 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systems biology approach to elucidation of contaminant biodegradation in complex samples – integration of high-resolution analytical and molecular tools†
Caroline
Gauchotte-Lindsay
 *a,
Thomas J.
Aspray‡
b,
Mara
Knapp
c and
Umer Z.
Ijaz
a
*a,
Thomas J.
Aspray‡
b,
Mara
Knapp
c and
Umer Z.
Ijaz
a
aInfrastructure and Environment Research Division, School of Engineering, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: caroline.gauchotte-lindsay@glasgow.ac.uk
bSchool of Energy, Geoscience, Infrastructure and Society, Heriot-Watt University, Edinburgh EH14 4AS, UK
cDepartment of Civil and Environmental Engineering, University of Strathclyde, Glasgow G1 1XQ, UK
First published on 29th March 2019
Abstract
We present here a data-driven systems biology framework for the rational design of biotechnological solutions for contaminated environments with the aim of understanding the interactions and mechanisms underpinning the role of microbial communities in the biodegradation of contaminated soils. We have considered a multi-omics approach that employs novel in silico tools to combine high-throughput sequencing data (16S rRNA amplicons) with chemical data including high-resolution analytical data generated by comprehensive two-dimensional gas chromatography (GC × GC). To assess this approach, we have considered a matching dataset with both microbiological and chemical signatures available for samples from two former manufactured gas plant sites. On this dataset, we applied the numerical procedures informed by ecological principles (predominantly diversity measures) as well as recently published statistical approaches that give discriminatory features and their correlations by maximizing the covariances between multiple datasets on the same sample space. In particular, we have utilized sparse projection to latent discriminant analysis and its derivative to multiple datasets, an N-integration algorithm called DIABLO. Our results indicate microbial community structure dependent on the contaminated environment and unravel promising interactions of some of the microbial species with biodegradation potential. To the best of our knowledge, this is the first study that incorporates with the microbiome an unprecedented high-level distribution of hydrocarbons obtained through GC × GC.
1. Introduction
Industrialisation has left behind a legacy of pollution that presents a risk to human health and the environment. Disposal to landfill has long been the preferred approach for disposal of contaminated soils, however rising landfill tax costs drive the development of novel, cheaper and safer remediation technologies.Newly developed sustainable and safe remediation approaches include ex situ (such as biopiles) and in situ (monitored natural attenuation (MNA) and enhanced natural attenuation (ENA)) bioremediation. MNA consists of the monitoring and testing of the progress of natural processes that degrade contaminants in situ, and biopiles and ENA consist of stimulating in situ microbial processes by either introducing new microorganisms (bioaugmentation) or modifying the environmental conditions to stimulate the growth of degrading organisms (biostimulation).1,2 They are advantageous practices because they produce minimal waste and disturbance to the site but also minimise contact between remediation engineers and the contaminated site.
The microbial organisms and catabolic genes involved in the biodegradation of organic contaminants have been well characterised for a large variety of contaminants including polycyclic aromatic hydrocarbons (PAHs). PAHs are recognised lipophilic legacy organic pollutants present in crude oil and are produced during the combustion of coal and organic matter – they are therefore ubiquitous soil contaminants. They are usually present in their thousands in complex environmental samples but only a few, such as naphthalene, phenanthrene or benzo(a)pyrene, are recognized as carcinogenic or mutagenic and are currently on priority pollutant lists. Therefore, the biodegradation of PAHs by microbes has up until now been almost exclusively studied in vitro in microcosms and is usually demonstrated for a single compound or a very limited number of compounds. This is, however, an overly simplistic view of what happens in situ where multiple contaminants are present and their fate likely to be interdependent.
The design of smart and efficient bioremediation solutions in soil requires extracting from the exhaustive knowledge on the degradation of complex samples the information that will enable enhancement of the degradation of listed contaminants. Eventually, the aim is also to optimise analysis carried out during site investigation to provide remediation practitioners with the necessary information to design remediation approaches. Before this is achievable, however, understanding what the relevant factors governing biodegradation of PAHs in soil are is crucial. These factors will include: soil characteristics, physicochemical characteristics (e.g. pH and organic matter content), microbial ecology and the nature of the contaminations (PAH distribution and concentration, but also the presence of organic and inorganic co-contaminants).
The recent rapid technological advances in nucleic acid sequencing have enabled the high-resolution characterisation of microbial communities in PAH contaminated soils. Specifically, metataxogenomic and metagenomic approaches have demonstrated that contamination induces a reduction in species richness and enriches the population with species that are adapted to hydrocarbon degradation.3 Consequently, initial microbial ecology could also be linked to the potential for PAH degradation,4 demonstrating that microbial ecology carries meaningful information on the potential for bioremediation in soil. However, without high resolution information on the contamination profile, notably the presence of other contaminants, this information might not be sufficient for the design of an efficient site-specific bioremediation. Comprehensive two-dimensional gas chromatography coupled with mass spectrometry (GC × GC-MS) has allowed the near comprehensive characterisation of semi-volatile organic carbons (SVOCs) in tars and contaminated soils from former manufactured gasworks (FMGs).5–7 The coupling of two columns allows for a two-dimensional separation across a retention plane rather than along a retention line. The peak capacity of GC × GC is several orders of magnitude higher than traditional techniques and thousands of compounds can be resolved; coupled with the resolution power of mass spectrometry, it generates ultra-resolution chemical signatures of environmental samples. It has been used for source apportionment,8 monitoring of bioremediation across SVOC classes6 and estimation of ecotoxicity.9
Integration of metataxogenomics and GC × GC analysis has the potential to unveil information on the interactions between complex mixtures of environmental contaminants and microbial ecology never accessed before. Integrative multivariate statistical approaches for multi-omics data are being developed in biomedical applications.10 For instance, the DIABLO10 method builds on the Generalised Canonical Correlation Analysis, which integrates multiple datasets by finding principal components (latent variables) that maximize the covariance of scores between different datasets and the categorical outcome of interest. The resulting loading vectors are then constrained to give discriminants that correlate between these datasets.
Here, through the comparison of soils from two different FMGs, we present an example of statistical integration of chemical and molecular data in contaminated soils. Chemical and DNA extracts were analysed and processed through robust methods and pipelines1–4 previously developed in our laboratories and statistical analysis was first carried out independently. Multivariate analysis enabled forensic characterisation of the site by exploring intra- and inter-site variations in distributions of PAHs and other compounds.2 16S rRNA sequencing data were explored for alpha and beta diversity analyses,5 inferred metabolic pathway analysis6 and differential abundance analysis of both species and metabolic pathways between sites.5 Then, the N-integration algorithm DIABLO7 was used to classify and discriminate features that correlate between the microbiome and chemical metadata including, for the first time, high-resolution distribution of SVOCs obtained by GC × GC-MS.
2. Experimental
2.1. Samples
Samples from two different former manufactured gas plants were provided by collaborators. Very little information was provided about the samples so no spatial resolution has been attempted in this work. The samples were stored in plastic tubes at 4 °C. Eighteen samples came from a site in the United Kingdom (COV site) and nine samples from a site in Switzerland (CH site). The COV soil samples, generally dark, compact and sticky in nature, were each sieved once with a 10 mm mesh. The CH samples, brown in colour and drier in nature, were each sieved through 10, 2.36 and 1.70 mm meshes. For each individual sample, the moisture content in percentage was measured by placing a subsample in an oven at 105 °C for 24 h, and loss on ignition (LOI), also in percentage, was measured by placing a subsample in a furnace at 550 °C for 2 h. No further soil characterisation was carried out at this stage.2.2. Semivolatile organic compound analysis
Approximately 0.25 g of each soil sample was extracted via pressurised liquid extraction (PLE) with on-line silica gel clean-up.5 The first fraction (hexane) and second fraction (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene) (v/v) (8:2) were collected together and concentrated to 1.15 mL. Quantification of 14 PAHs along with four surrogates was carried out using GC-MS. It was carried out in TIC or SIM mode depending on the concentrations. We first validated the PLE method by extracting two of the COV samples six times and quantifying the 14 PAHs in duplicates. The relative standard deviation for the quantification of an individual PAH varied between 1% and 16.2%, with only one value over 10% demonstrating that the extraction was repeatable. Consequently, all other samples were only extracted once.
:toluene) (v/v) (8:2) were collected together and concentrated to 1.15 mL. Quantification of 14 PAHs along with four surrogates was carried out using GC-MS. It was carried out in TIC or SIM mode depending on the concentrations. We first validated the PLE method by extracting two of the COV samples six times and quantifying the 14 PAHs in duplicates. The relative standard deviation for the quantification of an individual PAH varied between 1% and 16.2%, with only one value over 10% demonstrating that the extraction was repeatable. Consequently, all other samples were only extracted once.
