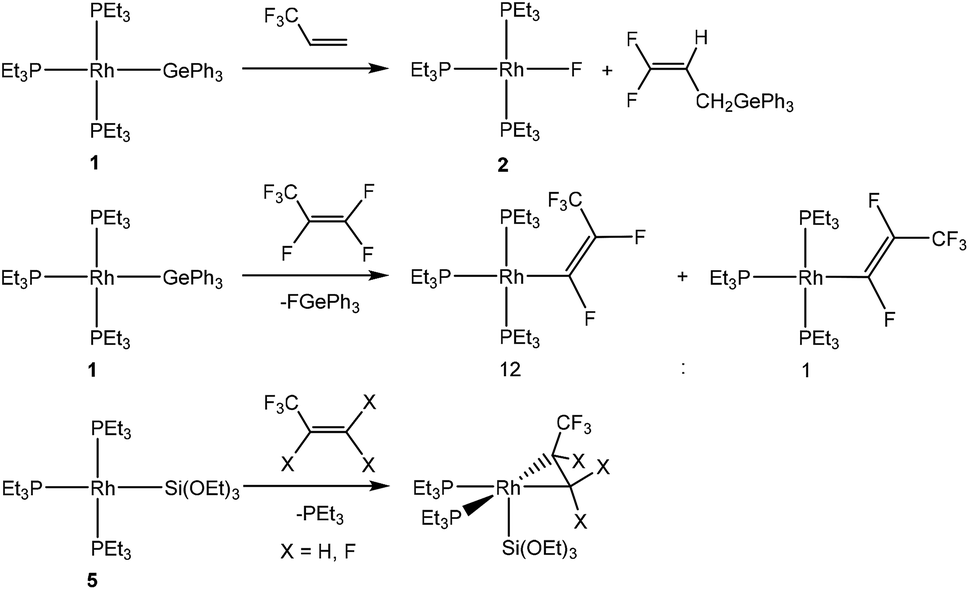 | ||
| Scheme 2 Known reactivity patterns of [Rh(E)(PEt3)3] (E = GePh3 (1), Si(OEt)3 (5)) towards fluoroolefins. | ||
Herein, we report the behavior of [Rh(GePh3)(PEt3)3] (1) and [Rh(Si(OEt)3)(PEt3)3] (5) towards the two isomers HFO-1234yf and HFO-1234ze as well as that of [Rh(H)(PEt3)3] (8) towards HFO-1234ze. Conceivable activation reactions include C(sp3)–F or C(sp2)–F bond cleavage or C–H bond activation, opening up several opportunities for derivatization pathways. The studies describe C–H bond activation, C–F bond activation and olefin coordination as well as catalytic C–F bond activation, germylation and silylation reactions.
Results and discussion
Reactions towards HFO-1234yf (2,3,3,3-tetrafluoropropene)
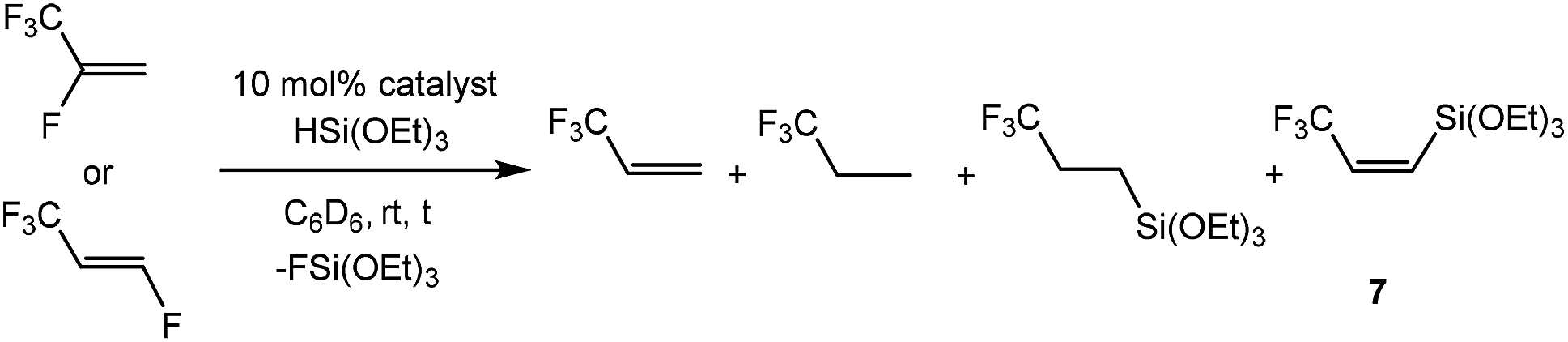
Treatment of the rhodium(I) germyl complex [Rh(GePh3)(PEt3)3] (1) with HFO-1234yf in benzene-d6 afforded as the main product the fluorido complex [Rh(F)(PEt3)3] (2) and 12% of the alkenyl complex [Rh{(E)-C(CF3)CHF}(PEt3)3] (3), both of which have been previously described.4,7c,11 Additionally, the germylated trifluoropropene Z-CF3CHCH(GePh3) (4a) as well as traces of an unknown rhodium complex (5%) were obtained (Scheme 3). Note that complex 3 might be formed from 2 by C–H activation which is mediated by a germane, as previously observed for boranes and silanes.4 The latter act as Lewis-acids and after fluoride abstraction, the C–H activation step at the coordinated olefin is induced by the fluoroborate or fluorosilicate, respectively, to yield 3 and HF.
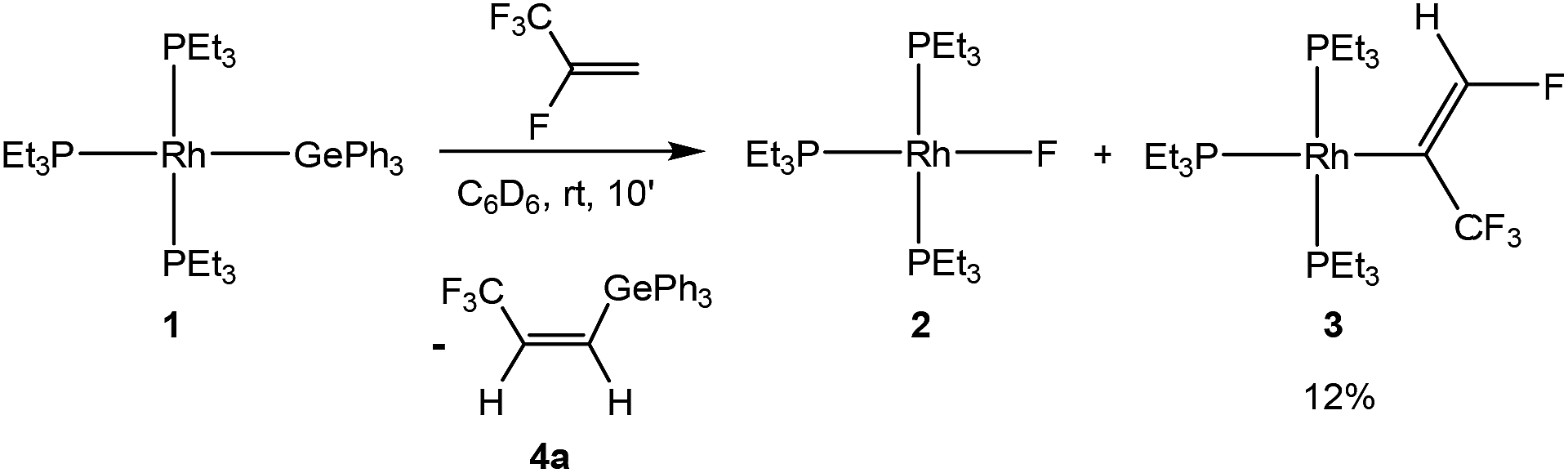 | ||
| Scheme 3 Reaction of the rhodium(I) germyl complex 1 with HFO-1234yf. | ||
The 19F NMR spectrum of 4a shows a doublet at δ −62.2 ppm (3J(F,H) = 7 Hz) for the CF3 group due to a coupling to the proton in the geminal position. The signals of the olefinic protons in the 1H NMR spectrum appear at δ 6.45 and 6.28 ppm as part of an ABX3 system (see ESI† for simulation) with a proton–proton cis coupling of 14.3 Hz and proton–fluorine couplings of 0.34 and 7.87 Hz for the trans and the geminal protons with respect to the CF3 group. A similar cis H,H coupling is reported for the vinylic protons in Z-CF3CHCH(SiMe3) with a trans H,F coupling constant of ca. 0.5 Hz,12 which supports the assignment of a cis configuration in 4a. Suitable crystals for the X-ray crystallography analysis have also been obtained and the structure, bond lengths and angles are shown in Fig. 1. The unit cell contains two independent molecules in the asymmetric unit, which exhibit comparable bond lengths and angles. Therefore, only one molecule will be discussed.
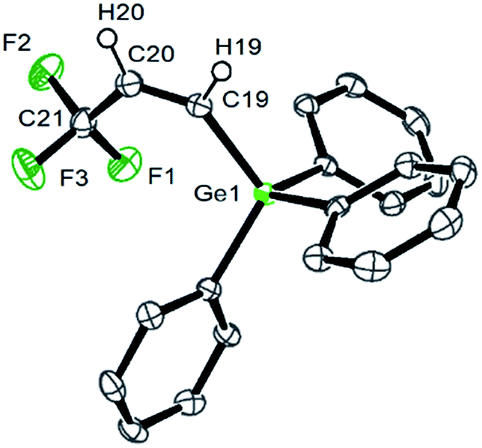 | ||
| Fig. 1 ORTEP diagram of one of the independent molecules in the unit cell of Z-CF3CHCH(GePh3) (4a) drawn at the 50% probability level. Hydrogen atoms of the phenyl rings are not shown for clarity. Selected bond lengths (Å) and angles (°): Ge(1)–C(19), 1.965(2); C(19)–C(20), 1.325(3); C(20)–C(21), 1.476(3); F(1)–C(21), 1.335(3); F(2)–C(21), 1.352(3); F(3)–C(21), 1.346(3); C(20)–C(19)–Ge(1), 133.07(19); C(19)–C(20)–C(21), 125.8(2). | ||
The molecular structure of 4a confirms the cis arrangement at the olefin moiety, and the C–F bond lengths of the trifluoropropene unit have typical values.13 The C–Ge bond distance (1.965(2) Å) is similar to that of C–Ge bonds in other germylated fluoroolefins such as CHFCF(GePh3) (1.958(9) Å),14 CF2CCl(GePh3) (1.954(5) Å)15 and CF3CFCF(GePh3) (1.993(5) Å).16 The Ge⋯F1 distance of 3.105 Å is ca. 0.4 Å less than the sum of the van-der-Waals radii.17 A comparable separation has been discussed for CF3CFCF(GePh3) as a short intramolecular contact.16
To assess the behavior of HFO-1234yf further, the reaction with [Rh(SiOEt)3(PEt3)3] (5) was studied. Treatment of complex 5 with HFO-1234yf in toluene-d8 yielded, in 10 minutes, the rhodium fluorido complex 2 and the cationic species [Rh(PEt3)4]+ (6)18 in a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio as well as 3,3,3-trifluoropropene and Z-CF3CHCH(Si(OEt)3) (7) (Scheme 4). Compound 7 exhibits a doublet for the CF3 group at δ −63.9 ppm in the 19F NMR spectrum with a coupling constant to the proton in the geminal position of 8 Hz. In the 1H NMR spectrum, an ABX3 system (see ESI† for simulation) for the olefinic protons appears at δ 6.07 and 5.96 ppm as an apparent doublet of quartets and a doublet, respectively. The proton–proton coupling constant of 15.54 Hz as well as the H,F coupling constant of less than 0.5 Hz to the non-vicinal proton suggests, as in the case of compound 4a, a cis configuration of the hydrogen atoms. Finally, the 1H, 29Si HMBC NMR spectrum confirms the presence of the silylated group with a correlation peak between its geminal proton and the silicon nucleus at −69.8 ppm in the silicon domain.
