 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Z-scheme photocatalyst systems employing Rh- and Ir-doped metal oxide materials for water splitting under visible light irradiation†
Akihiko
Kudo
 *ab,
Shunya
Yoshino
a,
Taichi
Tsuchiya
a,
Yuhei
Udagawa
a,
Yukihiro
Takahashi
a,
Masaharu
Yamaguchi
a,
Ikue
Ogasawara
a,
Hiroe
Matsumoto
a and
Akihide
Iwase
ab
*ab,
Shunya
Yoshino
a,
Taichi
Tsuchiya
a,
Yuhei
Udagawa
a,
Yukihiro
Takahashi
a,
Masaharu
Yamaguchi
a,
Ikue
Ogasawara
a,
Hiroe
Matsumoto
a and
Akihide
Iwase
ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: a-kudo@rs.kagu.tus.ac.jp
bPhotocatalysis International Research Center, Research Institute for Science and Technology, Tokyo University of Science, 2641, Yamazaki, Noda-shi, Chiba-ken 278-8510, Japan
First published on 25th January 2019
Abstract
Various types of Z-scheme systems for water splitting under visible light irradiation were successfully developed by employing Rh- and Ir-doped metal oxide powdered materials with relatively narrow energy gaps (EG): BaTa2O6:Ir,La (EG: 1.9–2.0 eV), NaTaO3:Ir,La (EG: 2.1–2.3 eV), SrTiO3:Ir (EG: 1.6–1.8 eV), NaNbO3:Rh,Ba (EG: 2.5 eV) and TiO2:Rh,Sb (EG: 2.1 eV), with conventional SrTiO3:Rh (an H2-evolving photocatalyst) or BiVO4 (an O2-evolving photocatalyst), and suitable electron mediators. The Z-scheme systems were classified into three groups depending on the combination of H2- and O2-evolving photocatalysts and electron mediator. The Z-scheme systems combining BaTa2O6:Ir,La with BiVO4, and NaTaO3:Ir,La with BiVO4 were active when a [Co(bpy)3]3+/2+ redox couple was used rather than an Fe3+/2+ one. The combination of SrTiO3:Ir with SrTiO3:Rh gave an activity when the [Co(bpy)3]3+/2+ and Fe3+/2+ redox couple ionic mediators were used. The Z-scheme systems combining NaNbO3:Rh,Ba and TiO2:Rh,Sb with SrTiO3:Rh showed activities by using the [Co(bpy)3]3+/2+ and Fe3+/2+ redox couples and also via interparticle electron transfer by just contact with/without reduced graphene oxide (RGO). These suitable combinations can be explained based on the impurity levels of doped Rh3+ and Ir3+ toward the redox potentials of the ionic mediators for the Z-scheme systems employing ionic mediators, and p-/n-type and onset potentials of the photocurrent in the photoelectrochemical properties of those photocatalyst materials for the Z-scheme systems working via interparticle electron transfer.
Introduction
Artificial photosynthesis has attracted attention from the view point of solar energy conversion to storable chemical energy. Because solar water splitting is one of the representative reactions, photocatalytic water splitting has extensively been studied.1–7 Powder-based metal oxide materials are attractive for photocatalytic water splitting because the cost will be low8–10 and stability will be high compared with other materials such as chalcogenides. Although the efficiency of powder-based photocatalyst systems is behind that of systems of photovoltaic + electrolysis at the present stage, there are some advantages to powder-based photocatalyst systems.It is crucial to demonstrate a solar water splitting system employing powder-based oxide photocatalysts. The present stage of research of artificial synthesis is along this topic. A reactor system including a gas separation system of evolved hydrogen from oxygen has been studied in addition to the development of photocatalyst materials aimed towards the practical use of solar water splitting.10 The separation system can be achieved by the use of a suitable separation membrane. Moreover, the safety issue of the co-evolved H2 and O2 has also been examined via the use of a suitable gas transportation tube. However, even if an excellent reactor system with a gas separation membrane system is established, which photocatalyst is employed for it is still a key issue as high efficient photocatalysts for real solar water splitting into H2 and O2 without any sacrificial electron donors and acceptors have not yet been developed. It is essential to develop an efficient photocatalyst for demonstrating a solar water splitting system taking gas separation and safety issues into account.
High efficiency of a photocatalyst can be brought about by a high quantum yield and a response to light with a long wavelength. From this viewpoint, it is vital to develop photocatalysts that can utilize up to 600–700 nm of the solar spectrum. Although the use of noble metals might prevent the photocatalyst materials from practical use, the usage would be allowed if the amount is small and the materials can be recycled.
There are single particulate and Z-scheme systems in powder-based photocatalysts.2,11 Z-scheme photocatalyst systems have the advantage that photocatalysts active for either H2 or O2 evolution can be employed. This means that a Z-scheme system can be constructed with various combinations of H2- and O2-evolving photocatalysts.12–15 SrTiO3:Rh (an H2-evolving photocatalyst) and BiVO4 (an O2-evolving photocatalyst) are representative metal oxide materials for the Z-scheme system.1,2,16 It should be stressed that photocatalyst sheets prepared by a particle transfer method using SrTiO3:Rh,La and BiVO4:Mo powders demonstrate a quantum efficiency of 30% at 420 nm and a solar to hydrogen energy conversion efficiency of 1%.17–19 This result suggests that the Z-scheme system is a promising photocatalyst system for practical solar water splitting. H2 evolution separated from O2 evolution is possible in the Z-scheme system if a suitable reactor is designed.8 However, this photocatalyst sheet consisting of the SrTiO3:Rh,La and BiVO4:Mo powders responds up to 520 nm because the energy gap (EG) of SrTiO3:Rh,La and the band gap (BG) of BiVO4:Mo are 2.3 eV and 2.4 eV, respectively. So, it is a key issue to develop Z-scheme photocatalyst systems consisting of metal oxide photocatalysts with a response at longer wavelengths than 520 nm.
Transition metal doping is one strategy to make wide band gap photocatalysts responsive to visible light.1,2 Rh and Ir are effective dopants in this strategy. We have reported that SrTiO3:Rh (EG: 2.3 eV),20 SrTiO3:Rh,Sb (EG: 2.2–2.4 eV),21 SrTiO3:Ir (EG: 1.6–1.8 eV),20,22 BaTa2O6:Ir,La (EG: 1.9–2.0 eV)23 and NaTaO3:Ir,La (EG: 2.1–2.3 eV)24 are active for sacrificial H2 evolution in the presence of electron donors such as methanol, while SrTiO3:Rh,Sb,21 SrTiO3:Ir20 and TiO2:Rh,Sb (EG: 2.1 eV)25 are active for sacrificial O2 evolution in the presence of electron acceptors such as Ag+. In these photocatalyst materials, the visible light responses are due to electronic transition from the impurity levels formed by the dopants to the conduction bands of the host materials. These energy and band gaps are close to, or narrower than, those of SrTiO3:Rh (EG: 2.3 eV)20 and BiVO4 (BG: 2.4 eV).26,27 So, it is attractive to utilize these Rh- and Ir-doped metal oxide photocatalysts for the construction of Z-scheme systems.
There are several types of Z-scheme system employing different electron mediators such as Fe3+/2+ and [Co(bpy)3]3+/2+ redox couples,16,28,29 and reduced graphene oxide (RGO)30–32 as shown in Fig. 1(a) and (b). Moreover, some Z-scheme systems work even without electron mediators, as shown in Fig. 1(c).33,34 In this case, electron transfer proceeds through an interface contacted between the particles of the H2- and O2-evolving photocatalysts. So, it is important to examine the electron mediator in the Z-scheme system.
