
Treatment of per- and polyfluoroalkyl substances in landfill leachate: status, chemistry and prospects
 †ab
Dongye
Zhao
*a
†ab
Dongye
Zhao
*a
Abstract
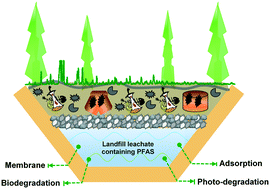
Per- and polyfluoroalkyl substances (PFAS) have been widely detected in municipal landfills due to the massive uses of PFAS in everyday consumer products. PFAS in landfill leachate can contaminate the neighbouring soil and groundwater and pose serious health concerns to human and ecosystems. Yet, information is lacking on the distribution, transformation and fate of various PFAS in landfills, and on the treatment technologies to remove or degrade PFAS in landfill leachate. As the relevant regulations are rapidly evolving, the research and technology development have been gaining momentum in recent years. Here, we present a comprehensive review of the state of the science on the occurrence and treatment technologies of PFAS in landfills. Specifically, this review aims to overview the following aspects: 1) the occurrence and transformation of PFAS under typical landfill environmental conditions, 2) the chemistry and state-of-art technologies for treating PFAS in landfill leachate, including adsorption, biodegradation, photo-degradation, and membrane processes, and 3) the key knowledge gaps and future research/technology needs for controlling PFAS from landfills.
- This article is part of the themed collections: PFAS and Best Papers 2019 – Environmental Science: Water Research & Technology
 Please wait while we load your content...
Please wait while we load your content...