:10 ratio as well as 3,3,3-trifluoropropene and Z-CF3CHCH(Si(OEt)3) (7) (Scheme 4). Compound 7 exhibits a doublet for the CF3 group at δ −63.9 ppm in the 19F NMR spectrum with a coupling constant to the proton in the geminal position of 8 Hz. In the 1H NMR spectrum, an ABX3 system (see ESI† for simulation) for the olefinic protons appears at δ 6.07 and 5.96 ppm as an apparent doublet of quartets and a doublet, respectively. The proton–proton coupling constant of 15.54 Hz as well as the H,F coupling constant of less than 0.5 Hz to the non-vicinal proton suggests, as in the case of compound 4a, a cis configuration of the hydrogen atoms. Finally, the 1H, 29Si HMBC NMR spectrum confirms the presence of the silylated group with a correlation peak between its geminal proton and the silicon nucleus at −69.8 ppm in the silicon domain.
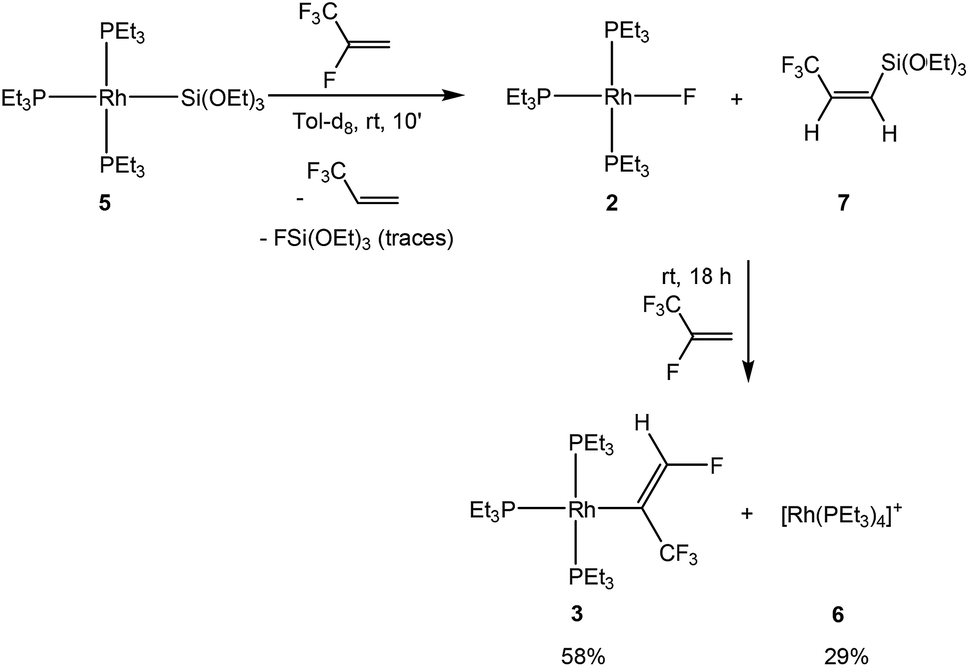 | ||
| Scheme 4 Reaction pathways of the rhodium(I) silyl complex 5 with HFO-1234yf. | ||
The mixture of 2 and the olefin 7 is not stable and it reacts further in the presence of HFO-1234yf to yield complex [Rh{(E)-C(CF3)CHF}(PEt3)3] (3) and approximately 13% of an unknown complex after 18 hours. The silylated olefin 7 presumably acts as a fluoride acceptor to activate the rhodium fluorido complex 2, forming complex 3 by the C–H activation of HFO-1234yf and a subsequent 1,2-fluorine shift. Such a reaction pattern was previously described for FSiPh3.4 Thus, mechanistically, as already indicated above, fluorosilicate generation from [Rh(F)(PEt3)3] (2) and silane followed by a C–H bond activation and a concomitant HF formation was postulated based on computational studies. Independent reactions suggested that the intermediate [Rh{(E)-C(H)CF(CF3)}(PEt3)3] then rearranges by a 1,2-fluorine shift to furnish 3. The latter rearrangement is distinctive and was also observed in the reactions of HFO-1234yf with [Rh(CH3)(PEt3)3] and [Rh(C6D7)(PEt3)3].4
A reasonable mechanism for the formation of the fluorido complex [Rh(F)(PEt3)3] (2) and the olefins Z-CF3CHCH(E) (E = GePh3 (4a), Si(OEt)3 (7)) from the germyl complex [Rh(GePh3)(PEt3)3] (1) or the silyl complex [Rh(Si(OEt)3)(PEt3)3] (5) would imply a two-step process of C–H and C–F bond activation reactions. Thus, C–H bond activation may occur via an initial coordination of HFO-1234yf at the rhodium(I) complex in the first step to form the complex A (Fig. 2 and 3, red line), followed by its insertion into the Rh–Ge or Rh–Si bond. An intramolecular C–H oxidative addition would subsequently yield a rhodacycle derivative C, which undergoes insertion into the rhodium–hydrogen bond (Fig. 2 and 3, blue line). Finally, C–F bond activation via a β-fluoride elimination would afford the rhodium fluorido complex 2 and the corresponding olefin Z-CH(CF3)CHE (E = GePh3 (4a), Si(OEt)3 (7)). Alternatively, intermolecular C–H activation in the initial reaction step would furnish the rhodium–alkenyl complex B (Fig. 2 and 3, green line). This complex could subsequently rearrange to C by a reductive elimination, again followed by an insertion into the Rh–H bond and subsequent β-fluoride elimination. Stereoselective insertion and elimination steps of the β-hydride and β-fluoride would determine the final cis configuration of the functionalized olefin. Note that in comparable conversions such reaction steps of fluorinated olefins showed a high stereoselectivity.19 In order to assess the feasibility of the proposed mechanism, DFT calculations were carried out for all reaction sequences depicted in Fig. 2 and 3. The overall reaction of complexes [Rh(E)(PEt3)3] (E = GePh3 (1), Si(OEt)3 (5)) with HFO-1234yf to afford 2 and 4a/7 is calculated to proceed exergonically by −23 kJ mol−1 and by −31 kJ mol−1 for 1 and 5, respectively.
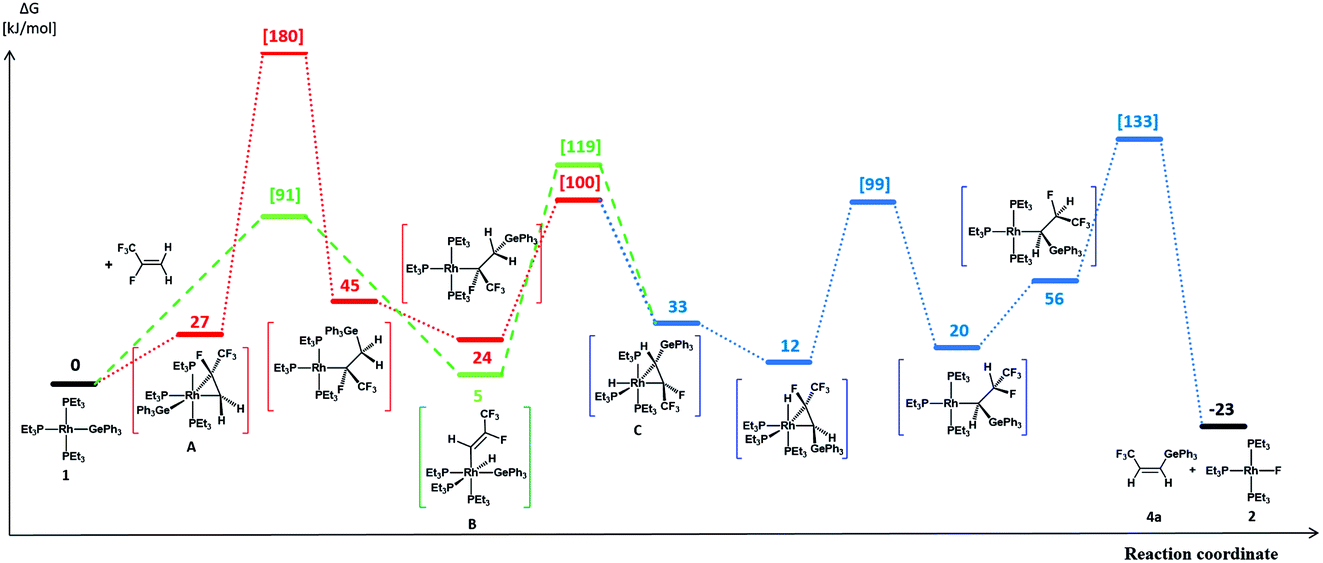 | ||
Fig. 2 Calculated profiles for the activation of HFO-1234yf with complex 1. All calculated reaction and activation free energies (298.15 K, 0.1 MPa; 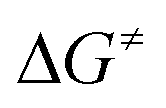 in square brackets) are given relative to the separate reactants 1 and HFO-1234yf (RI-BP86-D3(BJ)/def2-SVP/COSMO(toluene)). in square brackets) are given relative to the separate reactants 1 and HFO-1234yf (RI-BP86-D3(BJ)/def2-SVP/COSMO(toluene)). | ||
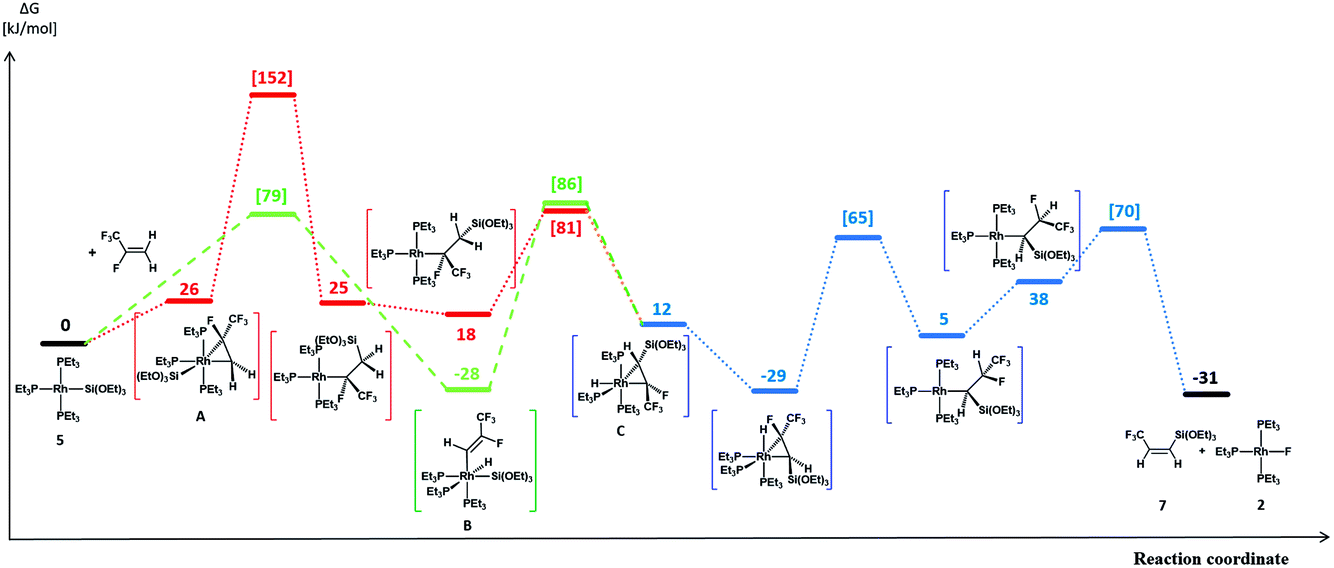 | ||
Fig. 3 Calculated profiles for the activation of HFO-1234yf with complex 5. All calculated reaction and activation free energies (298.15 K, 0.1 MPa;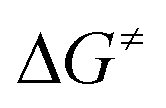 in square brackets) are given relative to the separate reactants 5 and HFO-1234yf (RI-BP86-D3(BJ)/def2-SVP/COSMO(toluene)). in square brackets) are given relative to the separate reactants 5 and HFO-1234yf (RI-BP86-D3(BJ)/def2-SVP/COSMO(toluene)). | ||
Formation of the prereactive complex A with subsequent insertion of HFO-1234yf into the Rh–E bond exhibits rather high barriers for both systems (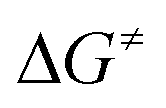 = +180 kJ mol−1 (1)/+152 kJ mol−1 (5)), which clearly renders this partial reaction the rate-determining step of this particular branch (1/5 → A → C → 2 + 4a/7, Fig. 2 and 3 red → blue lines). In contrast, an initial intermolecular C–H activation of HFO-1234yf at 1/5 to form intermediate B shows a much lower barrier (
= +180 kJ mol−1 (1)/+152 kJ mol−1 (5)), which clearly renders this partial reaction the rate-determining step of this particular branch (1/5 → A → C → 2 + 4a/7, Fig. 2 and 3 red → blue lines). In contrast, an initial intermolecular C–H activation of HFO-1234yf at 1/5 to form intermediate B shows a much lower barrier (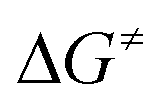 = +91 kJ mol−1 (1)/+79 kJ mol−1 (5)). In addition, the subsequent reductive elimination by migration of the functional group E (1/5 → B → C → 2 + 4a/7, Fig. 2 and 3 green → blue lines) also exhibits greatly reduced barriers (