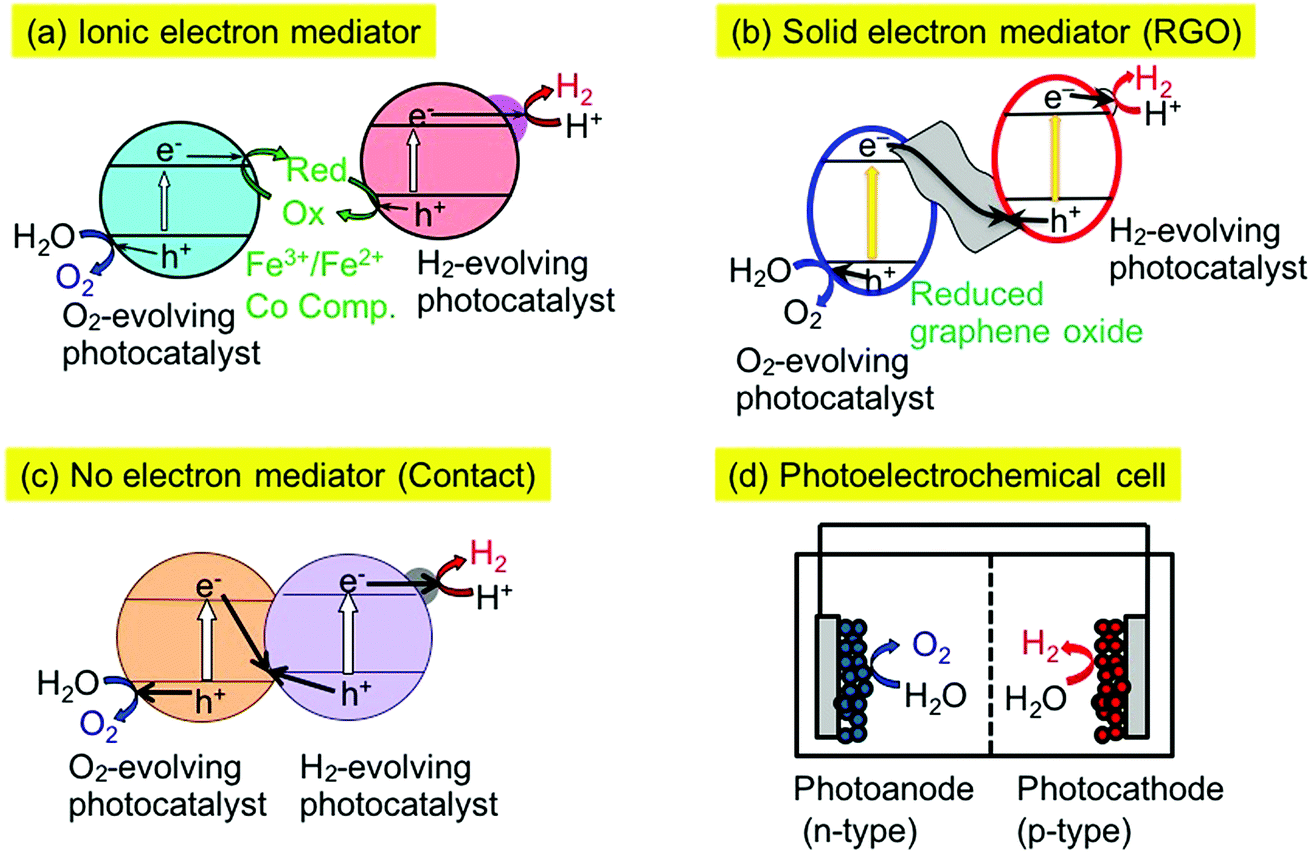 | ||
| Fig. 1 Various types of powder material-based Z-scheme systems for water splitting. | ||
In the present paper, sacrificial H2 and O2 evolutions over Rh- and Ir-doped metal oxide photocatalysts under visible light irradiation were examined first to see the relationship between the photocatalytic properties and their band structures. Then, these H2- and O2-evolving photocatalysts were employed for various types of Z-scheme system for water splitting into H2 and O2 in stoichiometric amounts under visible light irradiation, without any sacrificial reagents. The photocatalytic performances for the sacrificial H2 and O2 evolutions and the Z-schematic water splitting were discussed based on the band structures and photoelectrochemical properties of the photocatalyst materials such as p-/n-types and the onset potentials of photocurrents.
Experimental
Preparation of Rh- or Ir-doped metal oxide photocatalysts
SrTiO3:Rh(1%),29 SrTiO3:Ir(0.2%),22 TiO2:Rh(x%),Sb(2x%) (x = 0.5 or 1.3),25 NaTaO3:Ir(1%),La(2%),24 BaTa2O6:Ir(1%),La(2%)23 and BiVO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26,27 were prepared by a solid-state reaction, a borate-flux method, and a liquid–solid reaction according to previous reports. In addition to them, NaNbO3:Rh(x%),Ba(y%) (x, y) = (1.2, 1.44) or (1.0, 2.0) was newly prepared by a solid-state reaction. The starting materials, Na2CO3 (Kanto Chemical; 99.5 or 99.8%), Nb2O5 (Kanto Chemical; 99.99% or Kojundo Chemical; 99.99%), Rh2O3 (Wako Chemical; 98%), and BaCO3 (Kanto Chemical; 99%), were mixed at a molar ratio of Na/Nb/Rh/Ba = 1.05–1.05y:1 − x:x:y. An excess of sodium was added in the starting materials to compensate for volatilization. The starting materials mixture was calcined at 1173 K for 1 h, and then 1423–1473 K for 10 h once or twice. The excess sodium was washed out with water after the calcination. The obtained powders had nonspecific shapes with aggregations, judging from the SEM images (Jeol; JSM-6700F) (Fig. S1†). The obtained samples were identified using X-ray diffraction (Rigaku; MiniFlex, Cu Kα). Diffuse reflectance spectra were obtained by a UV-vis-NIR spectrometer (JASCO, V-570) equipped with an integrator sphere and were converted to absorbance measurements via the Kubelka–Munk method.
26,27 were prepared by a solid-state reaction, a borate-flux method, and a liquid–solid reaction according to previous reports. In addition to them, NaNbO3:Rh(x%),Ba(y%) (x, y) = (1.2, 1.44) or (1.0, 2.0) was newly prepared by a solid-state reaction. The starting materials, Na2CO3 (Kanto Chemical; 99.5 or 99.8%), Nb2O5 (Kanto Chemical; 99.99% or Kojundo Chemical; 99.99%), Rh2O3 (Wako Chemical; 98%), and BaCO3 (Kanto Chemical; 99%), were mixed at a molar ratio of Na/Nb/Rh/Ba = 1.05–1.05y:1 − x:x:y. An excess of sodium was added in the starting materials to compensate for volatilization. The starting materials mixture was calcined at 1173 K for 1 h, and then 1423–1473 K for 10 h once or twice. The excess sodium was washed out with water after the calcination. The obtained powders had nonspecific shapes with aggregations, judging from the SEM images (Jeol; JSM-6700F) (Fig. S1†). The obtained samples were identified using X-ray diffraction (Rigaku; MiniFlex, Cu Kα). Diffuse reflectance spectra were obtained by a UV-vis-NIR spectrometer (JASCO, V-570) equipped with an integrator sphere and were converted to absorbance measurements via the Kubelka–Munk method.