To optimise chromatographic resolution, we also employed a GC × GC method we had previously developed for the exhaustive characterisation of environmental coal tar samples.5 The method ensured optimal separation of PAH isomers but also separated alkanes and alkylated benzenes. Comprehensive semi-volatile signatures of the extracts5 were obtained using a LECO (St. Joseph, Michigan) time of flight mass spectrometer, model Pegasus 4D, connected to an Agilent 7890A gas chromatograph equipped with a LECO thermal modulator. The column set-up employed is known as reversed phase, with a first dimension capillary column (TR50-MS; 30 m × 0.25 mm i.d. × 0.25 μm film thickness; Thermo) that was more polar than the second capillary column (Rxi-5Sil; 2 m × 0.25 mm i.d. × 0.25 μm film thickness; Thames Restek). In this set-up, PAHs have a short retention time in the second dimension while alkanes are retained for longer. Alkylbenzenes, alkynes and alkenes, in order of polarity, elute in the retention space between PAHs and alkanes.5 To be input into multivariate analysis, the GC × GC data must first be aligned between samples. Alignment can be carried out either by aligning chromatograms or peak tables (the output of GC × GC data processing). Here, we employed a combination of peak picking using the LECO ChromaTOF software and peak alignment using the R code R2DGC.11 We optimised the data processing and R2DGC parameters but decided not to carry out any manual tidying up of the peak tables, which were left. Indeed, the peak tables contained over 500 compounds each, and a manual check of each peak for truly exhaustive analysis was not feasible. Two instrumental duplicates for each sample were analysed. For one COV soil sample, five replicate PL extracts were analysed in duplicate. Additional instrumental replicates were included for some samples because the second-dimension capillary column needed replacing during the study, shifting both retentions; hence samples were re-analysed after the change in columns. All peak tables were aligned to the CH peak table that contained the most peaks (top left in Fig. 1). The alignment of 68 peak tables was carried out twice with missing value limits equal to 0.1 i.e. a compound had to appear in at least 10% of the peak tables to be included in the alignment table, and 1 where a compound had to be present in all peak tables.
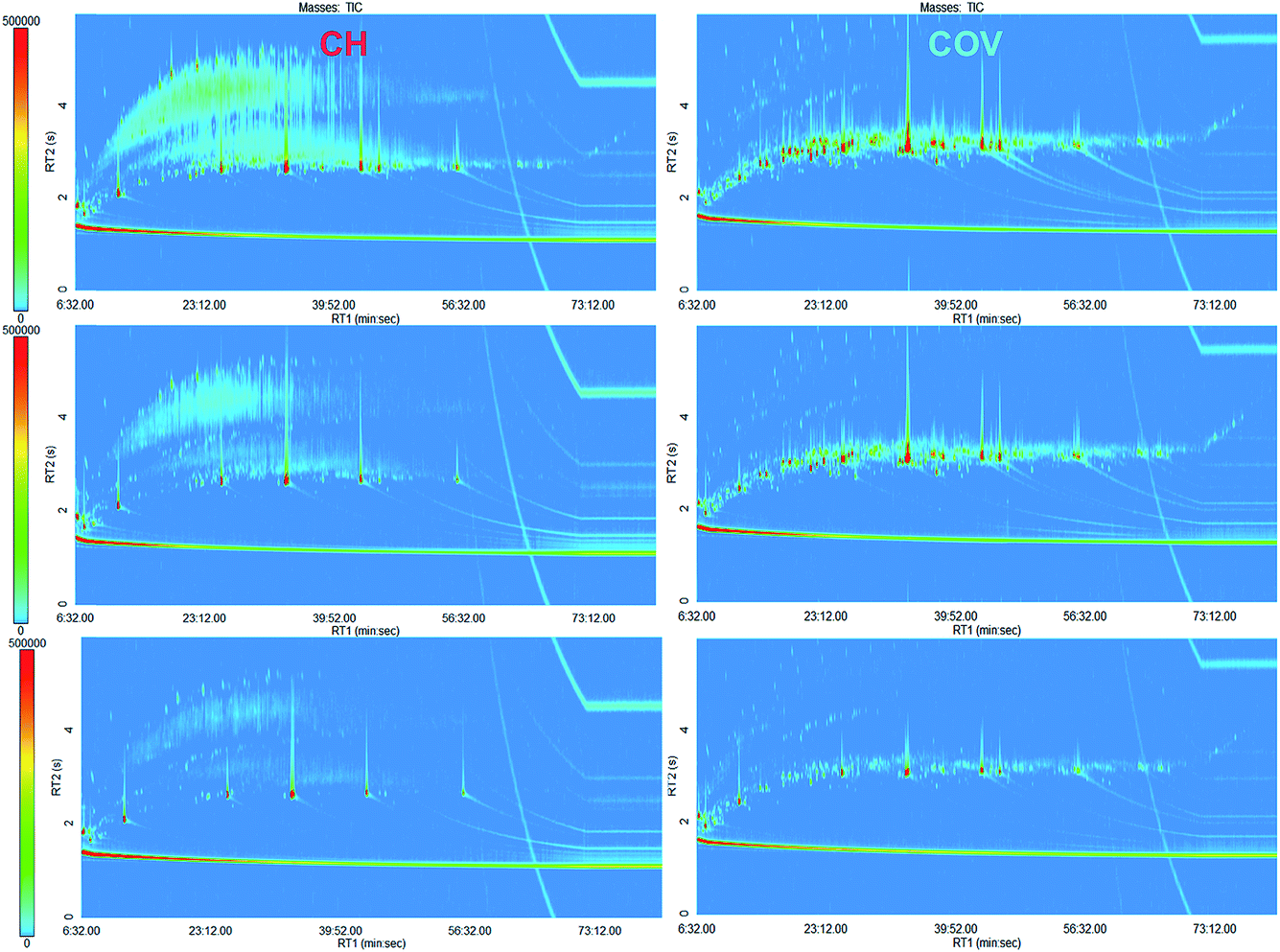 | ||
| Fig. 1 Representative GC × GC chromatograms for both sites. CH is on the left and COV is on the right. | ||
All methods are described in detail in the ESI.†
2.3. Transition metal analysis
The concentrations of lead, iron, cadmium, chromium, zinc, copper, and nickel were determined using inductively coupled plasma optical emission spectrometry (ICP-OES) and the methods are discussed in the ESI.†2.4. Genomic DNA analysis
Genomic DNA was extracted twice from soil samples (0.25 g fresh weight); once for quantitative PCR (qPCR) for the alkB12 and PAH RHD GN and PAH RHD GP genes, and a second time for 16S (V3 and V4 regions) metataxogenomic library preparation and sequencing. The detailed methods and bioinformatics workflow are presented in the ESI.†2.5. Statistical analysis
All statistical analyses were performed in R.Hierarchical clustering analysis (HCA) of the GC × GC alignment table was performed by calculating Manhattan distance between the samples each comprising 961 metabolites. Prior to analysis, zero values were replaced by one third of the smallest value in the table and each peak area was normalised to the calculated total peak areas for a given sample. Afterwards, we performed agglomerative clustering using complete linkages utilising R’s hclust() function. For visualisation, we used R’s dendextend package.13 We then used the color_branches() function from the same package to cluster both the terminal leaves of the dendrogram and the edges leading to the samples.
The vegan package was used for alpha and beta diversity analyses. For alpha diversity measures we have used: rarefied richness – the estimated number of species after rarefying the abundance table to minimum library size; Shannon entropy – a commonly used index to measure balance within a community; Simpson index – a measure of dominance that weighs towards the abundance of most common OTU and is less sensitive to rare OTUs, Pilou eveness – compares the actual diversity values to the maximum possible diversity value, is constrained between 0 and 1.0, and the more variation in abundance between different OTUs in the community, the lower its value; and Fisher’s alpha – a parametric index of diversity that assumes the abundance of OTUs following the log series distribution. Ordination of the OTU table in reduced space (beta diversity) was done using Principal Coordinate Analysis (PCoA) plots of OTUs using three different distance measures that were made using Vegan’s cmdscale() function: (1) Bray–Curtis is a distance metric that considers only OTU abundance counts, (2) unweighted UniFrac is a phylogenetic distance metric that calculates the distance between samples by taking the proportion of the sum of unshared branch lengths in the sum of all of the branch lengths of the phylogenetic tree for the OTUs observed in two samples, and without taking into account their abundances and, (3) weighted UniFrac is a phylogenetic distance metric combining phylogenetic distance with relative abundances. This places emphasis on dominant OTUs or taxa. UniFrac distances were calculated using the phyloseq package.14
Analysis of the variance for explanatory variables (or sources of variation) was performed using Vegan’s adonis() against distance matrices (Bray–Curtis/unweighted UniFrac/weighted UniFrac). This function, referred to as PERMANOVA, fits linear models to distance matrices and uses a permutation test with pseudo-F ratios. To give an account of environmental filtering (phylogenetic overdispersion versus clustering), phylogenetic distances within each sample were further characterised by calculating the nearest taxa index (NTI) and net relatedness index (NRI). This analysis aimed to determine whether the community structure was stochastic (overdispersion and driven by competition among taxa) or deterministic (clustering and driven by strong environmental pressure). The NTI was calculated using mntd() and ses.mntd(), and the mean phylogenetic diversity (MPD) and NRI were calculated using mpd() and ses.mpd() functions from the picante package.15 NTI and NRI represent the negatives of the output from ses.mntd() and ses.mpd(), respectively. Additionally, they quantify the number of standard deviations that separate the observed values from the mean of the null distribution (999 randomisation using null.model-‘richness’ in the ses.mntd() and ses.mpd() functions and only considering taxa as either present or absent regardless of their relative abundance). Based upon the recommendations,16 only the top 1000 most abundant OTUs were used for the calculations.