= +91 kJ mol−1 (1)/+79 kJ mol−1 (5)). In addition, the subsequent reductive elimination by migration of the functional group E (1/5 → B → C → 2 + 4a/7, Fig. 2 and 3 green → blue lines) also exhibits greatly reduced barriers (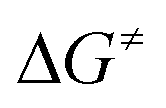 = +119 kJ mol−1 (1)/+86 kJ mol−1 (5)) compared to the migration of E to the RhCH2 moiety of A. Note that β-fluoride elimination is now rate-determining for the formation of 4a in this reaction sequence. It can be concluded that the reaction of 1/5 with HFO-1234yf most likely proceeds via a kinetically favored C–H oxidative addition in the first step, which in turn provides the basis for a kinetically feasible functionalization of the alkene by a reductive elimination step. For complex 5, the calculations further indicate that the proposed insertion and β-fluoride elimination steps, which follow the formation of C, exhibit comparable reaction barriers, supporting the plausibility of the proposed mechanism for a reaction at room temperature. However, despite expected effects due to the different steric and electronic natures of the Si(OEt)3 and GePh3 functional groups, the calculated barrier for β-fluoride elimination of the latter system appears too high for a reaction at room temperature. This might possibly indicate the existence of a lower-energy pathway for the final formation of 4a, which has not been considered here.
= +119 kJ mol−1 (1)/+86 kJ mol−1 (5)) compared to the migration of E to the RhCH2 moiety of A. Note that β-fluoride elimination is now rate-determining for the formation of 4a in this reaction sequence. It can be concluded that the reaction of 1/5 with HFO-1234yf most likely proceeds via a kinetically favored C–H oxidative addition in the first step, which in turn provides the basis for a kinetically feasible functionalization of the alkene by a reductive elimination step. For complex 5, the calculations further indicate that the proposed insertion and β-fluoride elimination steps, which follow the formation of C, exhibit comparable reaction barriers, supporting the plausibility of the proposed mechanism for a reaction at room temperature. However, despite expected effects due to the different steric and electronic natures of the Si(OEt)3 and GePh3 functional groups, the calculated barrier for β-fluoride elimination of the latter system appears too high for a reaction at room temperature. This might possibly indicate the existence of a lower-energy pathway for the final formation of 4a, which has not been considered here.
Reactions towards HFO-1234ze (E-1,3,3,3-tetrafluoropropene)
An interesting isomer of HFO-1234yf is the fluoroolefin HFO-1234ze (E-1,3,3,3-tetrafluoropropene), which exhibits some differences in reactivity towards the rhodium(I) hydrido complex [Rh(H)(PEt3)3] (8) when compared to the reaction of 8 with HFO-1234yf. In the latter case, complex 8 selectively activates the C(sp2)–F bond of the olefin to form the rhodium fluorido complex [Rh(F)(PEt3)3] (2) and 3,3,3-trifluoropropene (see Scheme 1).4 When HFO-1234ze is used, treatment of complex 8 with the fluorinated alkene gave within minutes at room temperature the rhodium fluorido complex 2 as well as 3,3,3-trifluoropropene, but also [Rh{(E)-CHCH(CF3)}(PEt3)3] (9)4 and HF through C–F bond activation (Scheme 5).
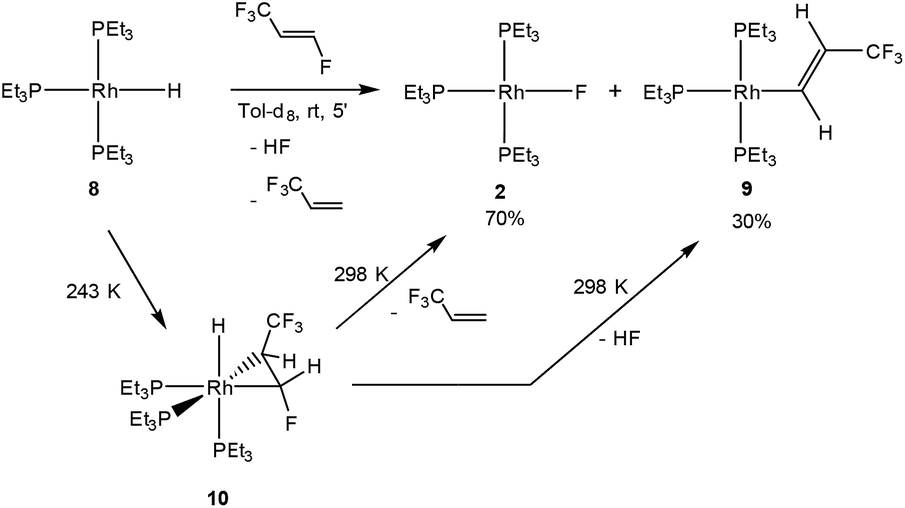 | ||
| Scheme 5 Reactivity of rhodium hydrido complex 8 towards HFO-1234ze. | ||
As has been described for the reaction of 8 with the isomer HFO-1234yf,4 low temperature NMR studies revealed that the fluoroolefin initially coordinates to the rhodium hydrido complex to give fac-[RhH(η2-CHFCHCF3)(PEt3)3] (10), which undergoes insertion and β-fluorine elimination to yield complex 2. Alternatively, C–F bond activation occurs to form 9, and HF is released (Scheme 5). The obtaining of the rhodium alkenyl complex 9 resembles the C–F bond activation at 8 observed with hexafluoropropene (see introduction, Scheme 1) and some conversions with fluoroaromatic compounds, although C–H bond activation is usually preferred.7a,20
Complex 10 was only observable in NMR measurements at low temperatures. The 31P{1H} NMR spectrum at 243 K shows three inequivalent resonances at δ 19.3, 10.2 and 6.1 ppm, with similar rhodium–phosphorus and phosphorus–phosphorus coupling constants as found for the previously described analogs [RhH(η2-olefin)(PEt3)3] (olefin = 3,3,3-trifluoropropene, HFO-1234yf).4,7d The signal at lower field, which corresponds to the phosphine in the trans position to the CF3 moiety, shows coupling to both fluorine groups of around 16 Hz. The signal for a phosphine at δ 10.2 ppm exhibits a larger P,F coupling of 49.3 Hz to the CFH group due to its trans arrangement. The resonance at δ 6.1 ppm also exhibits a P,F coupling to the CFH group of around 39 Hz. These couplings are confirmed by the 19F{1H} NMR spectrum. Thus, the resonance of the CF3 group appears at δ −53.6 ppm as a doublet of triplets due to its coupling to the phosphorus atom in the trans position and the phosphorus atom trans to the hydrido ligand as well as the other fluorine atom (4J = 5 Hz). Furthermore, the CFH group displays a resonance at δ −174.4 ppm which is shifted to higher field when compared to signals for fluorinated olefins bearing the trifluoromethyl moiety,7a,c,21 probably due to the single C–C bond character of the coordinated olefin. In the 1H NMR spectrum of 10, the rhodium bound hydrido resonance can be detected at δ −14.54 ppm as a doublet of triplets of doublets (J = 156.0, 18.9 and 10.9 Hz) due to the couplings to the rhodium and phosphorus atoms, where the coupling to the phosphine in the trans position displays the highest value and that to the rhodium nucleus the lowest. The olefinic protons appear at δ 2.88 and 6.40 ppm as a multiplet and a broad doublet of doublets of doublets, respectively. The latter, corresponding to the CHF moiety, shows a coupling to the fluorine in the geminal position of 69.5 Hz. However, the proton–proton coupling constant of 4.9 Hz is rather small due to the orientation of the protons. This structural assignment is confirmed by DFT structure optimization of complex 10. That is, the isomer with lower energy (favored by 16.7 kJ mol−1) has the CF3 group in the cis position to the hydrido ligand, while the fluorine atom of the CHF moiety remains in a trans arrangement. In addition, the C–C bond distance of the olefin is 1.448 Å, consistent with a metallacyclopropane moiety, suggesting the loss of the double bond character.