Preparation of an RGO–metal oxide composite
An RGO-incorporated O2-evolving photocatalyst was prepared by photocatalytic reduction of graphene oxide (GO) on the photocatalyst according to previous reports.30–32 GO prepared by the Hummers’ method35 and the O2-evolving photocatalyst were dispersed in an aqueous methanol (Kanto Chemical; 99.8%) solution (50 vol%). The suspension was irradiated with visible light from a 300 W Xe lamp (PerkinElmer; CERMAX PE300BF) with a long pass filter (HOYA; L42) under a N2 atmosphere with a pressure of 1 atm to obtain the RGO–photocatalyst composite. The methanol was carefully removed by washing with water. The RGO–photocatalyst composite was collected by filtration and was dried at room temperature in air.Sacrificial H2 and O2 evolutions (half reactions of water splitting)
H2 and O2 evolutions from aqueous solutions containing the sacrificial reagents CH3OH (Kanto Chemical; 99.8%) and AgNO3 (Kojima Chemical; 99.9% or Toyo Chemical; 99.9%) that were half reactions of water splitting were examined using a top-irradiation reaction cell with a Pyrex window and a 300 W Xe lamp (PerkinElmer; CERMAX PE300BF). NaTaO3:Ir,La, BaTa2O6:Ir,La, NaNbO3:Rh,Ba, and TiO2:Rh,Sb were used as prepared, whereas SrTiO3:Ir without a cocatalyst was reduced under 1 atm of H2 at 473 K for 2 h as a pretreatment for sacrificial O2 evolution. The photocatalyst powders (0.1–0.3 g) were suspended in aqueous solutions (120–150 mL) and irradiated with visible light. For the H2 evolution, Pt (0.3 wt%) cocatalyst, working as an H2 evolution site, was loaded on photocatalysts by photodeposition from an aqueous methanol solution containing H2PtCl6 (Tanaka Kikinzoku; 37.55% as Pt). The wavelength of the irradiation light was controlled to visible light using long-pass filters (HOYA; L42 and Y44). The amounts of evolved H2 and O2 were determined using an online gas chromatograph (Shimadzu; GC-8A, MS-5A column, TCD, Ar carrier).Z-schematic water splitting
Z-schematic water splitting was conducted using a gas-closed system with a top-irradiation cell with a Pyrex window. H2-evolving photocatalyst and O2-evolving photocatalyst powders (0.05 or 0.1 g, respectively) were suspended in 120 mL of water. For the interparticle Z-scheme systems without an electron mediator and with an RGO solid-state electron mediator, water not containing any ionic mediators was used. For the Z-scheme system with ionic mediator, an aqueous solution containing [Co(bpy)3]SO4 or FeCl3 as a mediator was used. The pH was adjusted with H2SO4 in each of the Z-scheme systems with and without electron mediator, if necessary. Ru (0.7 wt%) cocatalyst, functioning as an H2 evolution site, was loaded on SrTiO3:Rh (an H2-evolving photocatalyst) by photodeposition from an aqueous methanol solution containing RuCl3·nH2O (Tanaka Kikinzoku; 36% as Ru in RuCl3·nH2O). Pt (0.3–1 wt%) was loaded on the NaTaO3:Ir,La and BaTa2O6:Ir,La (H2-evolving photocatalysts), and SrTiO3:Ir (an O2-evolving photocatalyst) by an impregnation method. The photocatalyst powders and an aqueous H2PtCl6 solution were placed in a porcelain crucible and dried on a hot plate. The H2PtCl6-impregnated NaTaO3:Ir,La and BaTa2O6:Ir,La powders were calcined at 673 K for 2 h in air, whereas the SrTiO3:Ir was not. The Pt-loaded NaTaO3:Ir,La and Pt-loaded BaTa2O6:Ir,La were subsequently reduced at 673 K for 1 h under 1 atm of H2 as a pretreatment, while Pt-loaded SrTiO3:Ir was reduced at 573 K for 1 h. The light source and GC setup were the same as those for the sacrificial H2 and O2 evolutions.Photoelectrochemical measurements
A squeegee method was used to prepare the SrTiO3:Rh(1%) photoelectrode and a drop-casting method was used for the NaTaO3:Ir(1%),La(2%) and BaTa2O6:Ir(1%),La(2%) with and without H2-reduction; SrTiO3:Ir(0.2%) with H2-reduction; NaNbO3:Rh(1%),Ba(2%) and TiO2:Rh(0.5%),Sb(1%) without H2-reduction, and BiVO4 photoelectrodes using powdered photocatalyst materials. For the SrTiO3:Rh(1%) photoelectrode, a paste consisting of 20 mg of SrTiO3:Rh(1%) photocatalyst powder, 20 μL of acetylacetone (Kanto Chemical; 99.5%) and 40 μL of distilled water was coated on an indium tin oxide transparent electrode (ITO).36 For the other photoelectrodes, the photocatalyst powders were dispersed in ethanol (1–2 mg mL−1) by sonication. The suspensions were drop-cast onto a fluorine-doped tin oxide transparent electrode (FTO) to obtain 1–2 mg cm−2 of photocatalyst on the FTO. The H2-reduced SrTiO3:Ir-loaded FTO substrate was not calcined, whereas the other photocatalyst-loaded ITO and FTO substrates were calcined at 573–673 K for 2 h in air. The photoelectrochemical properties were evaluated with a three-electrode system consisting of working, Ag/AgCl reference, and Pt counter electrodes with a potentiostat (Hokuto Denko; HZ-series or HSV-110) using a conventional H-type cell with a Nafion membrane. The electrolyte was 0.1 mol L−1 K2SO4. 0.025 mol L−1 KH2PO4 + 0.025 mol L−1 Na2HPO4 pH buffer was added, if necessary. A 300 W Xe lamp (PerkinElmer; CERMAX PE300BF) with a long-pass filter (HOYA; L42) was employed as a light source.Results
Photocatalytic activities for sacrificial H2 and O2 evolutions over Rh- and Ir-doped metal oxide materials and their band structures
Sacrificial H2 and O2 evolutions of half reactions were carried out as test reactions of water splitting over Rh- and Ir-doped metal oxide photocatalysts using a sacrificial electron donor and acceptor to see the ability for photocatalytic H2 or O2 evolution, as shown in Table 1, prior to conducting water splitting.| Photocatalyst | Energy gap/eV | Incident light/nm | Activity/μmol h−1 | Ref. | |
|---|---|---|---|---|---|
| H2b | O2c | ||||
| a Photocatalyst: 0.1–0.3 g; light source: 300 W Xe lamp with long-pass filters (λ > 420 nm or λ > 440 nm); reaction cell: top-irradiation cell with a Pyrex window. b Cocatalyst: Pt (0.3 wt%, photodeposition); reactant solution: 10 vol% aqueous methanol solution. c Cocatalyst: none; reactant solution: 0.02–0.05 mol L−1 aqueous AgNO3 solution (120–150 mL). d Treatment: H2-reduction at 473 K for 2 h. | |||||
| Pt/NaTaO3:Ir(1%),La(2%) | 2.1–2.3 | λ > 420 | 3.2 | — | 24 |
| NaTaO3:Ir(1%),La(2%) | 2.1–2.3 | λ > 420 | — | 0 | 24 |
| Pt/BaTa2O6:Ir(1%),La(2%) | 1.9–2.0 | λ > 420 | 4.6 | — | 23 |
| BaTa2O6:Ir(1%),La(2%) | 1.9–2.0 | λ > 420 | — | 0 | 23 |
| Pt/SrTiO3:Ir(0.2%) | 1.6–1.8 | λ > 440 | 8.6 | — | 20 |
| SrTiO3:Ir(0.2%)d | 1.6–1.8 | λ > 420 | — | 4.3 | 20 |
| Pt/NaNbO3:Rh(1%),Ba(2%) | 2.5 | λ > 420 | 0.2 | — | This work |
| NaNbO3:Rh(1%),Ba(2%) | 2.5 | λ > 420 | — | 5.3 | This work |
| Pt/TiO2:Rh(0.5%),Sb(1%) | 2.1 | λ > 440 | 0 | — | This work |
| TiO2:Rh(0.5%),Sb(1%) | 2.1 | λ > 440 | — | 7.5 | 25 |
The energy gaps were determined from the diffuse reflectance spectra and wavelength dependence of the photocatalytic activities as shown in Fig. 2 and 3. In general, trivalent and tetravalent Rh and Ir species are doped at the Ti4+, Nb5+ and Ta5+ sites in metal oxide materials.20–25,37,38 Among the species, Rh3+ and Ir3+ contribute to the visible light response for metal oxide photocatalysts.20,24,37,38 Sb5+ was codoped with Rh3+ at the Ti4+ and Nb5+ sites for charge compensation to enhance the formation of Rh3+ and Ir3+ and suppress the formation of Rh4+ and Ir4+ as efficient recombination centers between photogenerated electrons and holes, while Ba2+ and La3+ were replaced at the alkali and alkaline earth metal sites for the same purpose. SrTiO3:Ir was reduced with H2 at 473 K to form the dopant Ir3+.