Discriminant analyses between the two sites were considered using microbiome data alone, and then together with the meta data (Table 1). For the former case, we used Sparse Projection to Latent Structure-Discriminant Analysis (sPLS-DA) with the R’s mixOmics package.10 The procedure constructs artificial latent components of the predicted dataset (OTUs table denoted X(N × P)) and the response variable (denoted Y with categorical information of samples, e.g. CH and COV) by factorizing these matrices into scores and loading vectors in a new space such that the covariance between the scores of these two matrices cov(Xhah,Yhbh) in this space is maximized under two constraints: ‖ah‖2 = 1; and ‖ah‖1 ≤ λ, where ah and bh are the corresponding loading vectors for X and Y, and h represents the number of components (akin to PCA analysis). To integrate meta data further, we utilised DIABLO from R’s mixOmics package. We have combined M = 2 datasets denoted X(1)(N × P1), X(2) (N × P2) where X(1) represents the microbiome data, and X(2) represents meta data (moisture content, LOI, 14 PAHs concentrations and GC × GC signatures whether considered or not) (Table 1). An additional matrix X(3) required for the algorithm to enable the discriminant analysis is a dummy matrix of the classes the samples belong to (whether COV or CH, and equivalent to matrix Y in the sPLS-DA case). The algorithm then constructs artificial latent components of the datasets by factorizing the datasets into scores and loading vectors in new space such that the covariance between the scores of these matrices in this space is maximized, i.e., for q = 1, 2, …, Q, DIABLO solves for each component h = 1, …, H:
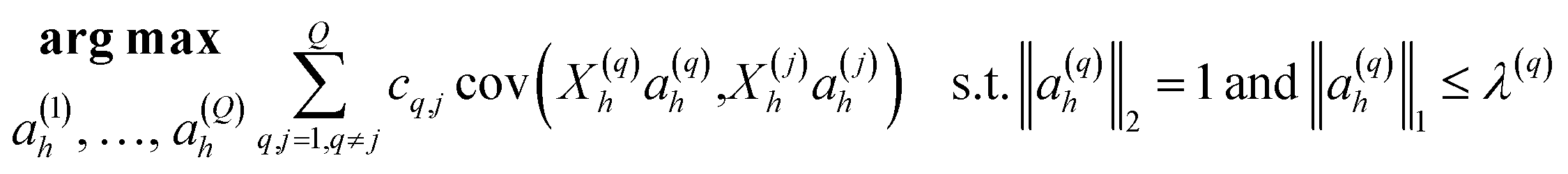
 .
.
| Chemical analysis | Molecular biology | Statistical analysis |
|---|---|---|
| Moisture content (A) | qPCR: alkB and PAH RHD GN and PAH RHD GP (I) | HCA of GC × GC signatures (meta: E) |
| LOI (B) | 16S metataxogenomic sequencing (II) | Alpha and beta diversity (rDNA: I) |
| Quantification of 14 PAHs by GC-MS (C) | Tax4Fun KEGG pathways analysis (rDNA: I) | |
| Quantification of 7 transition metals by ICP-OES (D) | Discriminant analysis: | |
| GC × GC-TOFMS signatures (missing values limit = 10%) (E) | PLS-DA (rDNA: I) | |
| GC × GC-TOFMS signatures (missing values limit = 100%) (F) | DIABLO 1 (rDNA: I; meta: II + A + B + C + D). | |
| DIABLO 2 (rDNA: I; meta: II + A + B + C + D + F). |
To predict the number of latent components (associated loading vectors) and the number of discriminants, for sPLS-DA, we used the perf.plsda() and tune.splsda() functions, whereas for DIABLO, block.splsda() and tune.block.splsda() functions were used, respectively. In both cases, we fine-tuned the model using leave-one-out cross-validation by splitting the data into training and testing sets and then finding the classification error rates employing two metrics, overall error rates and balanced error rates (BER), between the predicted latent variables with the centroid of the class labels (categories considered in this study) using the max.dist (which gave the minimal classification rate for the scenarios considered in this study). BER accounts for differences in the number of samples between different categories. Other than TSS + CLR normalisation for the abundance table, log10 normalisation for qPCR data was used, and GC × GC signatures were normalised using pareto scaling. When displaying boxplots, pair-wise ANOVA or Kruskal–Wallis were performed taking two categories at a time, and where significant (p ≤ 0.05), they were joined together by a line and significance was plotted on top (*: 0.01 ≤ p < 0.05; **: 0.05 ≤ p < 0.001; ***: p ≤ 0.001).
3. Results & discussion
3.1. Comprehensive SVOCs characterisations of contamination
Comprehensive two-dimensional analysis coupled with time-of-flight mass spectrometry analysis was carried out on the soil samples. GC × GC-TOFMS enables an exhaustive characterisation of the distribution of semi-volatile organic compounds (SVOCs) in the samples as a novel “omics” dimension in the study of bioremediation of complex contaminated soils. Fig. 1 shows the GC × GC chromatograms for representative samples of CH and COV. Visually, COV samples appeared more heavily contaminated with PAHs than the CH samples, which was confirmed using the one dimensional GC quantification (Fig. 2). The proportion of substituted PAHs was also higher in the COV samples. By comparison, the CH samples had higher proportions of alkanes, alkenes, alkynes and alkylbenzenes. This could possibly be explained by a difference in the processes involved in the coal gasification as we demonstrated previously in a forensic study of coal tar samples from FMGPs.8 Higher proportions of PAH parents and lower proportions of alkanes were associated with high temperature gasification, while high proportions of petroleum-like hydrocarbons can be associated with carburetted water gas plants.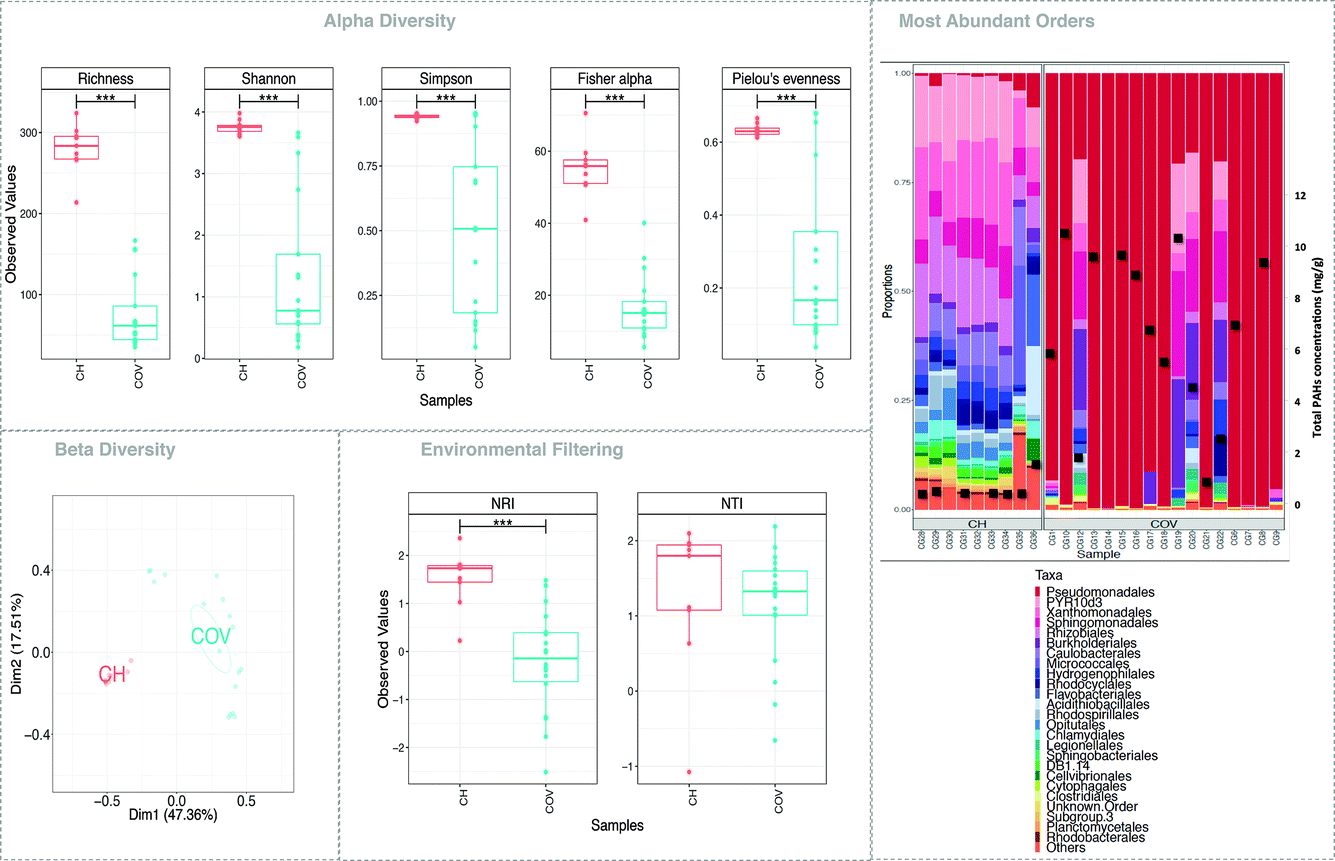 | ||
| Fig. 2 Microbial diversity and function. (a), (c), and (d) represent alpha diversity, beta diversity and environmental filtering indices, respectively. For beta diversity analysis, PERMANOVA suggested 39%, 42%, and 58% variability (R2; all significant p < 0.001) between COV and CH for Bray–Curtis [shown in (c)], UniFrac, and weighted UniFrac distances, respectively. In (c), the ellipses are drawn at a 95% confidence interval of standard error with the group labels located at the mean of the ordination. Note that in (d), the positive value of NTI indicates that species co-occur with more closely related species than expected by chance, with negative values suggesting otherwise. (b) shows community structure based on the relative abundance of the top-25 most abundant orders from across each site, where ‘others’ refers to all orders not included in the ‘top-25’. The grey bands indicate replicates of the same soil sample. The total concentration (mg g−1) of the 14 PAHs quantified was superimposed to this data. | ||
While the potential of GC × GC for comprehensive analysis of SVOCs in contaminated samples is evident in terms of the resolution of the analytical method, the availability of fully comprehensive, automated and reproducible data processing and alignment for GC × GC data is arguably the biggest bottleneck to its use in omics-like studies. The accuracy of the chosen processing method was evaluated using replicates and multivariate analysis. Alignment of 68 peak tables with a missing values limit of 10% returned 961 compounds. To evaluate the robustness of the data processing, HCA was carried out on the samples using the alignment table (Fig. S1†). The clustering clearly separated the two sites, with only one COV sample (both instrumental replicates) clustering with CH samples. The GC × GC chromatogram for this extract is presented in the bottom right of Fig. 1. One CH sample clustered away from both sites’ clusters; its chromatogram is presented in the top left of Fig. 1. In most cases, a sample nearest neighbour was its instrumental replicate. The five extracts from the same soil sample (COV-O5 in Fig. S1†) clustered altogether. Replicate runs before and after the column change, however, did not cluster near each other (e.g. see COV-18 and COV-17 on Fig. S1†). The data alignment was therefore deemed successful although it appeared somewhat susceptible to retention shift. Alignment with the missing values limit of 100% returned 58 compounds (Table S1†). While we were able to ascertain during GC-MS quantification that all of the samples contained the 14 quantified PAHs, the alignment only returned seven of those. Similarly, while five surrogates and one internal standard were added to each sample, only three were in the final alignment table and their peak areas showed great variations. Recovery quantification for the surrogates was carried out using GC-MS and showed good reproducibility (relative standard deviation around 10% for all surrogates) (data not shown). The errors in the GC × GC data, therefore, came from the data processing. Sources of errors could be related to a degree of misalignment but were also expected to be related to the peak finding algorithm. Deconvolution and reconstitution of peaks across multiple modulation times might lead to two types of errors: (1) one peak can be incorrectly split into two peaks and (2) two or more peaks can be artificially combined as one. The first error is acknowledged in most data processing pipelines for GC × GC data11,17 as it occurs in metabolomics samples. Here we used the PrecompressFiles function of R2DGC to correct the peak tables for the first error prior to alignment. The concentrations of surrogates, however, was very high compared to any other compound in the samples and their signals saturated the detectors, which increased the possibility of peak splitting and might explain the non-repeatability of their peak areas.