The rhodium silyl complex 5 was also treated with HFO-1234ze and after 10 min the formation of the complex [Rh{Si(OEt)3}(η2-CF3CHCFH)(PEt3)2] (11) together with [Rh(F)(PEt3)3] (2) in a 89:9 ratio, respectively, was observed. Additionally, free phosphine, small amounts of 3,3,3-trifluoropropene and FSi(OEt)3 were present in the reaction mixture (Scheme 6). Note that 3,3,3-trifluoropropene and hexafluoropropene also react with the rhodium silyl complex 5 by coordination and phosphine release (see Scheme 1).7d,10 After 2 h, the conversion of complex 11 into the rhodium fluorido complex 2 and 3,3,3-trifluoropropene was detected (with a ratio of 2:11 of 24:74). After 1 d, full conversion was observed to obtain an overall mixture of [Rh{(E)-CHCH(CF3)}(PEt3)3] (9), the cationic complex [Rh(PEt3)4]+ (6) and an unknown rhodium(I) complex. The formation of fluorosilicates was also observed. While the formation of the rhodium fluorido complex 2 and the rhodium alkenyl complex 9 involves a C–F bond activation step comparable to the one found for the hydrido complex 8 (see above), formation of the cationic complex can be explained by the reactivity of [Rh(F)(PEt3)3] (2) and FSi(OEt)3 where the fluorosilane acts as a fluorine acceptor, creating the vacant site that the phosphine occupies.4
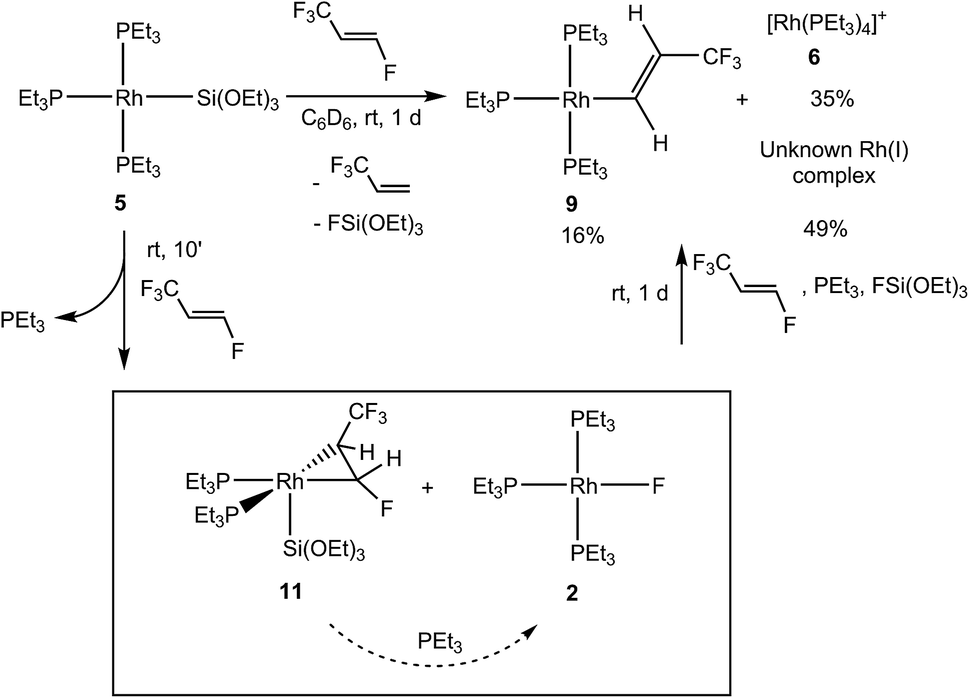 | ||
| Scheme 6 Reactivity of the rhodium(I) silyl complex 5 towards HFO-1234ze. | ||
The 31P{1H} NMR spectrum of complex 11 shows two resonances with an integral ratio of 1:1 for the two inequivalent phosphine ligands. At room temperature, those resonances can be distinguished as doublets of multiplets with rhodium–phosphorus coupling constants of 127.7 and 133.6 Hz, which are closer to the usual values for rhodium(I) complexes.8,9,22 When the NMR spectra are recorded at 233 K the coupling with other nuclei can be analyzed. Thus, a signal at δ 19.1 ppm appears as a doublet of doublets of doublets due to the coupling to rhodium, phosphorus (2J = 22.7 Hz) and the olefinic fluorine atom with a coupling constant of 36.3 Hz, suggesting a trans arrangement of the phosphine ligand to the CHF group. The second resonance appears at δ 21.1 ppm as a doublet of doublets of quartets of doublets. In this multiplicity, the phosphorus atom not only couples to the olefinic fluorine with a coupling constant of 15.8 Hz, but also shows a quartet due to the coupling to the CF3 group (4J = 18.4 Hz), which indicates that the CHCF3 is in the trans position. The 19F NMR spectrum depicts a doublet of doublets of pseudo triplets at δ −51.3 ppm, which can be assigned to the CF3 group. Apart from the already mentioned coupling to its phosphorus atom in the trans position, it shows coupling to both olefinic protons of 12 Hz and 3 Hz and the olefinic fluorine atom of also 3 Hz. The latter leads to a doublet of doublets of triplets of multiplets at high field. The coupling constants to the olefinic protons of 63 and 17 Hz suggest that the trans configuration of the fluorinated moieties of the olefin is maintained.21 The 1H NMR spectrum shows chemical shifts for the olefinic protons at δ 3.70 and 6.67 ppm, consistent with the presence of a certain double bond character of the carbon–carbon bond. These protons exhibit a mutual coupling of 6.3 Hz. Finally, the 1H, 29Si HMBC NMR spectrum confirms the coordination of the silyl ligand with a correlation peak at δ −54.2 ppm in the Si domain with a Si–Rh coupling constant of around 140 Hz. The chemical shift is similar to the values of previously described rhodium silyl complexes bearing a coordinated fluoroolefin.7d,10
Finally, treatment of [Rh(GePh3)(PEt3)3] (1) with HFO-1234ze yielded, after 16 hours, the rhodium fluorido complex 2, the C–F bond activation product [Rh{(E)-CHCH(CF3)}(PEt3)3] (9) and an unknown rhodium complex (15%) as well as 3,3,3-trifluoropropene. The reactivity pattern again resembles the ones found for the activation reactions at 5 and 8 (Scheme 7). The presence of the C–F bond activation product, CF2CHCH2GePh3, in the reaction mixtures suggests a competition between the C–F bond activation of HFO-1234ze and 3,3,3-trifluoropropene (see introduction, Schemes 1 and 2). Note that similar allylic C–F activations have been previously described.3c,5e,i,7d While the reaction mechanism is presumably the same, an initial coordination of the olefin was not observed using low-temperature NMR spectroscopy as with the hydrido complex 8 and the silyl complex 5. This is consistent with the reactivity of the germyl complex 1 with other fluorinated olefins, where no precoordination was observed in contrast to complexes 5 and 8.4,7d,8,10
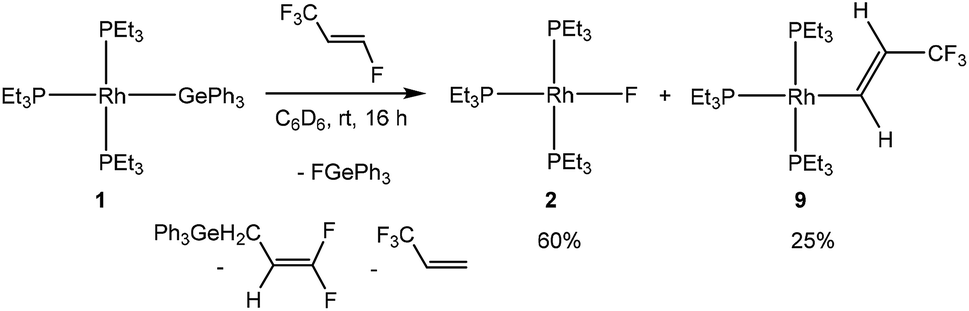 | ||
| Scheme 7 Reactivity of complex 1 towards HFO-1234ze. | ||
Catalytic studies
Based on the stoichiometric conversions, catalytic reactions with tertiary germanes were also investigated. With the rhodium germyl complex [Rh(GePh3)(PEt3)3] (1) (10 mol%) as a catalytic precursor, HFO-1234yf reacts in the presence of HGeR3 (R = Ph (a), Et (b), nBu (c)) to yield a mixture of the olefins Z-CF3CHCH(GeR3) (4) and 3,3,3-trifluoropropene as the main products (Scheme 8). After 1 day at room temperature the germanes were fully converted. Small amounts of CF3CH3CH3, E-CF3CHCH(GeR3) (12) (see below) and CF3CH3CH2GeR3,7d which is the hydrogermylation product of 3,3,3-trifluoropropene,7d were also generated. The hydrogermylation product of HFO-1234yf was not observed, which is in contrast to the catalytic conversions of 3,3,3-trifluoropropene with tertiary germanes using 1 as catalyst.7d
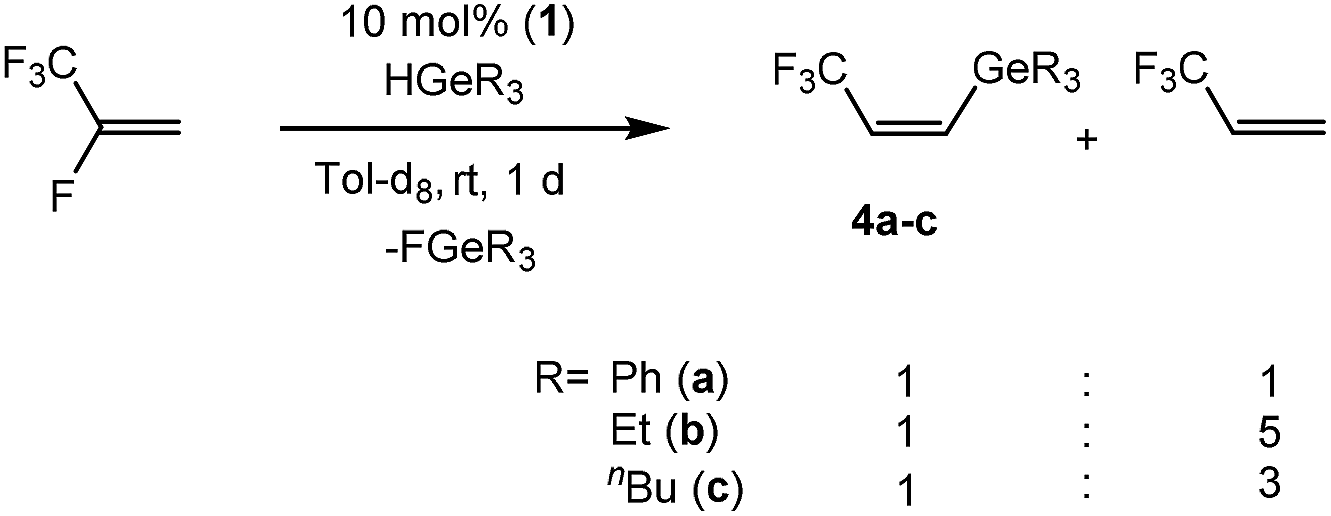 | ||
| Scheme 8 Catalytic reaction of HFO-1234yf with tertiary germanes. | ||
Mechanistically, the formation of Z-CF3CHCH(GeR3) (4) in catalytic amounts is consistent with the stoichiometric reaction of [Rh(GePh3)(PEt3)3] (1) with HFO-1234yf as an initial reaction (see above). For HGePh3 possible reaction steps are depicted in Scheme 9. A subsequent reaction of the initial C–F bond activation product [Rh(F)(PEt3)3] (2) with germane affords the known dihydrido complex [Rh(H)2(GePh3)(PEt3)3] (13)8 and a small amount of rhodium(I) germyl complex 1, as well as fluorogermane. The two germyl complexes are in equilibrium with each other. The generation of 3,3,3-trifluoropropene can be explained by a conversion of complex 13 and HFO-1234yf to form the fluorido complex 2 and 3,3,3-trifluoropropene (Scheme 9). Note that the latter reaction has been independently demonstrated.