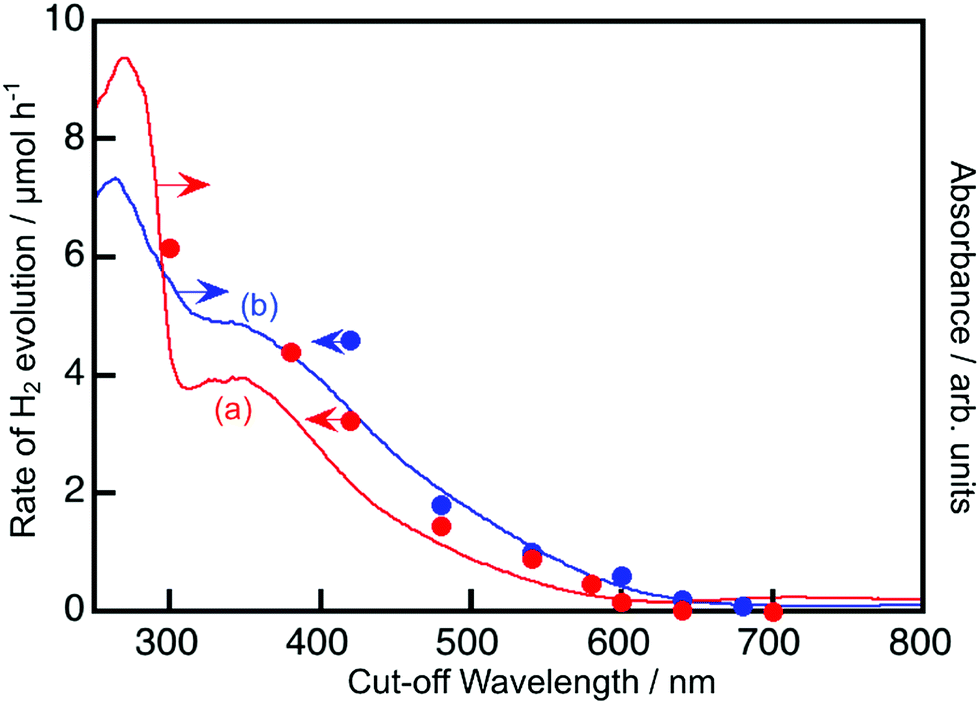 | ||
| Fig. 2 The wavelength dependence of H2 evolution from a 10 vol% aqueous methanol solution (closed circles) and diffuse reflectance spectra (solid line) of (a) NaTaO3:Ir(1%),La(2%) and (b) BaTa2O6:Ir(1%),La(2%). Photocatalyst: 0.1 g, cocatalyst: Pt (photodeposition) for H2 evolution, reactant solution: 120 mL, light source: 300 W Xe lamp with long-pass filters, and reaction cell: top-irradiation cell with a Pyrex window. Samples of diffuse reflectance spectra were reduced at 473 K. | ||
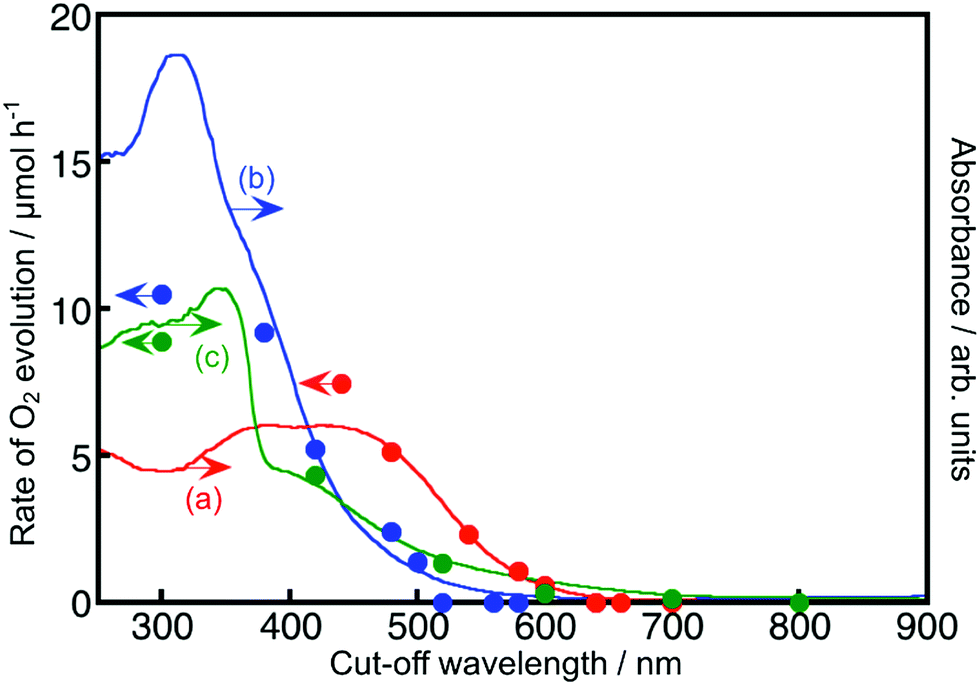 | ||
| Fig. 3 The wavelength dependence of O2 evolution from a 0.05 mol L−1 aqueous silver nitrate solution (closed circles) and diffuse reflectance spectra (solid line) of (a) TiO2:Rh(1.3%),Sb(2.6%), (b) NaNbO3:Rh(1.2%),Ba(1.44%) and (c) SrTiO3:Ir(0.2%) with H2 reduction at 473 K. Photocatalyst: 0.1–0.3 g, reactant solution: 150 mL, light source: 300 W Xe lamp with long-pass filters, and reaction cell: top-irradiation cell with a Pyrex window. | ||
In the case of the Ir-doped photocatalysts, SrTiO3:Ir was active for both the sacrificial H2 and O2 evolutions,20,22 whereas BaTa2O6:Ir,La and NaTaO3:Ir,La were active only for the sacrificial H2 evolution.23,24 BaTa2O6:Ir,La and NaTaO3:Ir,La were not active for sacrificial O2 evolution even if they were reduced with H2 at 673 K, as SrTiO3:Ir was, to reduce the Ir4+ species. The Rh-doped photocatalysts TiO2:Rh,Sb25 and NaNbO3:Rh,Ba were active for sacrificial O2 evolution using Ag+ as an electron acceptor. NaNbO3:Rh,Ba showed very low activity for the sacrificial H2 evolution. These properties will be discussed based on the band structure in the Discussion section. These results of the sacrificial H2 and O2 evolutions suggest that NaTaO3:Ir,La and BaTa2O6:Ir,La can be used as H2-evolving photocatalysts for the construction of Z-scheme systems, while SrTiO3:Ir, NaNbO3:Rh,Ba and TiO2:Rh,Sb are expected to be employed as O2-evolving photocatalysts.
Fig. 2 and 3 show the diffuse reflectance spectra and wavelength dependence of the photocatalytic H2 and O2 evolutions of the Rh- and Ir-doped metal oxide photocatalysts in the presence of sacrificial reagents. The wavelengths were controlled with long-pass filters. It is vital to see the wavelength dependency of the photocatalytic activity because it is not guaranteed that photocatalysts with visible light absorption bands always give activities under visible light irradiation. The onsets of the wavelength dependence agreed with those of the diffuse reflection spectra. The onset wavelengths for the H2 evolutions were 640 and 600 nm for BaTa2O6:Ir,La and NaTaO3:Ir,La, respectively. These onset wavelengths were longer than the 540 nm of SrTiO3:Rh (a conventional H2-evolving photocatalyst). The onset wavelengths for O2 evolution were 600, 500 and 700 nm for TiO2:Rh,Sb, NaNbO3:Rh,Ba and SrTiO3:Ir, respectively. It is noteworthy that TiO2:Rh,Sb and SrTiO3:Ir responded at longer wavelengths than the BiVO4 (BG: 2.4 eV) (a conventional O2-evolving photocatalyst).
Fig. 4 shows the band structures of the Rh- and Ir-doped metal oxide photocatalysts. The impurity levels of Rh3+ and Ir3+ were estimated from the energy gaps determined by diffuse reflection spectra supposing that the valence band consisting of O 2p located at +3 V vs. NHE at a pH of 0.39 The absorption bands in the visible light region shown in Fig. 2 and 3 are due to electronic transition from the impurity levels consisting of Rh3+ and Ir3+ to the conduction bands of the host materials. The impurity levels formed with electron-filled orbitals of Ir3+ were around 1.0–1.2 V for NaTaO3 and BaTa2O6, while Ir3+ in SrTiO3 formed an impurity level around 1.4–1.6 V that was deeper than those in the cases of the tantalates. The energy levels formed with electron-filled orbitals of Rh3+ located around 2.0–2.1 V that were similar to those of SrTiO3:Rh and SrTiO3:Rh,Sb.20,21 The reason Rh3+ forms a deeper impurity level than Ir3+ is due to Ir4+ being more stable than Rh4+ in metal oxides. Therefore, electronic transition from the Ir3+ impurity level to a conduction band is easier than that from Rh3+, resulting in the formation of the shallow impurity level by Ir3+.