The second error is less common in metabolomics as compounds have more distinct mass spectra. In hydrocarbon analysis, however, position isomers are very common, have very similar spectra and elute near each other and are therefore more likely to be integrated as a single peak. This can be minimised by optimising the parameters of data processing such as minimum peak widths in both dimensions and signal-to-noise ratio; it, however, remains an issue in samples with large dynamic ranges between compounds. The results therefore indicated that the probability of one of these errors to occur for any one compound in one of the 68 peak tables was high. Further optimisation of the data processing needs to be carried out for accurate and precise exhaustive characterisation of SVOCs.
3.2. Bacterial diversity in CH and COV soils
While 16 different samples were collected for the COV site and 9 for the CH sites, not all extracted DNA was successfully amplified. Additionally, some samples were extracted in replicates (see Fig. 2b) and variations in microbial communities between replicates was lower than the inter-sample variations.Five measures of alpha diversity clearly showed that the CH samples had significantly higher species richness than COV samples had (Fig. 2). While considering the top 25 most abundant orders, we can notice that the proportions of gammaproteobacteria, alphaproteobacteria and betaproteobacteria were consistent in the CH samples but varied in the COV samples. Comparison with the total concentrations of 14 PAHs suggested a possible connection between the levels of PAH contamination and the abundance of the dominant species in the samples (Fig. 2b), particularly the proportions of gammaproteobacteria. Two samples, however, showed contrary trends. One of them presented low total PAH concentration and a low proportion of gammaproteobacteria. In this sample, recovery values for the PAH surrogates were very low (between and 20 and 35%) indicating an issue during PLE extraction and therefore an underestimation of total PAH concentration.
NMDS (using Bray–Curtis distance) of sample dissimilarities showed separation between the two sites with more inter-sample variability in COV (Fig. 2c). PAH concentrations in COV were one order of magnitude higher on average than in CH samples and also presented a greater variance. This may explain the dispersion in the beta diversity space resulting in several ecological niches. The pathways analyses (not shown here) indicated that a significantly higher proportion of OTUs in the COV samples were available in the reference SilvaMod 123 database (for which reference pathways are available in the Tax4Fun package) than in CH samples, which suggested that the OTUs that inhabited the more contaminated sites are more ubiquitous in nature and thus well characterised with their pathways available. The gene content for each OTU in each sample was inferred from the closest sequenced genome using the Tax4Fun package and we investigated particular pathways from the KEGG database linked to contaminant degradation4 and related metabolic pathways. Out of the 12 tested pathways, seven presented significantly different average relative abundance between COV and CH. COV presented statistically more potential for degradation of hydrocarbon contaminants via the toluene (ko00623) and naphthalene (ko00626) pathways and CH via the drug metabolism (other pathways) (ko00983) and PAH (ko00624) degradation pathways (Fig. 3). Incidentally, COV samples also showed significantly more potential for the propanoate and pyruvate metabolisms and for glycolysis.
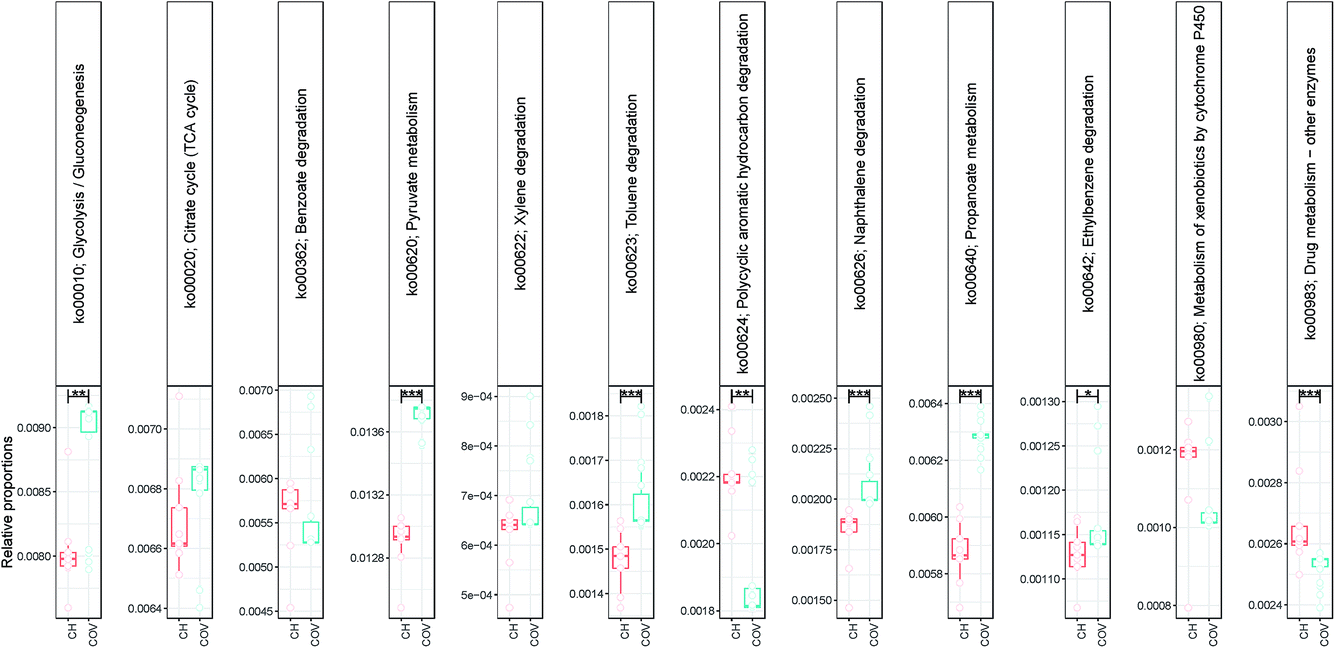 | ||
| Fig. 3 Relative abundance of KEGG pathways for hydrocarbon degradation and related metabolic pathways inferred using the Tax4Fun package. Lines connect two categories where the differences were significant (Kruskal–Wallis) with *** (p < 0.001). | ||
Next, we explored the influence of the environment on the assembly of microbial communities. We applied two phylogenetic alpha diversity indices, NTI and NRI (Fig. 2d), to explore phylogenetic clustering at both local and global scales. Positive values of NTI indicate that species co-occur with more closely related species than expected by chance, with negative values suggesting otherwise. This is mainly because NTI measures tip-level divergences (putting more emphasis on terminal clades and akin to “local” clustering) in phylogeny while NRI measures deeper divergences (akin to “global” clustering). For both NTI and NRI, values >+2 indicate strong environmental pressure, values <−2 indicate strong competition among species as the driver of community structure, and the values in between are a gradient between the two. The results indicated that the communities in CH samples are more deterministic, and influenced by the environment, whereas COV sites are driven by the competitive exclusion principle where the species that can outcompete others in a given niche dominate, leading to more dispersion in the phylogenetic tree. This phenomenon also supports the much higher variability in beta diversity space for COV samples (Fig. 2c).