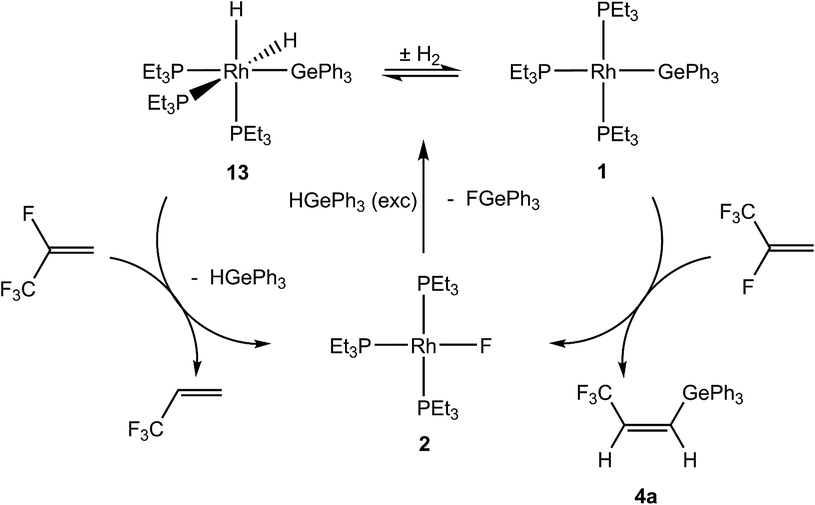 | ||
| Scheme 9 Catalytic mechanism of HFO-1234yf with tertiary germanes. | ||
When HFO-1234ze was used as a starting compound for catalytic conversions, full conversion of the germanes HGeR3 (R = Ph, nBu) was also achieved after 1 day with [Rh(GePh3)(PEt3)3] (1) (10 mol%) as catalyst. HGeEt3 required 2 days until full consumption was reached. However, the catalytic behavior of HFO-1234ze is in contrast to its isomer, HFO-1234yf, which seems to undergo more selective reactions. A hydrodefluorination reaction of HFO-1234ze to give 3,3,3-trifluoropropene occurred in every case, although the amount varies heavily depending on the germane used. For HGePh3, a subsequent hydrogermylation of 3,3,3-trifluoropropene gives a trifluoropropyl germane as the main product (Scheme 10). On the other hand, when HGeEt3 or HGenBu3 are the hydride source, the activation of 3,3,3-trifluoropropene is slower, leading to a lower selectivity of the reaction. Overall, up to seven different products have been identified including those from hydrogenation, hydrogermylation and C–F activation of products of 3,3,3-trifluoropropene, as well as compounds E-CF3CHCH(GeR3) (12) (R = Ph (a), Et (b), nBu (c)) from the defluorogermylation of HFO-1234ze (Scheme 10).
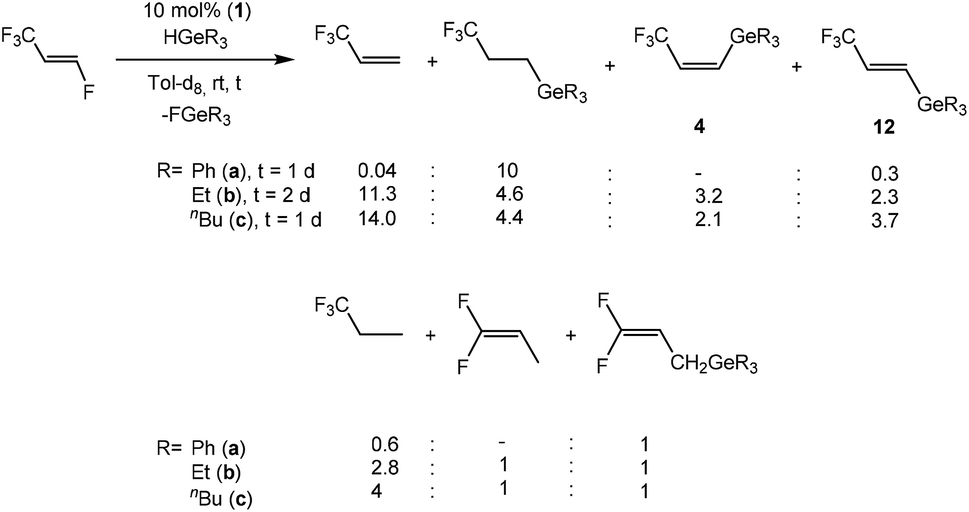 | ||
| Scheme 10 Catalytic reactions of HFO-1234ze with tertiary germanes. | ||
Compounds 12 show distinguishable NMR spectroscopic resonances from their cis isomers. For example, compound 12c exhibits a doublet of doublets in the 19F NMR spectrum at δ −66.0 ppm due to the coupling to the geminal and cis olefinic protons (3JF,H = 6, 4JF,H = 2 Hz). These couplings are also observed in the 1H NMR spectrum for resonances at δ 6.08 and 6.95 ppm, respectively, both as doublet of quartets with a proton–proton trans coupling constant of 18.9 Hz. The chemical shifts and coupling constant values are similar to data found for olefins with hydrogen atoms in a trans position such as E-CF3CHCH(Bpin) and E-CF3CHCH(SiPh3).3d,23
Finally, catalytic reactions involving the silane HSi(OEt)3 and the tetrafluoropropenes under study were performed using both [Rh{Si(OEt)3}(PEt3)3] (5) and [Rh(H)(PEt3)3] (8) as a catalyst (see Table 1). When HFO-1234yf is reacted together with HSi(OEt)3 and 10 mol% of complex 5, generation of 3,3,3-trifluoropropene and 3,3,3-trifluoropropane together with small amounts of the olefin Z-CF3CHCH(Si(OEt)3) (7) was observed, similar to the generation of vinyl germanes from tertiary germanes. However, in the case of silane and HFO-1234ze, a silylated olefin is not observed, but the silylated trifluoropropane is instead. When using complex 8 as catalyst under the same conditions, a faster consumption of silane is observed to provide similar products and ratios. In addition, reactions are generally more selective towards 3,3,3-trifluoropropene. The latter competes again with the tetrafluoropropenes as educt, leading also to the hydrogenation and the hydrosilylation products of 3,3,3-trifluoropropene.
|
|
|||
|---|---|---|---|
| Catalyst | Olefin | Timea | Ratio of productsb |
| a Time when full conversion was achieved based on the consumption of silane in 1H NMR spectra. b Based on 19F NMR measurements. | |||
| Complex 5 | HFO-1234yf | 8 h | 16.7:4.6:1.5:1 |
| Complex 8 | HFO-1234yf | 2 h | 16.8:9:1:0 |
| Complex 5 | HFO-1234ze | 8 h | 15:8.4:1:0 |
| Complex 8 | HFO-1234ze | 40 min | 9.5:5.5:1:0 |
Conclusions
In conclusion, it has been shown that the activity and selectivity of C–F bond activation reactions of the fluorinated olefins HFO-1234yf and HFO-1234ze at rhodium(I) complexes can be controlled by the anionic ligand. It was shown before that [Rh(GePh3)(PEt3)3] (1) performs a C–F bond activation towards hexafluoropropene and 3,3,3-trifluoropropene while [Rh(Si(OEt)3)(PEt3)3] (5) exhibits a different reactivity leading only to the coordination of the olefins.7d,8,10 It has been now demonstrated that both complexes show similar transformations with HFO-1234yf, yielding the rhodium fluorido complex 2 and the fluoroolefin Z-(CF3)CHCH(E) (E = GePh3 (4a), Si(OEt)3 (7)). DFT calculations of the reaction mechanism indicate an initial C–H oxidative addition of HFO-1234yf followed by C–E reductive elimination as a key step. For the silyl complex, the new olefin can subsequently undergo an insertion into the Rh–H bond followed by a β-fluoride elimination to give 7, whereas the computational results for a similar transformation in the case of the germyl system to afford 4a are not conclusive.
The reactivity of 1 and 5 differs with that previously reported for [Rh(H)(PEt3)3] (8), for which a reaction with HFO-1234yf yields the rhodium fluorido complex 2 and 3,3,3-trifluoropropene. On the other hand, the activation of HFO-1234ze proceeds in the same way for the three Rh starting compounds studied. Two different C–F activation pathways were identified, which involve either the generation of a Rh–C bond and FE (E = H, GePh3, Si(OEt)3) or the formation of the rhodium fluorido complex 2 and trifluoropropene. For the rhodium silyl complex 5 and rhodium hydrido complex 8, initial olefin coordination was observed. Finally, while the stoichiometric reactions provide a germyl/silyl substituted olefin, the catalytic reactions are less selective, yielding functionalized building blocks based on a competition between C–F activation/hydrodefluorination reactions and germylation or silylation reactions.