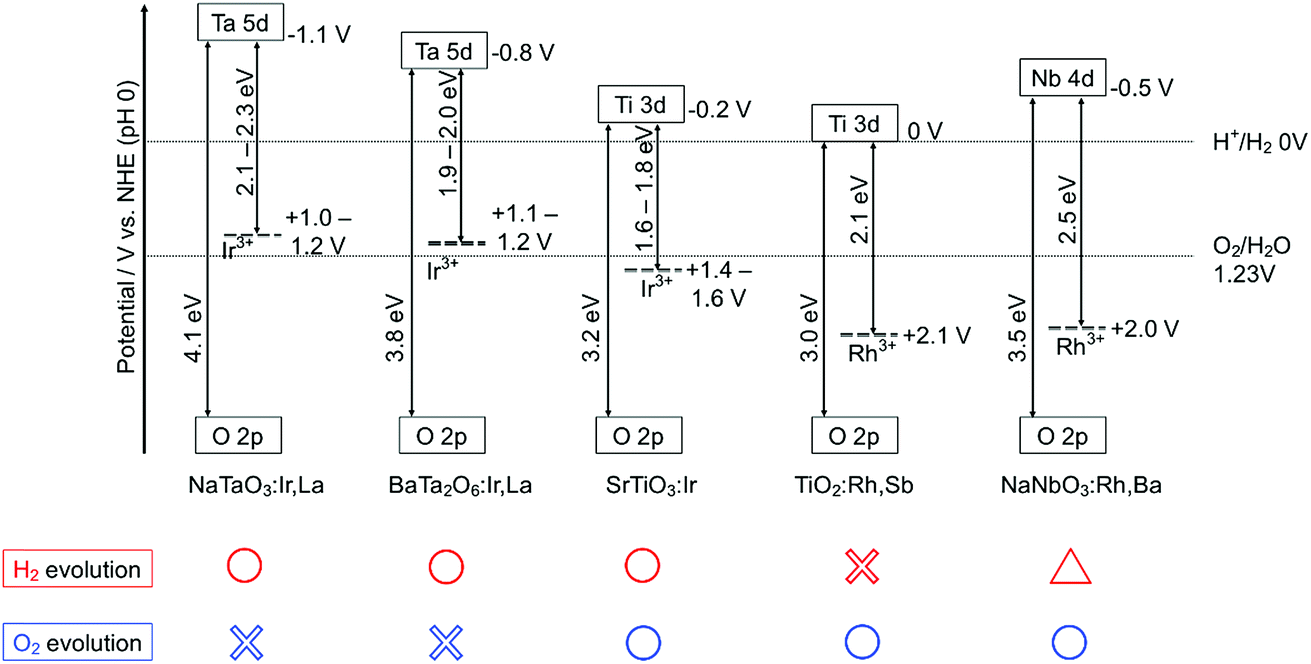 | ||
| Fig. 4 The band structures of Rh- or Ir-doped metal oxide photocatalysts at pH 0. | ||
Z-schematic systems for photocatalytic water splitting employing Rh- and Ir-doped metal oxide materials
The combination of SrTiO3:Rh and BiVO4 photocatalysts can be a benchmark of a Z-scheme photocatalyst system. The Z-schematic water splitting proceeds using Fe3+/2+ and [Co(bpy)3]3+/2+ redox couples as ionic mediators (Fig. 1(a))16,28,29 and also via interparticle electron transfer between SrTiO3:Rh and BiVO4 particles with and without RGO (Fig. 1(b) and (c)).30,33,34 In general, water splitting via Z-schematic interparticle electron transfer by contact between the particles of the H2- and O2-evolving photocatalysts with and without RGO can be achieved (Fig. 1(b) and (c)) when the H2- and O2-evolving photocatalysts satisfy the following two requirements; (i) H2- and O2-evolving photocatalysts possess p- and n-type semiconductor properties, respectively, (ii) there is a certain electrode potential at which the cathodic photocurrent of the p-type semiconductor overlaps with the anodic photocurrent of the n-type semiconductor, being similar to water splitting using a photoelectrochemical cell working with no applied external bias as shown in Fig. 1(d).31,32 Moreover, H2- and O2-evolving photocatalysts must have contact with each other for interparticle electron transfer. In contrast to this, a Z-scheme system employing an ionic electron mediator could work regardless of the p- or n-type properties of the H2- or O2-evolving photocatalysts, if the photocatalysts have potentials for the reduction or oxidation of redox couple ionic mediators and the adsorption abilities for the redox couples. The SrTiO3:Rh and BiVO4 photocatalysts satisfy these factors resulting in all of the Z-scheme systems showing activities for water splitting into H2 and O2 in stoichiometric amounts without any sacrificial reagents (Fig. 1(a)–(d)).Various types of Z-scheme photocatalyst systems employing Ir- and Rh-doped photocatalysts for water splitting into H2 and O2 under visible light irradiation are shown in Table 2. Here, the absolute evaluation of the performance of photocatalyst systems for water splitting is how much H2 and O2 is obtained under certain experimental conditions. In this sense, the activities of the different Z-scheme systems in Table 2 are comparable with each other, but not with those of Table 1, because of the almost identical experimental conditions for the water splitting, even if the kinetics would be different among the systems.
| H2-photocat. | O2-photocat. | Mediator | Initial pH | Activity/μmol h−1 | Ref. | |
|---|---|---|---|---|---|---|
| H2 | O2 | |||||
| a Photocatalyst: 0.05 or 0.1 g each; light source: 300 W Xe lamp with a long-pass filter (λ > 420 nm); reaction cell: top-irradiation cell with a Pyrex window. b Cocatalyst: Pt (0.3 wt%, impregnation at 673 K for 2 h and subsequent H2-reduction at 673 K for 1 h). c Cocatalyst: Ru (0.7 wt%, photodeposition). d Cocatalyst: Pt (1 wt%, impregnation without calcination and subsequent H2-reduction at 573 K for 1 h). e Reactant solution: H2SO4 solution. f Reactant solution: 2 mmol L−1 FeCl3 solution. g Reactant solution: 0.02 mmol L−1. h Reactant solution: 0.5 mmol L−1 [Co(bpy)3]SO4 solution (120 mL). | ||||||
| Pt/NaTaO3:Ir(1%),La(2%)b | BiVO4 | Nonee | 4.2 | Trace | Trace | This work |
| Pt/NaTaO3:Ir(1%),La(2%)b | BiVO4 | RGOe | 4.2 | Trace | Trace | This work |
| Pt/NaTaO3:Ir(1%),La(2%)b | BiVO4 | Fe3+/2+f | 2.4 | Trace | Trace | This work |
| Pt/NaTaO3:Ir(1%),La(2%)b | BiVO4 | [Co(bpy)3]3+/2+g | 4.2 | 0.8 | 0.3 | This work |
| Pt/BaTa2O6:Ir(1%),La(2%)b | BiVO4 | Nonee | 4.2 | 0.9 | 0.4 | This work |
| Pt/BaTa2O6:Ir(1%),La(2%)b | BiVO4 | RGOe | 4.2 | 0.9 | 0.4 | This work |
| Pt/BaTa2O6:Ir(1%),La(2%)b | BiVO4 | Fe3+/2+f | 2.4 | 0.1 | 0.4 | This work |
| Pt/BaTa2O6:Ir(1%),La(2%)b | BiVO4 | [Co(bpy)3]3+/2+g | 4.2 | 5.9 | 2.1 | 22 |
| Ru/SrTiO3:Rh(1%)c | Pt/SrTiO3:Ir(0.2%)d | Nonee | 3.5 | 0.4 | Trace | This work |
| Ru/SrTiO3:Rh(1%)c | Pt/SrTiO3:Ir(0.2%)d | RGOe | 3.5 | 0.8 | Trace | This work |
| Ru/SrTiO3:Rh(1%)c | Pt/SrTiO3:Ir(0.2%)d | Fe3+/2+f | 2.4 | 2.5 | 1.0 | This work |