3.3. Discriminant analysis of microbiome and chemical data
Through discriminant analysis based on the microbiome alone (sPLS-DA), we found a total of 10 discriminating OTUs (five for the first two components each). PLS-DA (Fig. 4a) shows ordination of the samples using all of the OTUs and sPLS-DA (Fig. 4b) shows ordination of the samples using only the discriminating OTUs. By comparing these two figures, it can be seen that in the reduced space, the sample dissimilarities (between COV and CH) are conserved. Three out of the ten OTUs were greater in abundance for CH, while the rest were greater for COV. Clustering of the samples according to these OTUs showed that four out of the five discriminating features from the first component (OTUs 146, 1214, 384 and 333; this group is henceforth referred to as Group A) were significantly more abundant in COV than in CH, and the last OTU (OTU_14) was significantly more abundant in the CH site. OTUs 333, 384 and 1214 belong to the Clostridiales order. OTUs 333 and 384 are both plant biopolymer degraders of the Ruminococcaceae family, which might relate to the organic matter present on the site, while OTU_1214 belongs to the genus Bacillus, which is known for both PAH and alkane degradation.18 OTU_146 belongs to the Desulfotomaculum genus, in which some species have been found to degrade cresol in sulfate-degrading condition19 and OTU_4 belongs to the genus Sulfuritalea, oxygen independent aromatic compound degrading bacteria.20 While none of the Group A OTUs are amongst the most abundant in the samples, OTU_14 is amongst the most abundant in the CH samples (results not shown). These 5 OTUs might be markers of the difference in hydrocarbon degradation mechanisms between the two sites because of differences in concentration and in soil properties and quality.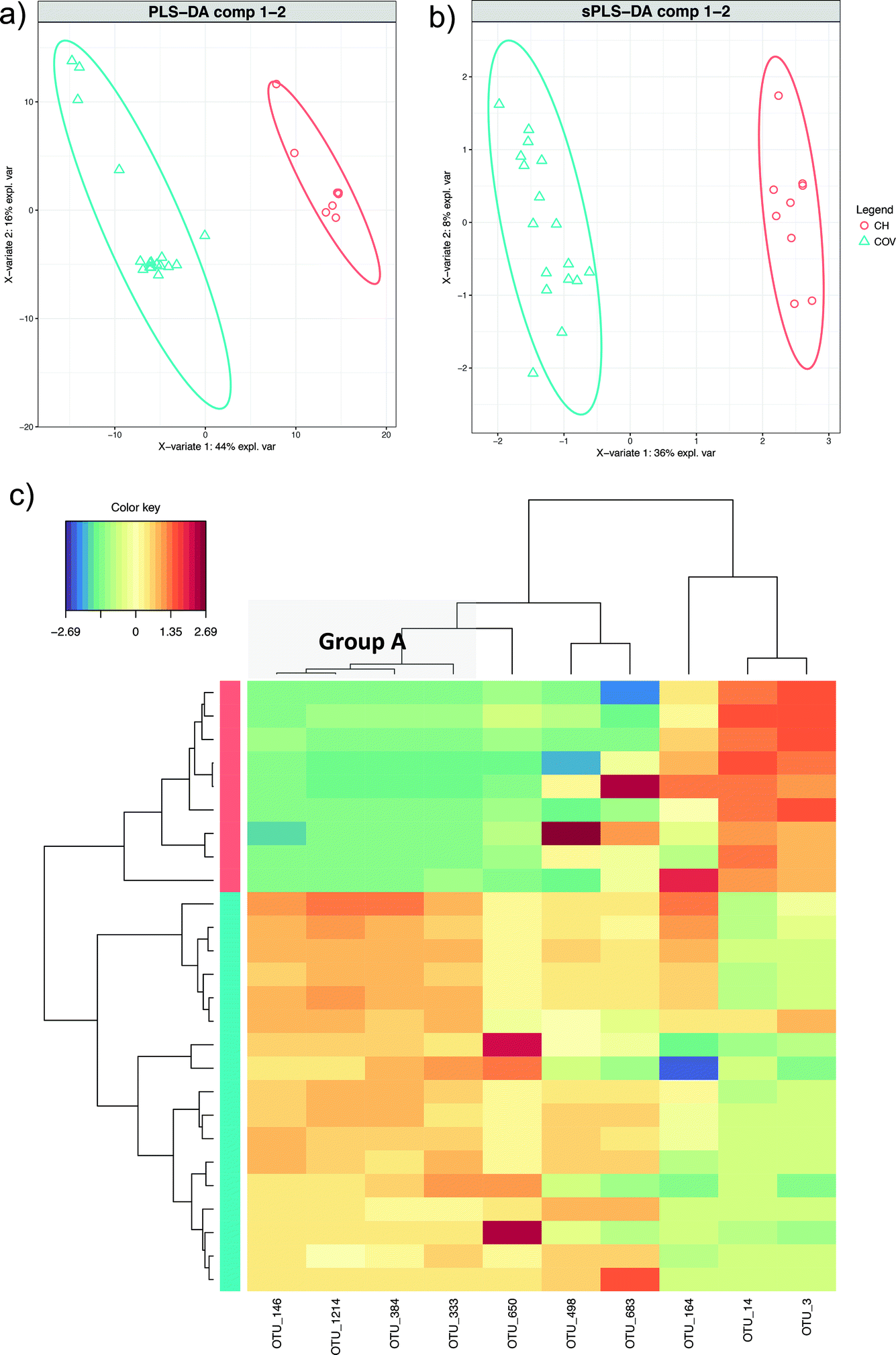 | ||
| Fig. 4 sPLS-DA of the microbiome data. (a) PLS-DA discriminant analysis and (b) sPLS-DA discriminant analysis – the ordination of the data is conserved when only the discriminating OTUs are employed. (c) shows the heatmaps of the 10 discriminant features selected in the sPLS-DA (see Table 2), with both rows and columns ordered using hierarchical (average linkage) clustering to identify blocks of features of interest. Group A (see Table 2) is indicated in a grey box. | ||
A first DIABLO analysis (hitherto referred to as DIABLO 1) (Fig. 5) was carried out by integrating additional metadata to the microbiome data: the concentrations of 14 PAHs, LOI, moisture, heavy metal concentrations and qPCR data for Gram negative PAH degrader (PAH RHD α GN), Gram positive PAH degrader (PAH RHD α GP) and alkane degrader (alkB) gene abundance (Table 1). We found two components to reduce the classification error rates resulting in 10 discriminating OTUs and 10 discriminating meta data features (five each for the two components). Group A represented four of the five OTUs that were selected in the first component (Table 2). This confirmed that in both cases, the first component isolated the OTUs that were the most representative of the differences between COV and CH and particularly the ones that were much more abundant in COV than in CH. The final OTU, OTU_147, was identified as the actinobacteria, Micromonospora sp. WMMB 894, for which little information is available in the literature. These five OTUs are further referred to as Group A′.