Experimental
General procedures, methods and materials
All experiments were carried out under an argon atmosphere using Schlenk techniques. Solvents were dried using the usual procedures24 and, prior to use, distilled under argon. The rhodium complexes [Rh(GePh3)(PEt3)3] (1),8 [Rh(Si(OEt)3)(PEt3)3] (5)9 and [Rh(H)(PEt3)3] (8)7e were prepared as described in the literature. All reagents were obtained from commercial sources. Complexes 2, 3 and 6 were identified by comparison of their analytical data with the literature.4,7a,c,18a Unless stated otherwise, NMR spectra were recorded at room temperature on a Bruker DPX-300 or a Bruker Avance 300 spectrometer using the solvent as the internal lock. 1H and 13C{1H} signals are referred to residual solvent signals, those of 31P{1H} to 85% H3PO4 and the 19F NMR spectra to external CFCl3. 1H and 13C{1H} NMR signal assignments were supported by 1H–1H COSY, 19F–1H COSY, 19F–19F COSY, 31P–31P COSY, 1H–13C HMQC, 1H–13C HMBC and 19F–13C HMBC NMR experiments.X-ray diffraction analysis
Suitable crystals for X-ray crystallography of Z-CF3CHCH(GePh3) (4a) were obtained from the reaction mixture of [Rh(F)(PEt3)3] (2), [Rh{(E)-C(CF3)CHF}(PEt3)3] (3) and the germylated olefin 4a. Recrystallization of a saturated n-hexane solution of 4a at 243 K was performed in an n-heptane solution at room temperature by slow evaporation of the solvent. Crystallographic data were collected on a Bruker D8 Venture diffractometer at 100 K. The structures were solved by intrinsic phasing (SHELXT-2013)25 and refined by full matrix least-squares procedures based on F2 with all measured reflections (SHELXL-2013).26 The SADABS program27 was used for multi-scan absorption corrections. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were included in idealized positions and refined using a riding model. Crystal data and structural refinement details for complex 4a are given in Table 2. CCDC 1912055 contains the crystallographic data.†
| 4a | |
|---|---|
| Empirical formula | C21H17F3Ge |
| Formula wt | 398.93 |
| Temp. (K) | 100(2) |
| Wavelength (Å) | 0.71073 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a (Å) | 21.1522(11) |
| b (Å) | 9.1254(4) |
| c (Å) | 19.5533(9) |
| β (°) | 107.649(2) |
| V (Å3) | 3596.6(3) |
| Z | 8 |
| Density (Mg m−3) | 1.474 |
| μ (mm−1) | 1.732 |
| F(000) | 1616 |
| Crystal size (mm) | 0.360 × 0.18 × 0.12 |
| θ range for data collection (°) | 2.45–26.41 |
| Index ranges | −26 ≤ h ≤ 24 |
| −11 ≤ k ≤ 11 | |
| −24 ≤ l ≤ 24 | |
| No. of rflns collected | 36723 |
| No. of indep rflns | 7353 [R(int) = 0.0405] |
| Data completeness | 0.995 |
| Absorption correction | Multi-scan |
| Max. and min. transmission | 0.614 and 0.745 |
| Refinement method | Full matrix least-squares on F2 |
| No. of data/restraints/params | 7353/0/451 |
| Goodness of fit on F2 | 1.016 |
| R indices [I > 2σ(I)] | R 1 = 0.0306, wR2 = 0.0671 |
| R indices (all data) | R 1 = 0.0402, wR2 = 0.0706 |
| Largest diff. peak and hole (eÅ−3) | 0.838 and −0.495 |
Computational details
Structure optimizations for all compounds except 10 (see below) were carried out with the Turbomole program, version 7.3.0,28 at the DFT-BP8629 level of theory in conjunction with all-electron def2-SVP basis sets for the main group atoms (H, C, O, F, Si, P, Ge)30 and a quasirelativistic energy-adjusted small-core pseudopotential (RECP) with a def2-SVP valence basis set for rhodium,31 using the resolution-of-identity (RI)31a,32 approximation as well as an atom-pairwise correction for dispersion forces via Grimme’s D3 model with Becke–Johnson damping (BJ)33 (RI-BP86-D3(BJ)/def2-SVP level). Solvent effects have been taken into account using the COSMO34 continuum solvent model, employing toluene with a dielectric constant of ε = 2.3741. The structures of the possible isomers of complex fac-[RhH(η2-CHFCHCF3)(PEt3)3] (10) were optimized in the gas phase using the Gaussian 09 (Revision D.01) program package35 at the B3LYP-D3(BJ) level, employing an RECP with associated cc-pVDZ valence basis set for rhodium36 and all-electron cc-pVDZ basis sets for the lighter atoms.37 All structures were characterized as true minima or transition states by analytical harmonic vibrational frequency analyses. Based on the frequency analyses, Gibbs free activation and reaction energies at 298.15 K and 0.1 MPa were evaluated within the rigid-rotor and harmonic-oscillator approximations. The calculated relative energy of the isomers of 10 is based on zero-point vibrational energy corrected total electronic energies from optimization in the gas phase. Cartesian coordinates for all optimized structures can be found in the ESI.†
Reaction of HFO-1234yf with [Rh(GePh3)(PEt3)3] (1)
In a Young NMR tube [Rh(GePh3)(PEt3)3] (1) (18.2 mg, 0.024 mmol) was dissolved in C6D6 or toluene-d8 (0.4 mL). The solution was cooled to 77 K, degassed in vacuo, and pressurized with HFO-1234yf to 0.2 bar. After warming up to room temperature, the NMR spectroscopic data for the reaction mixture revealed after 10 min the complete conversion of 1 into [Rh(F)(PEt3)3] (2) and [Rh{(E)-C(CF3)CHF}(PEt3)3] (3) in a 85:15 ratio, as well as a small amount of an unknown rhodium complex that evolves into complexes 2 and 3 after 1 day. The formation of Z-(CF3)CHCH(GePh3) (4a) and traces of trifluoropropene and another unknown CF3-containing derivative (10.7:1.8:1 ratio, respectively) as well as fluorogermane were also observed. Analytical data for 4a: 1H NMR (300.1 MHz, toluene-d8): δ = 7.46 (m, 6H, Ph); 7.13 (m, 9H, Ph); 6.45 (dq, 3JH–H = 14.3 Hz, 4JH–F = 0.34 Hz, 1H, CHGePh3); 6.285 (dq, 3JH–H = 14.3, 3JH–F = 7.87 Hz, 1H, CH) ppm. The data of the olefinic protons correspond to their simulation with the gNMR software.3819F NMR (282.4 MHz, toluene-d8): δ = −62.2 (d, 3JF–H = 7 Hz, 3F, CF3) ppm. 13C{1H} NMR (75.5 MHz, toluene-d8): δ = 139.0 (q, 3JC–F = 8 Hz, CGePh3); 135.2 (s, Ph); 135.0 (s, Ph); 134.2 (q, 2JC–F = 34 Hz, CH); 129.5 (s, Ph); 128.6 (s, Ph); 122.8 (q, 1JC–F = 273 Hz, CF3) ppm.
Reaction of HFO-1234yf with [Rh{Si(OEt)3}(PEt3)3] (5)
In a Young NMR tube [Rh{Si(OEt)3}(PEt3)3] (5) (20.4 mg, 0.033 mmol) was dissolved in C6D6 or toluene-d8 (0.5 mL). The solution was cooled to 77 K, degassed in vacuo, and pressurized with HFO-1234yf to 0.3 bar. After warming up to room temperature, the NMR spectroscopic data for the reaction mixture revealed after 10 min the complete conversion of 5 into [Rh(F)(PEt3)3] (2) and [Rh(PEt3)4]+ (6) in a 90:10 ratio, as well as the formation of Z-(CF3)CHCH(Si(OEt)3) (7), 3,3,3-trifluoropropene and traces of fluorosilane. When the mixture was reacted for 18 h, NMR spectroscopic data revealed the formation of [Rh{(E)-C(CF3)CHF}(PEt3)3] (3), complex 6 and an unidentified rhodium complex in a 58:29:13 ratio. Analytical data for Z-(CF3)CHCH(Si(OEt)3) (7): 1H NMR (300.1 MHz, toluene-d8): δ = 6.071 (dq, 3JH–H = 15.54, 3JH–F = 7.95 Hz, 1H, CH); 5.958 (d, 3JH–H = 15.54, 4JH–F = −0.22 Hz, 1H, CHSi(OEt)3); 4.13 (q, 3JH–H = 7.0 Hz, OCH2CH3); 1.40–1.52 (m, 9H, overlapped with PEt3 signals of 2 and 6, OCH2CH3); 3.78 (q, 3JH–H = 6.9 Hz, 6H, OCH2CH3) ppm. The olefinic protons correspond to their simulation with the gNMR software.3819F NMR (282.4 MHz, toluene-d8): δ = −63.9 (d, 2JF–H = 8 Hz, 3F, CF3) ppm. 1H, 29Si HMBC NMR (59.6 MHz, toluene-d8, 263 K): δ = −69.8 (s) ppm.
Reaction of HFO-1234ze with [Rh(H)(PEt3)3] (8)
In a Young NMR tube [Rh(H)(PEt3)3] (8) (50.1 mg, 0.109 mmol) was dissolved in toluene-d8 (0.5 mL). The solution was cooled to 77 K, degassed in vacuo, and pressurized with HFO-1234ze to 0.2 bar. After warming up to 213 K, the NMR spectroscopic data for the reaction mixture revealed the complete conversion of [Rh(H)(PEt3)3] into fac-[RhH(η2-CHFCHCF3)(PEt3)3] (10). The complex is only stable up to 283 K, when it fully converts into the rhodium fluorido complex [Rh(F)(PEt3)3] (2) and [Rh{(E)-CHCHCF3}(PEt3)3] (9) in a 70:30 ratio, respectively, with traces of an unknown rhodium complex. In addition, formation of HF and 3,3,3-trifluoropropene was also observed. Analytical data for 10: 1H NMR (300.1 MHz, toluene-d8, 243 K): δ = 6.40 (dddm, 2JH–F = 69.5, 3JH–P = 10.7, 3JH–H = 4.9 Hz, 1H, CHF); 2.88 (m br, dqdd in 1H{31P} NMR spectrum 3JH–F = 19.2, 3JH–F = 9.8, 3JH–H = 4.9, 2JH–Rh = 1.9 Hz, 1H, CHCF3); 1.70–1.50 (m, 6H, PCH2CH3); 1.50–1.23 (m, 12H, PCH2CH3); 1.02–0.86 (m, 18H, PCH2CH3); 0.85–0.65 (m, 9H, PCH2CH3); −14.54 (dtd, 2JH–P = 156.0, 2JH–P ≈ 2JH–P = 18.9, 1JH–Rh = 10.9 Hz, 1H, RhH) ppm. Although the shifts correspond to the experiments at 243 K, the 1H NMR spectra at different temperatures were analyzed to obtain all the coupling constant data. 19F NMR (282 MHz, toluene-d8, 243 K) δ = −53.6 (ddt br, 4JF–P = 17, 3JF–H = 10, 4JF–P ≈ 4JF–F = 5 Hz, 3F, CF3); −174.4 (m, dddp in 19F{1H} NMR spectrum 3JF–P = 49 Hz, 3JF–P = 34 Hz, 3JF–P = 17 Hz, 4JF–F ≈ 2JF–Rh = 4 Hz, 1F, CHF) ppm. 31P{1H} NMR (121.5 MHz, toluene-d8, 243 K): δ = 19.3 (dtqd, 1JP–Rh = 133.6, 2JP–P ≈ 2JP–P = 26.5, 4JP–F = 16.8, 3JP–F = 16.2 Hz, 1P, PEt3 trans to CHCF3); 10.2 (ddt, 1JP–Rh = 110.5, 3JP–F = 49.3, 2JP–P ≈ 2JP–P = 26.5, 1P, PEt3 trans to CHF); 6.1 (ddtm, 1JP–Rh = 96.1, 3JP–F = 39.4, 2JP–P ≈ 2JP–P = 26.5 Hz, 1P, PEt3 trans to H) ppm. 1H, 13C HMQC NMR (75.5 MHz, toluene-d8, 213 K): δ = 99 (dd, 1JC–F = 243, 1JC–Rh = 75 Hz, CHF), 28 (m, 1JC–Rh = 45 Hz, CHCF3) ppm. All the signals except for the ethyl groups have been simulated with the gNMR software (see ESI†).38
Reaction of HFO-1234ze with [Rh(GePh3)(PEt3)3] (1)
In a Young NMR tube [Rh(GePh3)(PEt3)3] (1) (21.3 mg, 0.028 mmol) was dissolved in C6D6 (0.5 mL). The solution was cooled to 77 K, degassed in vacuo, and pressurized with HFO-1234ze to 0.2 bar. After warming up to room temperature, the NMR spectroscopic data for the reaction mixture revealed after 16 h the complete conversion of 1 into [Rh(F)(PEt3)3] (2), [Rh{(E)-CHCH(CF3)}(PEt3)3] (9) and an unidentified rhodium complex in a 54:24:22 mole ratio, as well as the formation of 3,3,3-trifluoropropene, (Ph3Ge)CH2CHCF2, fluorogermane and some unknown compounds.