| Ru/SrTiO3:Rh(1%)c | Pt/SrTiO3:Ir(0.2%)d | [Co(bpy)3]3+/2+h | 7.8 | 5.1 | 2.4 | This work |
| Ru/SrTiO3:Rh(1%)c | NaNbO3:Rh(1%),Ba(2%) | Nonee | 2.4 | 2.1 | 1.1 | This work |
| Ru/SrTiO3:Rh(1%)c | NaNbO3:Rh(1%),Ba(2%) | Fe3+/2+f | 2.4 | 5.7 | 2.3 | This work |
| Ru/SrTiO3:Rh(1%)c | NaNbO3:Rh(1%),Ba(2%) | [Co(bpy)3]3+/2+h | 3.8 | 4.1 | 1.7 | This work |
| Ru/SrTiO3:Rh(1%)c | TiO2:Rh(0.5%),Sb(1%) | Nonee | 3.5 | 5.4 | 2.4 | This work |
| Ru/SrTiO3:Rh(1%)c | TiO2:Rh(0.5%),Sb(1%) | Fe3+/2+f | 2.4 | 12 | 5.3 | This work |
| Ru/SrTiO3:Rh(1%)c | TiO2:Rh(0.5%),Sb(1%) | [Co(bpy)3]3+/2+h | 3.5 | 28 | 13 | This work |
For the construction of Z-scheme systems, BiVO4 (an O2-evolving photocatalyst) was combined with BaTa2O6:Ir,La and NaTaO3:Ir,La (H2-evolving photocatalysts), while SrTiO3:Rh (an H2-evolving photocatalyst) was combined with TiO2:Rh,Sb, NaNbO3:Rh,Ba and SrTiO3:Ir (O2-evolving photocatalysts) as suggested by their H2 and O2 evolution abilities. In addition to the suspension system, the photoelectrochemical properties of photocatalyst powders immobilized on a conducting substrate as shown in Fig. 1(d) were examined using a Pt counter electrode without any sacrificial reagents to see the cathodic or anodic photocurrent and the onset potentials of semiconductor properties, not as a water splitting device, as shown in Fig. 5, in order to consider the potential overlap for Z-schematic water splitting via interparticle electron transfer with and without RGO. It has been reported that SrTiO3:Rh36,40 and BiVO432,41–44 function as a photocathode and photoanode, respectively. NaTaO3:Ir,La showed only an anodic photocurrent. Although BaTa2O6:Ir,La and SrTiO3:Ir showed cathodic and anodic photocurrents, the photocurrents were poor and the onset potentials shifted with the sweeping direction of the CV curves. This result implies that those photocurrents might not be due to H2 and O2 evolutions, but possibly due to redox reactions of the doped Ir species. In contrast, TiO2:Rh,Sb,45 and NaNbO3:Rh,Ba gave clear anodic photocurrents indicating an n-type semiconductor character. The onset potential of NaNbO3:Rh,Ba was more negative than that of TiO2:Rh,Sb, whereas the anodic photocurrent of NaNbO3:Rh,Ba was much smaller than that of TiO2:Rh,Sb. These anodic photocurrents overlapped with the cathodic photocurrent of SrTiO3:Rh at a certain electrode potential.
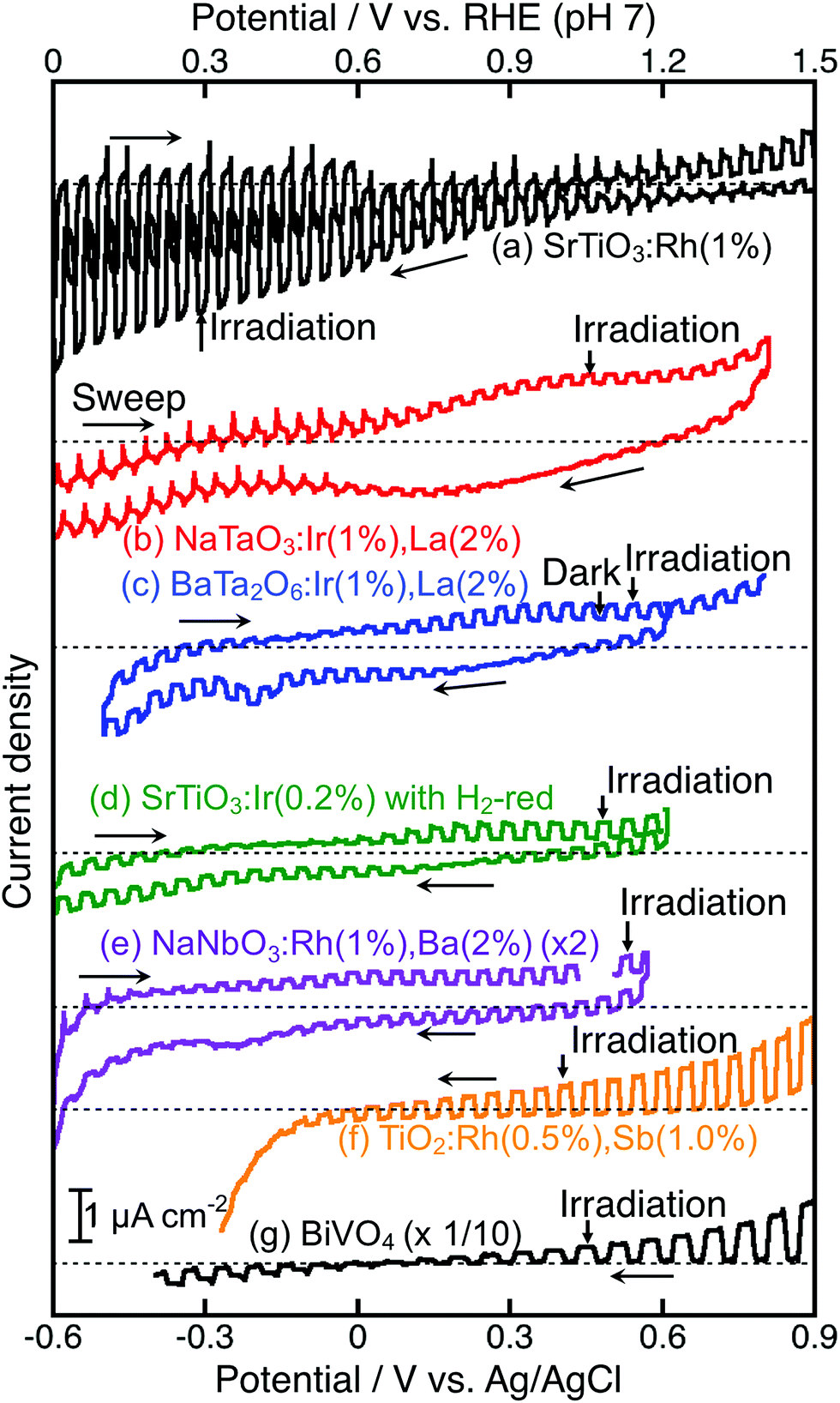 | ||
| Fig. 5 Current vs. potential curves of (a) SrTiO3:Rh(1%), (b) NaTaO3:Ir(1%),La(2%), (c) BaTa2O6:Ir(1%),La(2%), (d) SrTiO3:Ir(0.2%) with H2 reduction, (e) NaNbO3:Rh(1%),Ba(2%), (f) TiO2:Rh(0.5%),Sb(1%), and (g) BiVO4. Electrolyte: 0.1 mol L−1 K2SO4 aqueous solution (pH 7, phosphate buffer was added if necessary), light source: 300 W Xe lamp (λ > 420 nm). | ||
The Z-scheme systems were classified into three groups depending on the combination of H2- and O2-evolving photocatalysts and an electron mediator; being active for only a [Co(bpy)3]3+/2+ redox couple, active for [Co(bpy)3]3+/2+ and Fe3+/2+ redox couple ionic mediators, active not only for [Co(bpy)3]3+/2+ and Fe3+/2+ redox couples but also via interparticle electron transfer with and without RGO. NaTaO3:Ir,La + BiVO4 and BaTa2O6:Ir,La + BiVO4 were active when not Fe3+/2+ but a [Co(bpy)3]3+/2+ redox couple was used. Although the BaTa2O6:Ir,La + BiVO4 showed activities via interparticle electron transfer at a pH of 4.2 with and without RGO, the activities were smaller than that with the ionic electron mediator [Co(bpy)3]3+/2+. SrTiO3:Rh + SrTiO3:Ir was active when the [Co(bpy)3]3+/2+ and Fe3+/2+ redox couple ionic mediators were used, whereas it was not active via interparticle electron transfer with and without RGO at pH 3.5. SrTiO3:Rh + NaNbO3:Rh,Ba and SrTiO3:Rh + TiO2:Rh,Sb were active for the [Co(bpy)3]3+/2+ and Fe3+/2+ redox couples and via interparticle electron transfer without RGO. These Z-scheme systems with RGO using NaNbO3:Rh,Ba and TiO2:Rh,Sb were not examined in detail, because the GOs were not suitably photoreduced on the NaNbO3:Rh,Ba and TiO2:Rh,Sb due to their poor reducing activities, as expected from their poor H2 evolution activities as shown in Table 1. SrTiO3:Rh + TiO2:Rh,Sb showed the best performances among the Z-scheme systems in Table 2.