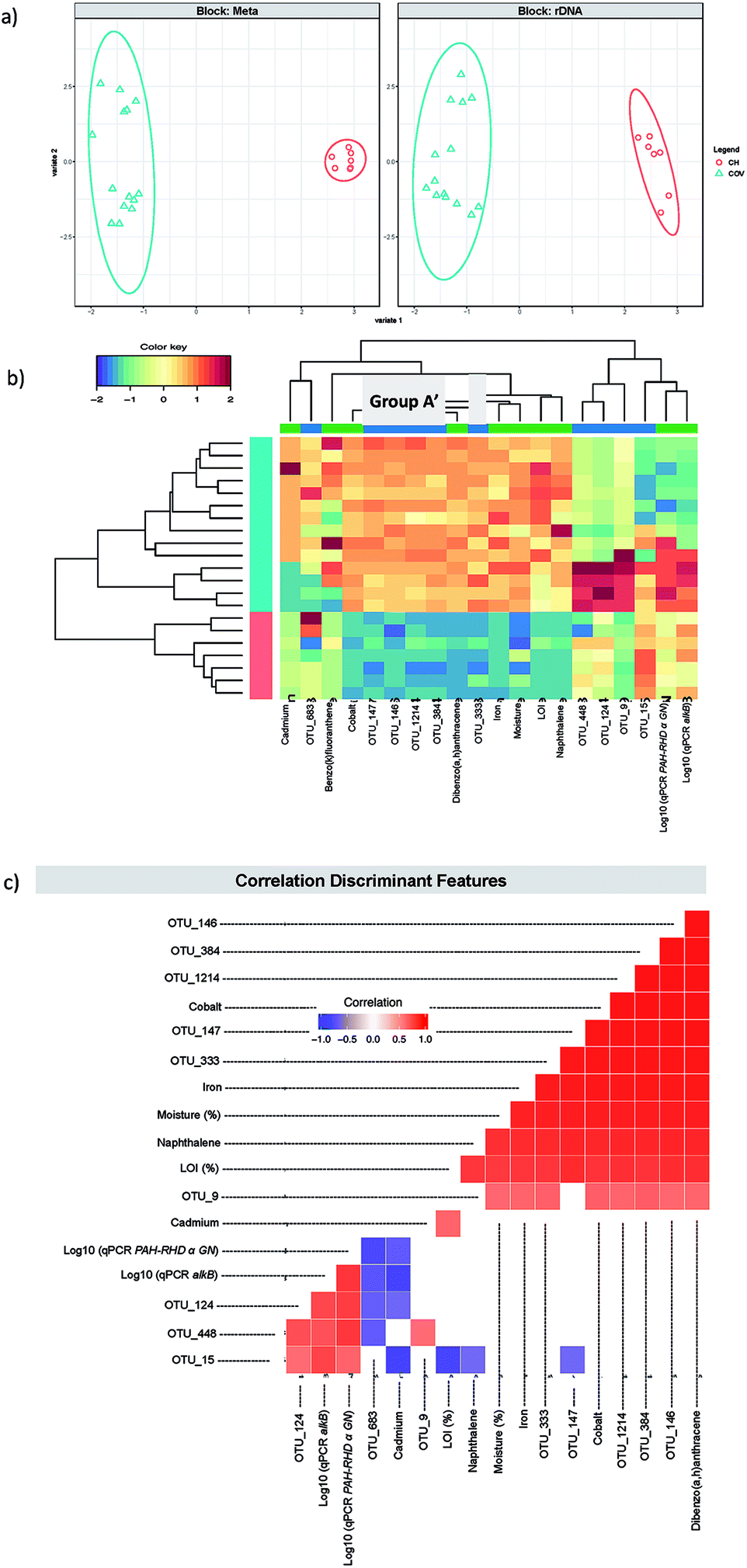 | ||
| Fig. 5 DIABLO 1. (a) The algorithm found two components reducing the classification error rates in the DIABLO algorithm and shows the ordination of samples with ellipses representing 95% confidence interval and percentage variations explained by these components in axes labels for both microbiome (Block: rDNA) and meta data (Block: Meta). (b) shows the heatmaps of the discriminant features in DIABLO 1 (see Table 2), with both rows and columns ordered using hierarchical (average linkage) clustering to identify blocks of features of interest. Group A′ (Table 1) is indicated in a grey box. (c) shows the significant correlations (−0.6 < R > 0.6) between the features as calculated by the algorithm. | ||
| Multivariate analysis | Discriminating OTUs | ||||||
|---|---|---|---|---|---|---|---|
| ID | Kingdom | Phylum | Class | Order | Family | ||
| sPLS-DA (OTUs) | First Component | OTU_146 | Bacteria | Firmicutes | Bacilli | Bacillales | Bacillaceae |
| OTU_333 | Bacteria | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | ||
| OTU_384 | Bacteria | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | ||
| OTU_1214 | Bacteria | Firmicutes | Clostridia | Clostridiales | Peptococcaceae | ||
| OTU_14 | Bacteria | Proteobacteria | Alphaproteobacteria | Sphingomonadales | Sphingomonadaceae | ||
| Second Component | OTU_164 | Archaea | Euryarchaeota | Methanomicrobia | Methanosarcinales | Methanosarcinaceae | |
| OTU_683 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhizobiales | Methylobacteriaceae | ||
| OTU_650 | Bacteria | Proteobacteria | Gammaproteobacteria | Xanthomonadales | Xanthomonadaceae | ||
| OTU_498 | Bacteria | Proteobacteria | Deltaproteobacteria | Bdellovibrionales | Bdellovibrionaceae | ||
| OTU_3 | Bacteria | Proteobacteria | Betaproteobacteria | Rhodocyclales | Rhodocyclaceae | ||
| Multivariate analysis | Discriminating OTUs | Disciminating Metadata Features | ||
|---|---|---|---|---|
| Genus | OTUs | ID | ||
| sPLS-DA (OTUs) | First Component | Bacillus | n.a. | |
| Ruminiclostridium 1 | [Clostridium] termitidis CT1112 | n.a. | ||
| Acetivibrio | Acetivibrio cellulolyticus CD2 | n.a. | ||
| Desulfotomaculum | n.a. | |||
| Sandaracinobacter | Y | n.a. | ||
| Second Component | n.a. | |||
| n.a. | ||||
| Stenotrophomonas | n.a. | |||
| Bdellovibrio | n.a. | |||
| Sulfuritalea | n.a. | |||
| Multivariate analysis | Discriminating OTUs | ||||||
|---|---|---|---|---|---|---|---|
| ID | Kingdom | Phylum | Class | Order | Family | ||
| DIABLO 1 | First Component | OTU_146 | Bacteria | Firmicutes | Bacilli | Bacillales | Bacillaceae |
| OTU_384 | Bacteria | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | ||
| OTU_333 | Bacteria | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | ||
| OTU_147 | Bacteria | Actinobacteria | Actinobacteria | Micromonosporales | Micromonosporaceae | ||
| OTU_1214 | Bacteria | Firmicutes | Clostridia | Clostridiales | Peptococcaceae | ||
| Second Component | OTU_15 | Bacteria | Proteobacteria | Gammaproteobacteria | Xanthomonadales | Xanthomonadaceae | |
| OTU_683 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhizobiales | Methylobacteriaceae | ||
| OTU_448 | Bacteria | Proteobacteria | Betaproteobacteria | Burkholderiales | Alcaligenaceae | ||
| OTU_9 | Bacteria | Proteobacteria | Betaproteobacteria | Burkholderiales | Alcaligenaceae | ||
| OTU_124 | Bacteria | Proteobacteria | Alphaproteobacteria | Rhizobiales | Rhizobiaceae | ||
| Multivariate analysis | Discriminating OTUs | Disciminating Metadata Features | ||
|---|---|---|---|---|
| Genus | OTUs | ID | ||
| DIABLO 1 | First Component | Bacillus | Dibenzo(a,h)anthracene | |
| Acetivibrio | Acetivibrio cellulolyticus CD2 | Cobalt | ||
| Ruminiclostridium 1 | [Clostridium] termitidis CT1112 | Iron | ||
| Plantactinospora | Micromonospora sp. WMMB 894 | Moisture | ||
| Desulfotomaculum | Naphthalene | |||
| Second Component | Pseudoxanthomonas | Pseudoxanthomonas spadix BD-a59 | Log10 (qPCR PAH-RHD _ GN) | |
| Log10 (qPCR alkB) | ||||
| Achromobacter | Cadmium | |||
| Pusillimonas | Benzo(k)fluoranthene | |||
| Shinella | Sinorhizobium sp. enrichment culture clone Van49 | LOI | ||
| Multivariate analysis | Discriminating OTUs | ||||||
|---|---|---|---|---|---|---|---|
| ID | Kingdom | Phylum | Class | Order | Family | ||
| DIABLO 2 | First Component | OTU_384 | Bacteria | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae |
| OTU_146 | Bacteria | Firmicutes | Bacilli | Bacillales | Bacillaceae | ||
| OTU_1214 | Bacteria | Firmicutes | Clostridia | Clostridiales | Peptococcaceae | ||
| OTU_333 | Bacteria | Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | ||
| OTU_147 | Bacteria | Actinobacteria | Actinobacteria | Micromonosporales | Micromonosporaceae | ||
| Second Component | OTU_116 | Bacteria | Proteobacteria | Alphaproteobacteria | Sphingomonadales | Erythrobacteraceae | |
| OTU_255 | Bacteria | Proteobacteria | Alphaproteobacteria | Sphingomonadales | Erythrobacteraceae | ||
| OTU_635 | Bacteria | Proteobacteria | Alphaproteobacteria | Rickettsiales | Holosporaceae | ||
| OTU_344 | Bacteria | Proteobacteria | Betaproteobacteria | Burkholderiales | Comamonadaceae | ||
| OTU_1397 | Bacteria | Proteobacteria | Alphaproteobacteria | Sphingomonadales | Sphingomonadaceae | ||
| Multivariate analysis | Discriminating OTUs | Disciminating Metadata Features | ||
|---|---|---|---|---|
| Genus | OTUs | ID | ||
| DIABLO 2 | First Component | Acetivibrio | Acetivibrio cellulolyticus CD2 | Dibenzo(a,h)anthracene |
| Bacillus | Cobalt | |||
| Desulfotomaculum | Iron | |||
| Ruminiclostridium 1 | [Clostridium] termitidis CT1112 | Moisture | ||
| Plantactinospora | Micromonospora sp. WMMB 894 | Naphthalene | ||
| Second Component | 1-Butyloctylbenzene | |||
| 1-Butylheptylbenzene | ||||
| 1-Methylundecylbenzene | ||||
| Extensimonas | 2,4-Di-tert-butylphenol | |||
| Sphingopyxis | 1-Pentyloctylbenzene | |||
Consequently, this overlap between the first component OTUs for the sPLS-DA and DIABLO 1 allowed us to interpret the discriminating metadata selected for the first component in DIABLO 1 as the ones that were the most representative of the differences between CH and COV. These included moisture, naphthalene and dibenzo(a,h)anthracene, cobalt and iron. Given that moisture content between the two sites is significantly different – the average moisture in COV samples was 19.5% ± 0.8 (95% confidence interval) and the CH average moisture was 8.1% ± 1.8 (95% confidence interval) – it is likely to be a strong driver for bacterial community composition. Naphthalene and dibenzo(a,h)anthracene were not the PAHs with the highest concentrations in the samples but were selected statistically to represent low molecular weight and high molecular weight PAHs, respectively, as markers of the difference in PAH concentration between the two sites. Similarly, the heavy metal concentrations were much higher in COV samples than in CH samples and this is reflected in the presence of iron and cobalt as significant contributors to the first component. Noteworthy was the fact that D8-naphthalene was the surrogate with the lowest repeatability and therefore, although naphthalene in an aged contaminated soil is less likely to evaporate than a freshly spiked surrogate during extraction, the concentration of naphthalene was likely to be underestimated in our sample and this should be addressed in the future considering its importance in the analysis.