Reaction of HFO-1234ze with [Rh{Si(OEt)3}(PEt3)3] (5)
In a Young NMR tube [Rh{Si(OEt)3}(PEt3)3] (5) (30 mg, 0.048 mmol) was dissolved in C6D6 (0.5 mL). The solution was cooled to 77 K, degassed in vacuo, and pressurized with HFO-1234ze to 0.2 bar. After warming up to room temperature, the NMR spectroscopic data for the reaction mixture revealed after 10 min the complete conversion of 5 into [Rh(F)(PEt3)3] (2), the coordination product [Rh{Si(OEt)3}(η2-CHFCHCF3)(PEt3)2] (11) and an unknown rhodium(I) complex in a 18:71:11 mole ratio, as well as the release of PEt3. The reaction mixture evolved after 1 d to give the complexes [Rh{(E)-CHCH(CF3)}(PEt3)3] (9), [Rh(PEt3)4]+ (6) and an unidentified rhodium complex in a 22:48:30 mole ratio, as well as the formation of 3,3,3-trifluoropropene and fluorosilane. Analytical data for 11: 1H NMR (300.1 MHz, C6D6): δ = 1.10 (t, 3JH–H = 7.1 Hz, 9H, Si(OCH2CH3)3); 3.4 (m br, 1H, CF3CH); 3.76 (pseudo quint, 3JH–H ≈ 4JH–Rh = 6.9 Hz, 6H, Si(OCH2CH3)3); 6.79 (d br, 2JH–F = 64 Hz, 1H, CFH) ppm. 1H NMR (300.1 MHz, toluene-d8, 233 K): δ = 6.67 (dm, ddd in 1H{31P} and pseudo td in 1H{19F} NMR spectra, 2JH–F = 63.4, 3JH–H ≈ 3JH–P = 6.3, 2JH–Rh = 2.5 Hz, 1H, CFH); 3.70 (m, 3JH–H = 7.0 Hz, 6H, Si(OCH2CH3)3); 3.32 (m, dqd in 1H{31P} NMR spectrum, 3JH–F = 17.5, 3JH–F = 11.8, 3JH–H = 6.3 Hz, 1H, CF3CH); 1.09 (t, 3JH–H = 7.0 Hz, 9H, Si(OCH2CH3)3) ppm. 19F NMR (282.4 MHz, C6D6): δ = −51.2 (s br, 3F, CF3); −185.5 (s br, 1F, CFH) ppm. 19F NMR (282.4 MHz, toluene-d8, 233 K): δ = −51.3 (ddt, 4JF–P = 18, 3JF–H = 12, 4JF–H ≈ 4JF–F = 3 Hz, 3F, CF3); −185.5 (ddtm, 2JF–H = 63, 3JF–P = 36, 3JF–H ≈ 3JF–P = 17 Hz, 1F, CFH) ppm. 31P{1H} NMR (121.5 MHz, C6D6): δ = 18.0 (dm, 1JP–Rh = 127.7 Hz, 1P); 21.5 (dm, 1JP–Rh = 133.6 Hz, 1P) ppm. 31P{1H} NMR (121.5 MHz, toluene-d8, 233 K): δ = 19.1 (ddd, 1JP–Rh = 123.2, 3JP–F = 36.3, 2JP–P = 22.7 Hz, 1P, PEt3 trans to CHF); 21.1 (ddqd, 1JP–Rh = 133.9, 2JP–P = 22.7, 4JP–F = 18.4, 3JP–F = 15.8 Hz, 1P, PEt3 trans to CHCF3) ppm. Phosphorus resonances have been simulated with the gNMR software.381H, 29Si HMBC NMR (59.6 MHz, toluene-d8): δ = −54.2 (d, 1JSi–Rh ≈ 140 Hz) ppm.
General procedure for the catalytic conversion of the fluorinated olefins using [Rh(GePh3)(PEt3)3] (1) and tertiary germanes
In a Young NMR tube [Rh(GePh3)(PEt3)3] (1) (10.0 mg, 0.013 mmol) was dissolved in toluene-d8 (0.4 mL) and HGeR3 (0.13 mmol) was added to the solution. The solution was cooled to 77 K, degassed in vacuo, and pressurized with HFO-1234yf or HFO-1234ze to 0.2 bar. After warming up to room temperature the reaction was monitored using NMR spectroscopy. After the reaction time at room temperature the 1H and 19F NMR spectroscopic data revealed the complete conversion of HGeR3, and the formation of the corresponding fluorogermane as well as the products.CH(GePh3) (4a), CH2CHCF3, Ph3GeCH2CH2CF3, E-(CF3)CHCH(GePh3) (12a) and CF3CH2CH3 in a ratio of 1:1:0.5:0.1:0.05.
CH(GeEt3) (4b), CH2CHCF3, Et3GeCH2CH2CF3, E-(CF3)CHCH(GeEt3) (12b) and CF2CHCH3 in a ratio of 1:5:0.6:0.35:0.3. Spectroscopic data for Z-(CF3)CHCH(GeEt3) (4b): 1H NMR (300.1 MHz, toluene-d8): δ = 6.46 (d, 3JH–H = 14.3 Hz, 1H, CHGeEt3); 6.29 (dq, 3JH–H = 14.3, 3JH–F = 8.0 Hz, 1H, CHCF3) ppm. The signals for hydrogen atoms of the ethyl groups of 4b could not be assigned due to overlap with the signals of complexes 2 and other germylated compounds. 19F NMR (282.4 MHz, toluene-d8): δ = −62.4 (d, 3JF–H = 8 Hz, 3F, CF3) ppm.
CH{Ge(nBu)3} (4c), CH2CHCF3, nBu3GeCH2CH2CF3, E-(CF3)CHCH(Ge(nBu)3) (12c), CF3CH2CH3 and CF2CHCH3 in a ratio of 1:3:0.14:0.14:0.05:0.33, respectively. Spectroscopic data for Z-(CF3)CHCH{Ge(nBu)3} (4c): 1H NMR (300.1 MHz, toluene-d8): δ = 6.58 (d, 3JH–H = 14.3 Hz, 1H, CHGe(nBu)3); 6.41 (dq, 3JH–H = 14.3, 3JH–F = 7.5 Hz, 1H, CHCF3) ppm. The signals for hydrogen atoms of the butyl groups of 4c could not be assigned due to overlap with the signals of complex 2 and other germylated compounds. 19F NMR (282.4 MHz, toluene-d8): δ = −62.4 (d, 3JF–H = 7 Hz, 3F, CF3) ppm.
CHCF3, CH3CH2CF3, Ph3GeCH2CH2CF3, CF2CHCH2GePh3,7d and E-(CF3)CHCH(GePh3) (12a) in a ratio of 0.04:0.6:10:1:0.3, respectively. Analytical data for E-(CF3)CHCH(GePh3) (12a): 1H NMR (300.1 MHz, toluene-d8): δ = 6.26 (dq, 3JH–H = 18.2, 3JH–F = 5.6 Hz, 1H, CHCF3) ppm. The signals for hydrogen atoms of the phenyl groups as well as the second olefinic proton of 12a could not be assigned due to overlap with the signals of the other germylated derivatives. 19F NMR (282.4 MHz, toluene-d8): δ = −65.9 (dd, 3JF–H = 6, 4JF–H = 2 Hz, 3F, CF3) ppm.
CH2, Et3GeCH2CH2CF3, Z-(CF3)CHCH(GeEt3) (4b), E-(CF3)CHCH(GeEt3) (12b), CF2CHCH2GePh3, CF3CH2CH3 and CF2CHCH3 in a ratio of 11.3:4.6:3.2:2.3:1:2.8:1, respectively. Analytical data for E-(CF3)CHCH(GeEt3) (12b): 1H NMR (300.1 MHz, toluene-d8): δ = 6.74 (dq, 3JH–H = 18.8, 4JH–F = 2.0 Hz, 1H, CHGeEt3), 5.92 (dq, 3JH–H = 18.8, 3JH–F = 5.8 Hz, 1H, CHCF3) ppm. The signals for hydrogen atoms of the ethyl groups of 12b could not be assigned due to overlap with the signals of the other germylated derivatives. 19F NMR (282.4 MHz, toluene-d8): δ = −66.1 (dd, 3JF–H = 6, 4JF–H = 2 Hz, 3F, CF3) ppm.
CH2, (nBu)3GeCH2CH2CF3, Z-(CF3)CHCH(Ge(nBu)3) (4c), E-(CF3)CHCH(Ge(nBu)3) (12c), CF2CHCH2GePh3, CF3CH2CH3 and CF2CHCH3 in a ratio of 14:4.4:2.1:3.7:1:4:1, respectively. Analytical data for E-(CF3)CHCH(Ge(nBu)3) (12c): 1H NMR (300.1 MHz, toluene-d8): δ = 6.95 (dq, 3JH–H = 18.9, 4JH–F = 2.0 Hz, 1H, CHGe(nBu)3), 6.08 (dq, 3JH–H = 18.9, 3JH–F = 5.7 Hz, 1H, CHCF3) ppm. The signals for hydrogen atoms of the butyl groups of 12c could not be assigned due to overlap with the signals of the other germylated derivatives. 19F NMR (282.4 MHz, toluene-d8): δ = −66.0 (dd, 3JF–H = 6, 4JF–H = 2 Hz, 3F, CF3) ppm. 13C{1H} NMR (75.5 MHz, toluene-d8): δ = 144.8 (m, observed by 1H–13C HMQC NMR experiment, CHGe(nBu)3); 130.6 (m, observed by 1H–13C HMQC NMR experiment, CH(CF3)) ppm.
General procedure for the catalytic conversion of fluorinated olefins with [Rh(Si(OEt)3)(PEt3)3] (5) or [Rh(H)(PEt3)3] (8) and HSi(OEt)3
In a Young NMR tube [Rh(Si(OEt)3)(PEt3)3] (5) (8.0 mg, 0.013 mmol) or [Rh(H)(PEt3)3] (8) (6.0 mg, 0.013 mmol) was dissolved in C6D6 (0.4 mL) and HSi(OEt)3 (25 μL, 0.13 mmol) was added to the solution. The solution was cooled to 77 K, degassed in vacuo, and pressurized with HFO-1234yf or HFO-1234ze to 0.2 bar. After warming up to room temperature the reaction was monitored using NMR spectroscopy. After the reaction time at room temperature the 1H and 19F NMR spectroscopic data revealed the complete conversion of HSi(OEt)3, and the formation of the FSi(OEt)3 as well as the products.CH(Si(OEt)3) (7), (OEt)3SiCH2CH2CF3 (ref. 39), CH2CHCF3, and CH3CH2CF3 in a ratio of 1:1.5:16.7:4.6, respectively.
CHCF3, CH3CH2CF3, (OEt)3SiCH2CH2CF3 (ref. 39) and an unknown CF3CH2 containing compound in a ratio of 15:8.4:1:4.1, respectively.