Discussion
H2 and O2 evolution abilities (shown in Table 1) can be considered based on the band structure, as shown in Fig. 4. Photogenerated holes in the Ir3+ levels in NaTaO3:Ir,La and BaTa2O6:Ir,La have no potentials for water oxidation. In contrast, the Ir3+ level of SrTiO3:Ir possesses the water oxidation potential, though the driving force is not so large. Conduction bands consisting of Ta 5d orbitals in BaTa2O6:Ir,La and NaTaO3:Ir,La possess thermodynamically enough potentials for water reduction to form H2. On the other hand, the photogenerated holes in the Rh3+ levels in NaNbO3:Rh,Ba and TiO2:Rh,Sb have enough potential for water oxidation to form O2. These energy levels were consistent with the H2 and O2 evolution abilities, as shown in Table 1. Of course, other kinetic factors of the active sites would exist for the H2 and O2 evolution abilities in addition to the energy levels of the thermodynamic factor.Let us consider the reason why the Z-scheme systems shown in Table 2 are classified into three groups, based on the band structure and photoelectrochemical properties. Fig. 6 shows energy diagrams for the Z-schemes employing Fe3+/2+ and [Co(bpy)3]3+/2+ redox couples at pH 2.4 and 4.0, respectively. It is assumed that the band levels shift with −0.059 V pH−1 because those materials are metal oxides.
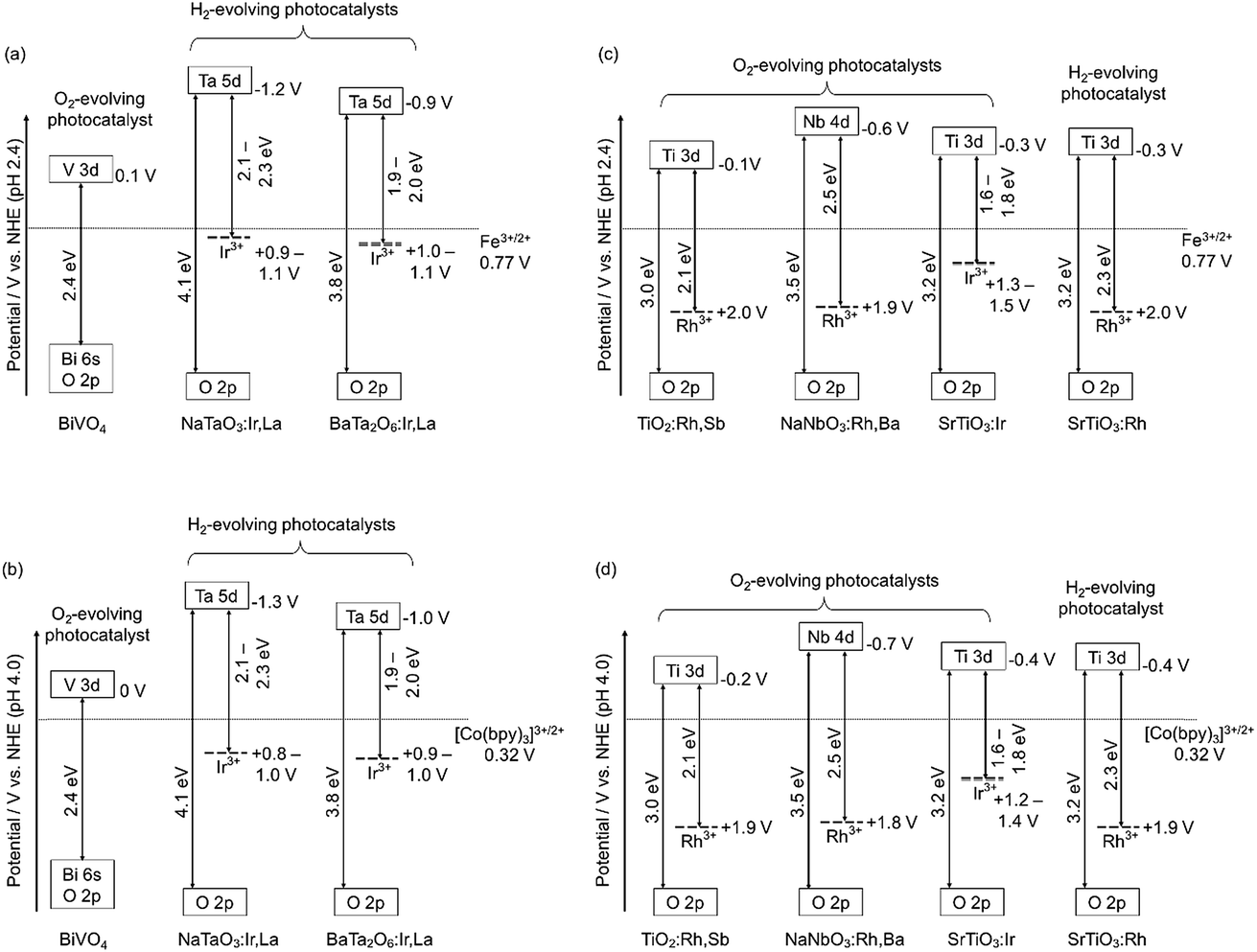 | ||
| Fig. 6 The band structures of Rh- or Ir-doped metal oxide photocatalysts, and redox potentials of Fe and Co-complex ionic electron mediators at pH 2.4 and 4.0. (a) and (b) represent Z-scheme systems of Ir-doped metal oxides as H2-evolving photocatalysts combined with BiVO4. (c) and (d) represent Z-scheme systems of Rh-doped metal oxides and SrTiO3:Ir as O2-evolving photocatalysts combined with SrTiO3:Rh. | ||
NaTaO3:Ir,La + BiVO4 and BaTa2O6:Ir,La + BiVO4 were active with a [Co(bpy)3]3+/2+ redox couple at pH 4.2, but not with Fe3+/2+ at pH 2.4. Photogenerated holes in the Ir3+ levels in NaTaO3:Ir,La and BaTa2O6:Ir,La (an H2-evolving photocatalyst) possess small driving forces for oxidation of Fe2+ (an electron mediator) at pH 2.4 as shown in Fig. 6(a), whereas they possess enough potential for oxidation of [Co(bpy)3]2+ at pH 4.2 as shown in Fig. 6(b). The activities from the interparticle electron transfer were very small or negligible because of poor photoresponse and poor overlaps of the potentials giving cathodic photocurrents of NaTaO3:Ir,La and BaTa2O6:Ir,La and an anodic photocurrent of BiVO4 as shown in Fig. 5, and also may be due to poor contact between NaTaO3:Ir,La or BaTa2O6:Ir,La (an H2-evolving photocatalyst) and BiVO4 (an O2-evolving photocatalyst) in the suspension at pH 4.2. In contrast to them, SrTiO3:Rh + SrTiO3:Ir was active when the [Co(bpy)3]3+/2+ and Fe3+/2+ redox couple ionic mediators were used, because the conduction band of SrTiO3:Ir (an O2-evolving photocatalyst) has enough potential for the reduction of Fe3+ and [Co(bpy)3]3+ (Fig. 6(c) and (d)). However, because the anodic photocurrent of SrTiO3:Ir hardly overlapped with the cathodic photocurrent of SrTiO3:Rh, the activities by interparticle electron transfer with and without RGO were negligible. The conduction bands of TiO2:Rh,Sb and NaNbO3:Rh,Ba (O2-evolving photocatalysts) also possessed enough potential for the reduction of Fe3+ and [Co(bpy)3]3+ as well as SrTiO3:Ir (Fig. 6(c) and (d)). Moreover, in these cases, the anodic photocurrents of the TiO2:Rh,Sb and NaNbO3:Rh,Ba photoelectrodes overlapped enough with the cathodic photocurrent of SrTiO3:Rh. So, it is reasonable that SrTiO3:Rh + NaNbO3:Rh,Ba and SrTiO3:Rh + TiO2:Rh,Sb were active not only for the [Co(bpy)3]3+/2+ and Fe3+/2+ redox couples but also via interparticle electron transfer with and without RGO. The activity of SrTiO3:Rh + TiO2:Rh,Sb was higher than that of SrTiO3:Rh + NaNbO3:Rh,Ba for all of the types of Z-scheme system. In the cases with the use of ionic electron mediators, it is probably due to the higher activity for O2 evolution and the narrower energy gaps of TiO2:Rh,Sb than those of NaNbO3:Rh,Ba, as shown in Table 1. The reason why SrTiO3:Rh + TiO2:Rh,Sb showed a higher activity than SrTiO3:Rh + NaNbO3:Rh,Ba via interparticle electron transfer is that TiO2:Rh,Sb gave much larger anodic photocurrents than NaNbO3:Rh,Ba, as shown in Fig. 5.