A second DIABLO analysis (DIABLO 2) (Fig. 6) was carried out introducing to the previous dataset, the 58 compounds that were found in all samples by alignment of the GC × GC data. For the first component, once again Group A′ was selected as discriminant features along with the same metadata features. Two-dimensional hierarchical clustering (2D-HCA) of the samples and variables for the first and the second components in all three cases (Fig. 4c, 5b and 6b) presented ideal site separation of the samples and the Group A′ OTUs and the metadata that got selected for the first component drove this clustering.
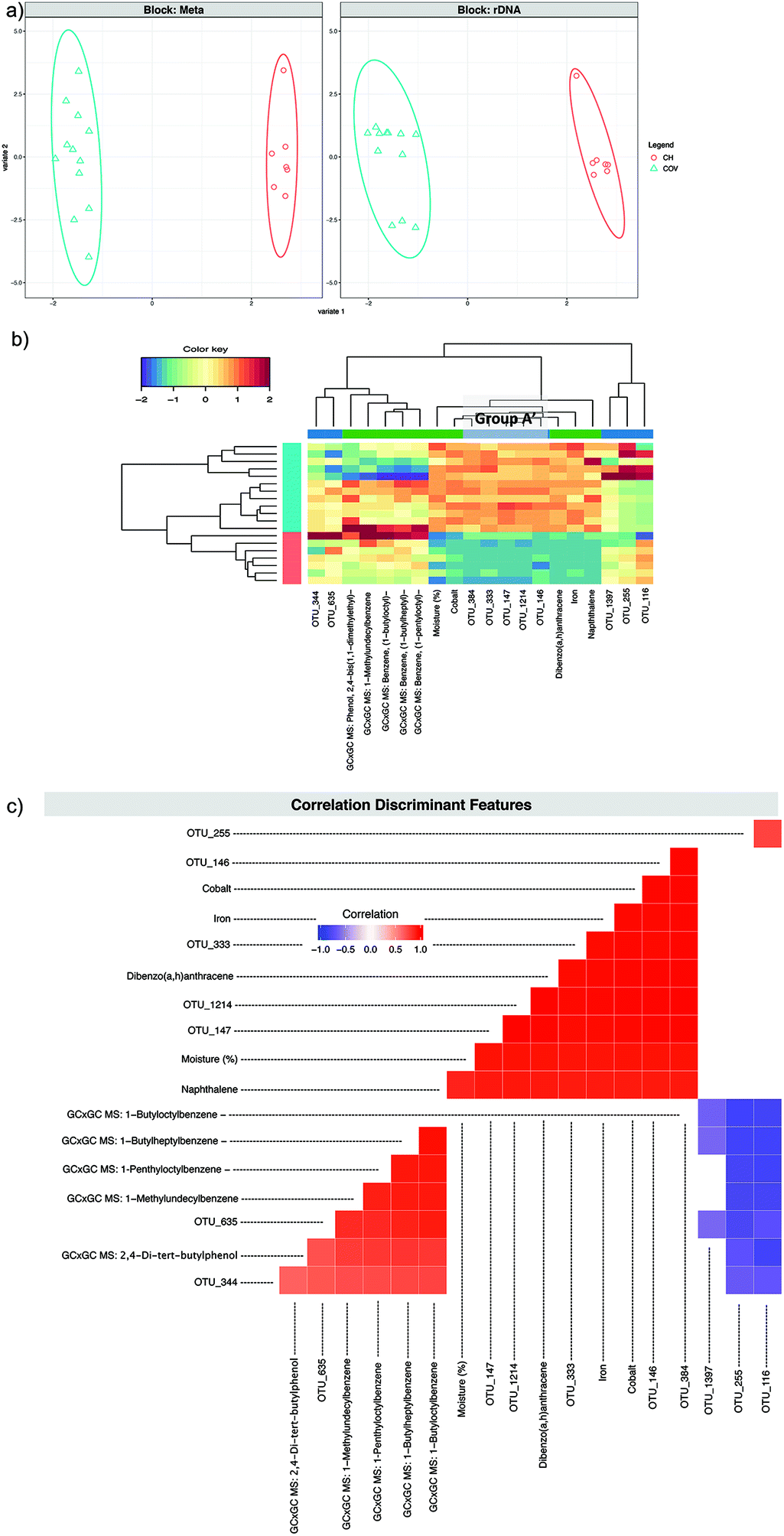 | ||
| Fig. 6 DIABLO 2. (a) The algorithm found two components reducing the classification error rates in the DIABLO algorithm and shows the ordination of samples with ellipses representing 95% confidence interval and percentage variations explained by these components in axes labels for both microbiome (Block: rDNA) and meta data (Block: Meta). (b) shows the heatmaps of these discriminant features, with both rows and columns ordered using hierarchical (average linkage) clustering to identify blocks of features of interest. Group A′ (Table 2) is indicated in a grey box. (c) shows the significant correlations (−0.6 < R > 0.6) between the features as calculated by the algorithm. | ||
Pairwise correlations (Fig. 5c and 6c) showed that the discriminating features from the first component (in both DIABLO 1 and DIABLO 2) were also highly correlated to each other, demonstrating the link between the microbiome and the stressors most responsible for the difference between these sites.
Since all three discriminatory analyses above rank the components based on percentage variability explained by the components, the features selected in lower components (i.e. the second component) serve as a cue to elucidate finer differences between samples as opposed to the first component.
In the sPLS-DA analysis, while two of the OTUs amongst the five OTUs selected for the second component (Table 2) are clearly more abundant in one site than the others (OTU_3 in CH and OTU_650 in COV), the three others have more nuanced distribution. OTUs 683 and 498 are more abundant in COV overall but are also present in high abundance in some CH samples, while OTU_164 is more abundant in CH on average but is abundant in some COV samples. These three OTUs that are significantly present in both samples might be indicative of change of principal function of the communities as the hydrocarbon distribution changes. OTU_650 was identified as belonging to the genus of Stenotrophomonas (gammaproteobacteria), which have been found to be PAH degraders.21 OTU_3 belongs to the genus Sandaracinobacter, a genus of aerobic anoxygenic phototrophic extremophiles.22 OTU_683 is a member of the Methylobacteriaceae family, some strains of which are known to exhibit high tolerance to heavy metal contamination and have been used beneficially in the bioremediation of contaminated environment.23,24
OTU_683 was the only OTU from the second component of the sPLS-DA to also be selected for the second component in DIABLO 1. Three other OTUs (124, 15 and 448) selected for the second component in DIABLO 1 were significantly and positively correlated with each other and with the (log10) qPCR results for Gram-negative PAH and alkane degraders. The same three OTUs correlated negatively with OTU_683 and with cadmium (Fig. 5c). The abundance of the aforementioned OTUs seems to drive the clustering within the COV samples in the 2D-HCA, demonstrating two possible regimes of hydrocarbon degradation, which, however, do not appear to be related to the concentration of PAHs. OTUs 124, 15 and 448 all belong to known aromatic hydrocarbon degrading genera of Achromobacter, Pseudoxanthomonas and Sinorhizobium.25–29 The abundance of these OTUs was low in samples from COV where cadmium concentration was high, which appeared to favour OTU_683. In the DIABLO 2 analysis, the discriminating features of the second component were entirely different than those of DIABLO 1. The five OTUs formed two distinct groups negatively correlated to each other: OTUs 635 and 344 lying in one group and OTUs 116, 255 and 1397 lying in the other group. The latter all belong to the Sphingomonadales and were also negatively correlated to the discriminating metadata features: four long chain branched alkylbenzenes and 2,4-di-tert-butylphenol (Fig. 6c). The abundance of these chemicals also appeared to upregulate the abundance of OTUs 635 and 344. OTU_344 was identified as belonging to the Extensimonas genus, aerobic chemoorganotrophs that have been isolated before from wastewater.30 These highlighted again two different regimes in the COV samples, depending this time on the abundance of hydrocarbons that were not PAHs. Samples in COV with high abundance of these hydrocarbons clustered closer to the CH samples (in which the abundances were high too) than to other COV samples. The integration of the GC × GC data does not affect the first component, demonstrating that the differences in moisture level, heavy metals and PAH concentrations explains the differences between the two sites more than the differences in the exhaustive SVOC signatures. It highlighted, however, through the second component, the influence on the autochthonous microbiome of compounds that would not have normally been measured or taken in consideration during site investigation.