CHCF3, CH3CH2CF3 and (OEt)3SiCH2CH2CF3 (ref. 39) in a ratio of 16.8:9:1, respectively.
CHCF3, CH3CH2CF3 and (OEt)3SiCH2CH2CF3 (ref. 39) in a ratio of 9.5:5.5:1, respectively.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the AvH Foundation and the CRC 1349 “Fluorine-Specific Interactions” funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, project 387284271) for financial support. We also thank Dr Mike Ahrens for his support with DFT calculations.Notes and references
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed; (b) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity and Applications, Wiley-VCH, Weinheim, 2nd edn, 2013 CrossRef; (c) P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS; (d) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS; (e) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) P. Javidmand and K. A. Hoffmann, Int. J. Refrig., 2016, 69, 114–135 CrossRef CAS; (b) G. Righetti, C. Zilio and G. A. Longo, Int. J. Refrig., 2015, 54, 1–9 CrossRef CAS.
- (a) M. R. Crimmin, C. Bakewell and A. White, Angew. Chem., Int. Ed., 2018, 57, 6638–6642 CrossRef; (b) H. Sakaguchi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2018, 57, 328–332 CrossRef CAS; (c) T. Braun, G. Meißner, E. Kemnitz and K. Kretschmar, Angew. Chem., Int. Ed., 2017, 56, 16338–16341 CrossRef PubMed; (d) H. Sakaguchi, Y. Uetake, M. Ohashi, T. Niwa, S. Ogoshi and T. Hosoya, J. Am. Chem. Soc., 2017, 139, 12855–12862 CrossRef CAS PubMed; (e) Y. Li, D.-H. Tu, Y.-J. Gu, B. Wang, Y.-Y. Wang, Z.-T. Liu, Z.-W. Liu and J. Lu, Eur. J. Org. Chem., 2015, 2015, 4340–4343 CrossRef CAS; (f) Y. Hiraoka, T. Kawasaki-Takasuka, Y. Morizawa and T. Yamazaki, J. Fluorine Chem., 2015, 179, 71–76 CrossRef CAS; (g) Y. L. Yagupolskii, N. V. Pavlenko, S. V. Shelyazhenko, A. A. Filatov, M. M. Kremlev, A. I. Mushta, I. I. Gerus, S. Peng, V. A. Petrov and M. Nappa, J. Fluorine Chem., 2015, 179, 134–141 CrossRef CAS.
- M. Talavera, C. N. Von Hahmann, R. Müller, M. Kaupp and T. Braun, Angew. Chem., Int. Ed., 2019, 58, 10688–10692 CrossRef CAS PubMed.
- (a) N. A. LaBerge and J. A. Love, in Organometallic Fluorine Chemistry, ed. T. Braun and R. Hughes, Springer, Cham, 2015, vol. 52 Search PubMed; (b) M. E. Slaney, M. J. Ferguson, R. McDonald and M. Cowie, Organometallics, 2012, 31, 1384–1396 CrossRef CAS; (c) N. O. Andrella, K. Liu, B. Gabidullin, M. Vasiliu, D. A. Dixon and R. T. Baker, Organometallics, 2018, 37, 422–432 CrossRef CAS; (d) R. Kojima, S. Akiyama and H. Ito, Angew. Chem., Int. Ed., 2018, 57, 7196–7199 CrossRef CAS PubMed; (e) T. Fujita, R. Morioka, T. Arita and J. Ichikawa, Chem. Commun., 2018, 54, 12938–12941 RSC; (f) L. M. Milner, L. M. Hall, N. E. Pridmore, M. K. Skeats, A. C. Whitwood, J. M. Lynam and J. M. Slattery, Dalton Trans., 2016, 45, 1717–1726 RSC; (g) O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed; (h) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (i) J.-D. Hamel and J.-F. Paquin, Chem. Commun., 2018, 54, 10224–10239 RSC; (j) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS PubMed; (k) T. Braun and R. N. Perutz, Chem. Commun., 2002, 2749–2757 RSC; (l) S. A. Macgregor, Chem. Soc. Rev., 2007, 36, 67–76 RSC; (m) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (n) A. D. Sun and J. A. Love, Dalton Trans., 2010, 39, 10362–10374 RSC; (o) J.-F. Paquin, Synlett, 2011, 2011, 289–293 CrossRef; (p) M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef; (q) P. A. Champagne, Y. Benhassine, J. Desroches and J.-F. Paquin, Angew. Chem., Int. Ed., 2014, 53, 13835–13839 CrossRef CAS PubMed; (r) W. Chen, C. Bakewell and M. R. Crimmin, Synthesis, 2017, 49, 810–821 CrossRef CAS; (s) E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333–348 CrossRef CAS PubMed; (t) S. A. Johnson, J. A. Hatnean and M. E. Doster, in Prog. Inorg. Chem., Wiley, 2011, vol. 57, pp. 255–352 Search PubMed; (u) J. Weaver and S. Senaweera, Tetrahedron, 2014, 70, 7413–7428 CrossRef CAS; (v) U. Mazurek and H. Schwarz, Chem. Commun., 2003, 1321–1326 RSC; (w) W. D. Jones, Dalton Trans., 2003, 3991–3995, 10.1039/b307232k; (x) R. P. Hughes, Eur. J. Inorg. Chem., 2009, 2009, 4591–4606 CrossRef; (y) H. Torrens, Coord. Chem. Rev., 2005, 249, 1957–1985 CrossRef CAS.
- (a) G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 48, 1546–1548 CrossRef CAS PubMed; (b) R. J. Lindup, T. B. Marder, R. N. Perutz and A. C. Whitwood, Chem. Commun., 2007, 3664–3666 RSC; (c) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS; (d) Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 CrossRef; (e) N. A. Phillips, A. J. P. White and M. R. Crimmin, Adv. Synth. Catal., 2019, 361, 3351–3358 CrossRef CAS.
- (a) T. Braun, D. Noveski, B. Neumann and H.-G. Stammler, Angew. Chem., Int. Ed., 2002, 41, 2745–2748 CrossRef CAS; (b) T. Braun, F. Wehmeier and K. Altenhöner, Angew. Chem., Int. Ed., 2007, 46, 5321–5324 CrossRef CAS PubMed; (c) D. Noveski, T. Braun, M. Schulte, B. Neumann and H.-G. Stammler, Dalton Trans., 2003, 4075–4083 RSC; (d) T. Ahrens, M. Teltewskoi, M. Ahrens, T. Braun and R. Laubenstein, Dalton Trans., 2016, 45, 17495–17507 RSC; (e) T. Braun, D. Noveski, M. Ahijado and F. Wehmeier, Dalton Trans., 2007, 3820–3825 RSC.
- T. Ahrens, M. Ahrens, T. Braun, B. Braun and R. Herrmann, Dalton Trans., 2016, 45, 4716–4728 RSC.
- A. L. Raza, J. A. Panetier, M. Teltewskoi, S. A. Macgregor and T. Braun, Organometallics, 2013, 32, 3795–3807 CrossRef CAS.
- A. L. Raza, M. F. Kuehnel, M. Talavera, M. Teltewskoi, M. Ahrens, P. Kläring, T. Braun and D. Lentz, J. Fluorine Chem., 2018, 214, 80–85 CrossRef CAS.
- D. Noveski, T. Braun and S. Krückemeier, J. Fluorine Chem., 2004, 125, 959–966 CrossRef CAS.
- R. N. Haszeldine, C. R. Pool and A. E. Tipping, J. Chem. Soc., Perkin Trans. 1, 1974, 2293–2296 RSC.
- R. Lide, Handbook of Chemistry and Physics, CRC Press, New York, 76th edn, 1995 Search PubMed.
- A. K. Brisdon, I. R. Crossley, R. G. Pritchard and J. E. Warren, Inorg. Chem., 2002, 41, 4748–4755 CrossRef CAS.
- A. K. Brisdon, I. R. Crossley, J. A. Greenall, R. G. Pritchard and J. E. Warren, J. Fluorine Chem., 2004, 125, 1099–1103 CrossRef CAS.
- A. K. Brisdon, R. G. Pritchard and A. Thomas, Organometallics, 2012, 31, 1341–1348 CrossRef CAS.
- (a) M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed; (b) R. Chauvin, J. Phys. Chem., 1992, 96, 9194–9197 CrossRef CAS.
- (a) L. Zámostná and T. Braun, Angew. Chem., Int. Ed., 2015, 54, 10652–10656 CrossRef; (b) J. J. Gambaro, W. H. Hohman and D. W. Meek, Inorg. Chem., 1989, 28, 4154–4159 CrossRef CAS.
- (a) M. F. Kuehnel, P. Holstein, M. Kliche, J. Krüger, S. Matthies, D. Nitsch, J. Schutt, M. Sparenberg and D. Lentz, Chem.–Eur. J., 2012, 18, 10701–10714 CrossRef CAS PubMed; (b) B. M. Kraft and W. D. Jones, J. Am. Chem. Soc., 2002, 124, 8681–8689 CrossRef CAS PubMed; (c) C. J. Bourgeois, R. P. Hughes, J. Yuan, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2006, 25, 2908–2910 CrossRef CAS; (d) R. T. Thornbury and F. D. Toste, Angew. Chem., Int. Ed., 2016, 55, 11629–11632 CrossRef CAS.
- (a) D. Noveski, T. Braun, B. Neumann, A. Stammler and H.-G. Stammler, Dalton Trans., 2004, 4106–4119 RSC; (b) S. I. Kalläne, M. Teltewskoi, T. Braun and B. Braun, Organometallics, 2015, 34, 1156–1169 CrossRef.
- A. Foris, Magn. Reson. Chem., 2004, 42, 534–555 CrossRef CAS.
- M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947–3951 CrossRef CAS PubMed.
- A. K. Brisdon, I. R. Crossley, R. G. Pritchard, G. Sadiq and J. E. Warren, Organometallics, 2003, 22, 5534–5542 CrossRef CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Butterworth/Heinemann, London/Oxford, 3rd edn, 1988 Search PubMed.
- G. M. Sheldrick, SHELXT-2014, Program for the Solution of Crystal Structures from X-Ray Data, University of Göttingen, Germany, 2013 Search PubMed.
- G. M. Sheldrick, SHELXL-2013, Program for the Refinement of Crystal Structures from X-Ray Data, University of Göttingen, Germany, 2013 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- (a) R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS; (b) O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS; (c) M. Von Arnim and R. Ahlrichs, J. Comput. Chem., 1998, 19, 1746–1757 CrossRef CAS; (d) P. Deglmann, K. May, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2004, 384, 103–107 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- (a) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed; (b) D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- (a) K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC; (c) M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC; (b) A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101 CrossRef PubMed.
- (a) T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef; (b) D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- P. H. M. Budzelaar, gNMR, Version 4.1, Adept Scientific plc, Letchworth, 2001 Search PubMed.
- I. Ojima, T. Fuchikami and M. Yatabe, J. Organomet. Chem., 1984, 260, 335–346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all reactions, additional DFT details and xyz coordinates for all calculated compounds. CCDC 1912055. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9fd00059c |
| This journal is © The Royal Society of Chemistry 2019 |