Conclusions
We have successfully developed Z-scheme photocatalyst systems for water splitting under visible light irradiation employing Rh- and Ir-doped metal oxide photocatalysts with longer wavelength responses than conventional SrTiO3:Rh and BiVO4. The impurity levels of doped Ir3+ and Rh3+ that contributed to the visible light responses for the photocatalytic reactions were determined from diffuse reflectance spectra and supposing +3.0 V vs. NHE of a valence band of the oxide photocatalyst materials. The impurity levels of Ir3+ in NaTaO3:Ir,La and BaTa2O6:Ir,La have sufficient potentials for the oxidation of the electron mediator [Co(bpy)3]2+, but insufficient potentials for water oxidation to form O2 and oxidation of the electron mediator Fe2+. The conduction bands of the NaTaO3:Ir,La and BaTa2O6:Ir,La have enough potential for water reduction to form H2. Therefore, the NaTaO3:Ir,La and BaTa2O6:Ir,La could be used as H2-evolving photocatalysts only when a [Co(bpy)3]3+/2+ redox couple was used. The impurity levels of Ir3+ in SrTiO3:Ir and Rh3+ in TiO2:Rh,Sb and NaNbO3:Rh,Ba have potentials for water oxidation and their conduction bands possess the potentials for reduction of the electron mediators Fe3+ and [Co(bpy)3]3+. This property means that SrTiO3:Ir, TiO2:Rh,Sb and NaNbO3:Rh,Ba could be employed as O2-evolving photocatalysts for the construction of Z-scheme systems employing ionic electron mediators. Moreover, photoelectrochemical measurements using photocatalyst powders immobilized on conducting substrate revealed that the n-type characters and relatively negative onset potentials of the TiO2:Rh,Sb and NaNbO3:Rh,Ba photoanodes enabled the Z-scheme systems to work by interparticle electron transfer. Although the efficiencies of the present Z-scheme systems are low at the present stage, these will be improved by interfacial controls from a kinetic point of view in basic research. These results and discussion will contribute to the design of a highly active photocatalyst system for water splitting into H2 and O2, aiming for the demonstration of actual solar water splitting using a suitable reactor.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 17H06440 and 17H06433 in Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC)”.References
- A. Kudo, H. Kato and I. Tsuji, Chem. Lett., 2004, 33, 1534 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35 CrossRef CAS.
- Y. Inoue, Energy Environ. Sci., 2009, 2, 364 RSC.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- K. Maeda and K. Domen, Bull. Chem. Soc. Jpn., 2016, 89, 627 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983 RSC.
- D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterloh and S. Ardo, Energy Environ. Sci., 2015, 8, 2825 RSC.
- T. Yamada and K. Domen, Chem. Eng., 2018, 2, 36 Search PubMed.
- A. Kudo, in “Photocatalysis”, Contemporary Catalysis; Science, Technology and Applications, ed. P. Kamer, D. Vogt and J. Thybaut, Royal Society of Chemistry, Cambridge, 2017, ch. 3.7, pp. 326–343 Search PubMed.
- A. J. Bard, J. Photochem., 1979, 10, 59 CrossRef CAS.
- A. Kudo, MRS Bull., 2011, 36, 32 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201 CrossRef CAS PubMed.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348 CrossRef CAS.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Y. Suzuki, Z. Pan, J. Seo, M. Katayama, T. Minegishi, H. Nishiyama, T. Takata, K. Seki, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2017, 139, 1675 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, M. Katayama, T. Takata, T. Minegishi, A. Kudo, T. Yamada and K. Domen, Faraday Discuss., 2017, 197, 491 RSC.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992 CrossRef CAS.
- R. Niishiro, S. Tanaka and A. Kudo, Appl. Catal., B, 2014, 150, 187 CrossRef.
- S. Suzuki, H. Matsumoto, A. Iwase and A. Kudo, Chem. Commun., 2018, 54, 10606 RSC.
- A. Iwase and A. Kudo, Chem. Commun., 2017, 53, 6156 RSC.
- A. Iwase, K. Saito and A. Kudo, Bull. Chem. Soc. Jpn., 2009, 82, 514 CrossRef CAS.
- R. Niishiro, R. Konta, H. Kato, W. J. Chun, K. Asakura and A. Kudo, J. Phys. Chem. C, 2007, 111, 17420 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459 CrossRef CAS.
- A. Iwase, H. Kato and A. Kudo, J. Sol. Energy Eng., 2010, 132, 21106 CrossRef.
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, J. Catal., 2008, 259, 133 CrossRef CAS.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441 CrossRef CAS PubMed.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054 CrossRef CAS.
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604 CrossRef CAS PubMed.
- A. Iwase, S. Yoshino, T. Takayama, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2016, 138, 10260 CrossRef CAS PubMed.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536 CrossRef CAS.
- Q. Jia, A. Iwase and A. Kudo, Chem. Sci., 2014, 5, 1513 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- K. Iwashina and A. Kudo, J. Am. Chem. Soc., 2011, 133, 13272 CrossRef CAS PubMed.
- S. Kawasaki, K. Akagi, K. Nakatsuji, S. Yamamoto, I. Matsuda, Y. Harada, J. Yoshinobu, F. Komori, R. Takahashi, M. Lippmaa, C. Sakai, H. Niwa, M. Oshima, K. Iwashina and A. Kudo, J. Phys. Chem. C, 2012, 116, 24445 CrossRef CAS.
- S. Kawasaki, R. Takahashi, K. Akagi, J. Yoshinobu, F. Komori, K. Horiba, H. Kumigashira, K. Iwashina, A. Kudo and M. Lippmaa, J. Phys. Chem. C, 2014, 118, 20222 CrossRef CAS.
- D. E. Scaife, Sol. Energy, 1980, 25, 41 CrossRef CAS.
- S. Kawasaki, K. Nakatsuji, J. Yoshinobu, F. Komori, R. Takahashi, M. Lippmaa, K. Mase and A. Kudo, Appl. Phys. Lett., 2012, 101, 033910 CrossRef.
- K. Sayama, A. Nomura, Z. Zou, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2003, 2908 RSC.
- K. Sayama, A. Nomura, T. Arai, T. Sugita, R. Abe, M. Yanagida, T. Oi, Y. Iwasaki, Y. Abe and H. Sugihara, J. Phys. Chem. B, 2006, 110, 2908 CrossRef PubMed; F. F. Adbi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 CrossRef PubMed.
- Y. Park, K. J. McDonald and K.-S. Choi, Chem. Soc. Rev., 2013, 42, 2321 RSC.
- Q. Jia, K. Iwashina and A. Kudo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11564 CrossRef CAS PubMed.
- M. Yamaguchi, R. Niishiro, Q. Jia, Y. Kuang, K. Kitamura, A. Iwase, T. Minegishi, T. Yamada, K. Domen and A. Kudo, to be submitted.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8fd00209f |
| This journal is © The Royal Society of Chemistry 2019 |