4. Conclusions
Our results demonstrated the usefulness of “multi-omics” approaches in the context of contaminated soils to correlate the abundance of chemical contaminants to the abundance of microbial OTUs. At the same time, utilising the ecological principles, we have highlighted the deterministic nature of microbial communities, dependent on the presence of chemical contaminants. Whilst the discriminant analysis approaches, sPLS-DA and DIABLO, adopted in this study give a reduction of large microbial feature space to the subset of species that form an association with the chemical contaminants and other meta data, care must be taken to interpret causality, primarily because we have only considered sites with spatial variabilities and the patterns found were predominantly inter-site discriminants. It was already successful, however, in demonstrating the efficacy of qPCR analysis, where causality is already known, as a rapid screening tool for hydrocarbon biodegradation potential on a site. This preliminary study has been useful in firstly validating our experimental methods such as exhaustive SVOC and DNA extraction from highly contaminated soil samples and evaluating our in silico pipelines for data processing, highlighting notably the potential of the R2DGC free package for alignment of GC × GC peak tables but also its limitations and those of peak picking algorithms, which will need improving for true comprehensive studies. To give the mechanistic underpinnings, a thorough exploration such as in the context of time series microcosms is required as well as subsequent carefully designed experiments that are informed by the findings of this study. Furthermore, the metabolic potential of the microbial community in this study is explored through a proxy method (Tax4Fun) dependent on the availability of the reference pathway database. Although approaches such as shotgun metagenomics give the actual metabolic potential, we have demonstrated that with high representation of taxa from contaminated soils in the reference database, Tax4Fun offers a cost-effective solution with reasonable resolution for biodegradation pathways. Thus, for contaminated sites, coupling microbial community surveys using 16S rRNA with GC × GC MS offers a whole that is greater than the sum of its parts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Felipe Sepúlveda Olea and Gillian MacKinnon from the Scottish Universities Environmental Research Centre for transition metal quantification and Diana Guillen Ferrari from Heriot-Watt University for carrying out the qPCR analysis. We are also thankful to Scottish Crucible and the RSE for funding through the project “Describing multiple contaminants biodegradation in soil using comprehensive two-dimensional gas chromatography coupled with intelligent data analysis” and to NERC for Umer Z. Ijaz’s Independent Research Fellowship NE/L011956/1. EPSRC is also acknowledged for financial support with grant EP/K038885/1.References
- T. J. Aspray, D. J. C. Carvalho and J. C. Philp, Application of soil slurry respirometry to optimise and subsequently monitor ex situ bioremediation of hydrocarbon-contaminated soils, Int. Biodeterior. Biodegrad., 2007, 60, 279–284 CrossRef.
- T. Aspray, A. Gluszek and D. Carvalho, Effect of nitrogen amendment on respiration and respiratory quotient (RQ) in three hydrocarbon contaminated soils of different type, Chemosphere, 2008, 72, 947–951 CrossRef CAS PubMed.
- S. Yang, et al., Hydrocarbon degraders establish at the costs of microbial richness, abundance and keystone taxa after crude oil contamination in permafrost environments, Sci. Rep., 2016, 6, 37473 CrossRef CAS PubMed.
- M. Crampon, J. Bodilis and F. Portet-Koltalo, Linking initial soil bacterial diversity and polycyclic aromatic hydrocarbons (PAHs) degradation potential, J. Hazard. Mater., 2018, 359, 500–509 CrossRef CAS PubMed.
- L. A. McGregor, et al., Ultra resolution chemical fingerprinting of dense non-aqueous phase liquids from manufactured gas plants by reversed phase comprehensive two-dimensional gas chromatography, J. Chromatogr. A, 2011, 1218, 4755–4763 CrossRef CAS PubMed.
- D. Mao, et al., Detailed analysis of petroleum hydrocarbon attenuation in biopiles by high-performance liquid chromatography followed by comprehensive two-dimensional gas chromatography, J. Chromatogr. A, 2009, 1216, 1524–1527 CrossRef CAS PubMed.
- C. Gauchotte-Lindsay, P. Richards, L. A. McGregor, R. Thomas and R. M. Kalin, A one-step method for priority compounds of concern in tar from former industrial sites: Trimethylsilyl derivatisation with comprehensive two-dimensional gas chromatography, J. Chromatogr. A, 2012, 1253, 154–163 CrossRef CAS PubMed.
- L. A. McGregor, C. Gauchotte-Lindsay, N. Nic Daeid, R. Thomas and R. M. Kalin, Multivariate Statistical Methods for the Environmental Forensic Classification of Coal Tars from Former Manufactured Gas Plants, Environ. Sci. Technol., 2012, 46, 3744–3752 CrossRef CAS PubMed.
- D. Mao, et al., Estimation of ecotoxicity of petroleum hydrocarbon mixtures in soil based on HPLC–GC × GC analysis, Chemosphere, 2009, 77, 1508–1513 CrossRef CAS PubMed.
- F. Rohart, B. Gautier, A. Singh and K.-A. L. Cao, mixOmics: An R package for ‘omics feature selection and multiple data integration, PLoS Comput. Biol., 2017, 13, e1005752 CrossRef PubMed.
- R. C. Ramaker, E. R. Gordon and S. J. Cooper, R2DGC: threshold-free peak alignment and identification for 2D gas chromatography-mass spectrometry in R, Bioinformatics, 2018, 34(10), 1789–1791 CrossRef CAS PubMed.
- K. Kloos, J. C. Munch and M. Schloter, A new method for the detection of alkane-monooxygenase homologous genes (alkB) in soils based on PCR-hybridization, J. Microbiol. Methods, 2006, 66, 486–496 CrossRef CAS PubMed.
- dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering|Bioinformatics|Oxford Academic, available at https://academic.oup.com/bioinformatics/article/31/22/3718/240978, accessed 13th February 2019.
- phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data, available at https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0061217, accessed 13th February 2019.
- S. W. Kembel, et al., Picante: R tools for integrating phylogenies and ecology, Bioinformatics, 2010, 26, 1463–1464 CrossRef CAS PubMed.
- J. C. Stegen, X. Lin, A. E. Konopka and J. K. Fredrickson, Stochastic and deterministic assembly processes in subsurface microbial communities, ISME J., 2012, 6, 1653–1664 CrossRef CAS PubMed.
- X. Wei, et al., MetPP: a computational platform for comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry-based metabolomics, Bioinformatics, 2013, 29, 1786–1792 CrossRef CAS PubMed.
- D. Ghosal, S. Ghosh, T. K. Dutta and Y. Ahn, Current State of Knowledge in Microbial Degradation of Polycyclic Aromatic Hydrocarbons (PAHs): A Review, Front. Microbiol., 2016, 7, 1369 Search PubMed.
- K. L. Londry, P. M. Fedorak and J. M. Suflita, Anaerobic Degradation of m-Cresol by a Sulfate-Reducing Bacterium, Appl. Environ. Microbiol., 1997, 63, 6 Search PubMed.
- M. Sperfeld, C. Rauschenbach, G. Diekert and S. Studenik, Microbial community of a gasworks aquifer and identification of nitrate-reducing Azoarcus and Georgfuchsia as key players in BTEX degradation, Water Res., 2018, 132, 146–157 CrossRef CAS PubMed.
- S. Gao, et al., Multiple degradation pathways of phenanthrene by Stenotrophomonas maltophilia C6, Int. Biodeterior. Biodegrad., 2013, 79, 98–104 CrossRef CAS PubMed.
- V. Yurkov and E. Hughes, Aerobic Anoxygenic Phototrophs: Four Decades of Mystery, in Modern Topics in the Phototrophic Prokaryotes: Environmental and Applied Aspects, ed. P. C. Hallenbeck, Springer International Publishing, 2017, pp. 193–214, DOI:10.1007/978-3-319-46261-5_6.
- D. P. Kelly, I. R. McDonald and A. P. Wood, The Family Methylobacteriaceae, in The Prokaryotes: Alphaproteobacteria and Betaproteobacteria, ed. E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt and F. Thompson, pp. 313–340, Springer Berlin Heidelberg, 2014, DOI:10.1007/978-3-642-30197-1_256.
- P. De Marco, C. C. Pacheco, A. R. Figueiredo and P. Moradas-Ferreira, Novel pollutant-resistant methylotrophic bacteria for use in bioremediation, FEMS Microbiol. Lett., 2004, 234, 75–80 CrossRef CAS PubMed.
- J.-S. Seo, Y.-S. Keum, R. M. Harada and Q. X. Li, Isolation and Characterization of Bacteria Capable of Degrading Polycyclic Aromatic Hydrocarbons (PAHs) and Organophosphorus Pesticides from PAH-Contaminated Soil in Hilo, Hawaii, J. Agric. Food Chem., 2007, 55, 5383–5389 CrossRef CAS PubMed.
- A.-M. Tanase, R. Ionescu, I. Chiciudean, T. Vassu and I. Stoica, Characterization of hydrocarbon-degrading bacterial strains isolated from oil-polluted soil, Int. Biodeterior. Biodegrad., 2013, 84, 150–154 CrossRef CAS.
- Y.-S. Keum, J.-S. Seo, Y. Hu and Q. X. Li, Degradation pathways of phenanthrene by Sinorhizobium sp. C4, Appl. Microbiol. Biotechnol., 2006, 71, 935–941 CrossRef CAS PubMed.
- J. M. Kim, et al., Influence of Soil Components on the Biodegradation of Benzene, Toluene, Ethylbenzene, and o-, m-, and p-Xylenes by the Newly Isolated Bacterium Pseudoxanthomonas spadix BD-a59, Appl. Environ. Microbiol., 2008, 74, 7313–7320 CrossRef CAS PubMed.
- D. R. Nielsen, P. J. McLellan and A. J. Daugulis, Direct estimation of the oxygen requirements of Achromobacter xylosoxidans for aerobic degradation of monoaromatic hydrocarbons (BTEX) in a bioscrubber, Biotechnol. Lett., 2006, 28, 1293–1298 CrossRef CAS PubMed.
- A. Willems, The Family Comamonadaceae, in The Prokaryotes: Alphaproteobacteria and Betaproteobacteria, ed. E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt and F. Thompson, Springer Berlin Heidelberg, 2014, pp. 777–851, DOI:10.1007/978-3-642-30197-1_238.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9fd00020h |
| ‡ Now at: ERS Ltd, Westerhill Road, Bishopbriggs, Glasgow G64 2QH, UK. |
| This journal is © The Royal Society of Chemistry 2019 |